Current Challenges in Understanding the Cellular and Molecular Mechanisms in Niemann–Pick Disease Type C1

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
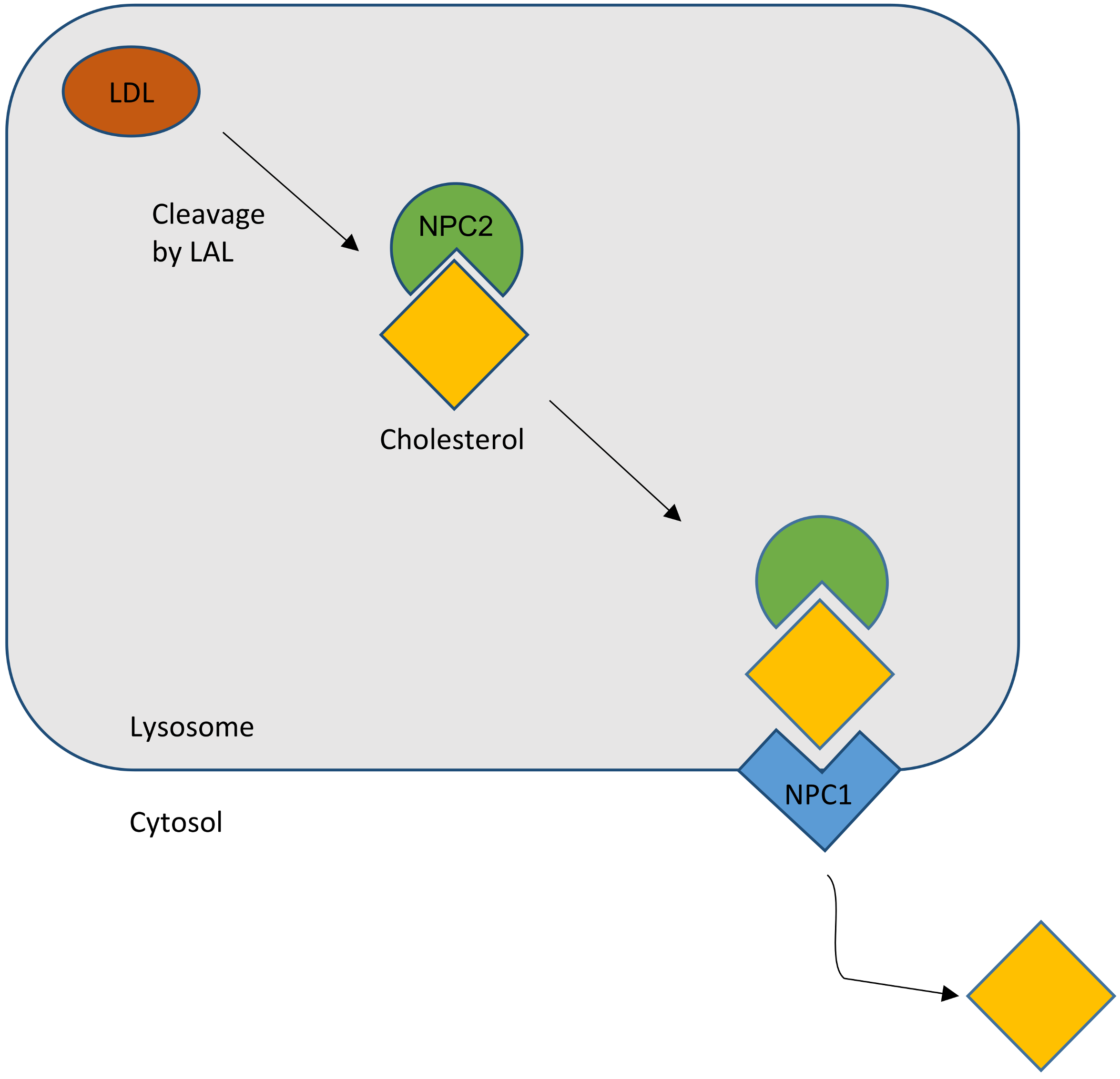
2. Lipid Trafficking and NPC1 (Niemann–Pick Disease Type C1)
2.1. Cholesterol Transport
2.2. NPC Protein Function
2.2.1. NPC1
2.2.2. NPC2
2.3. Diagnostic Tools
2.4. Therapies
3. Pathology of NPC1 in Humans and Mice
3.1. Behavior
3.2. Sensory Systems
3.2.1. Hearing
3.2.2. Vision
3.2.3. Olfaction
3.2.4. Peripheral Nervous System
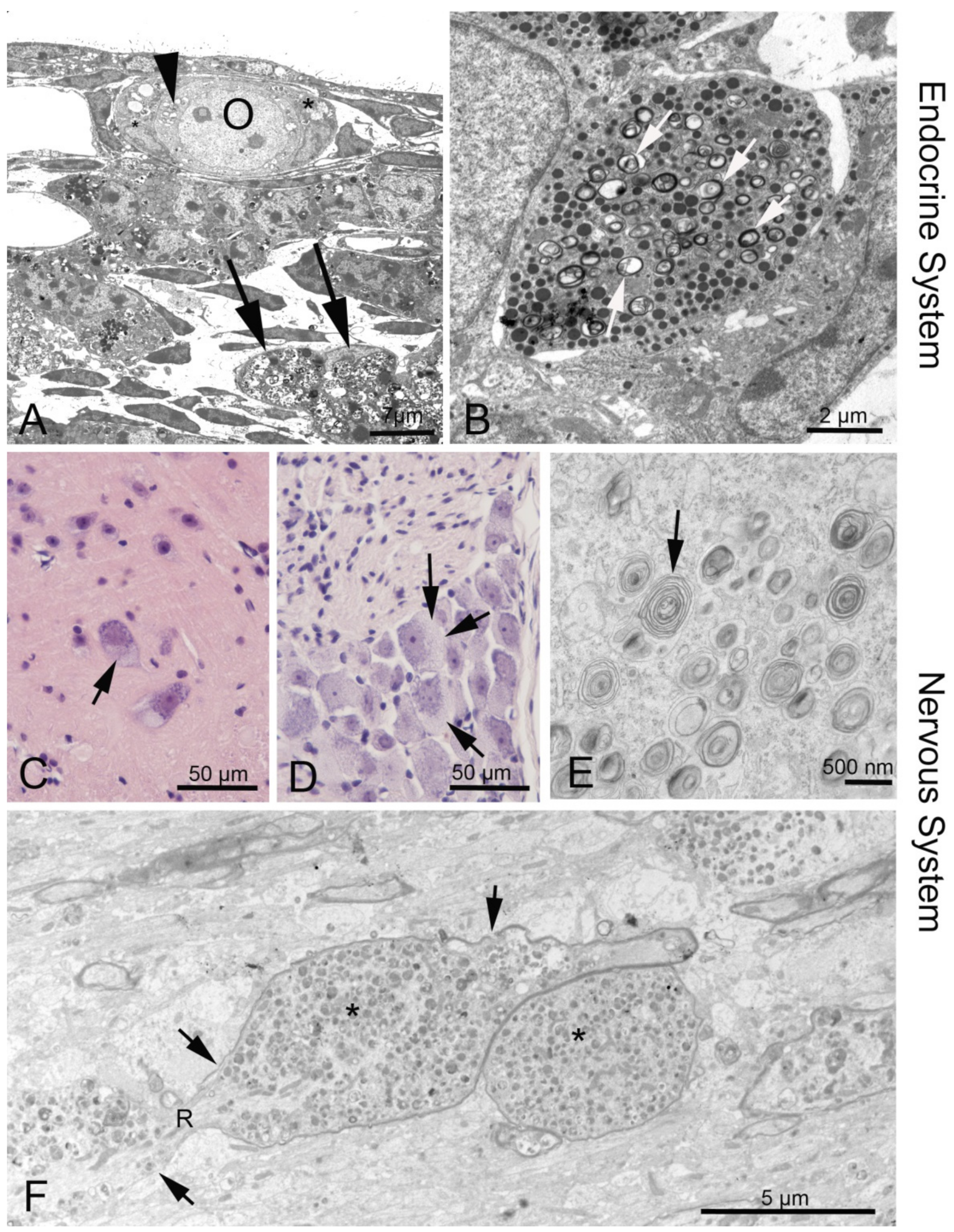
3.3. Endocrine Organs and Reproductive System
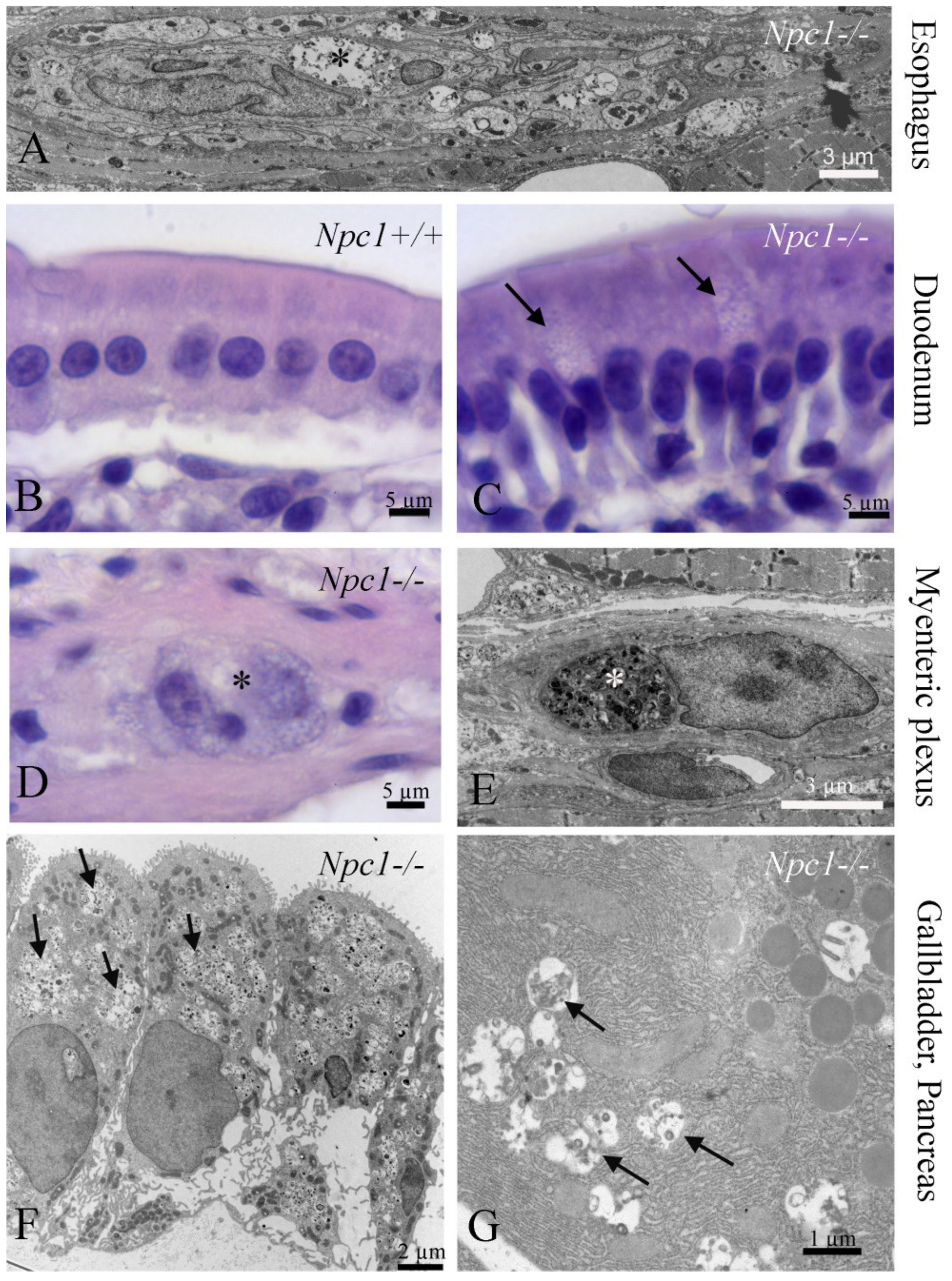
3.4. Gastrointestinal Tract (GI)
3.5. Pancreas
3.6. Lung
3.7. Kidney
3.8. Liver and Biliary Tract
3.9. Spleen and Lymphatic System
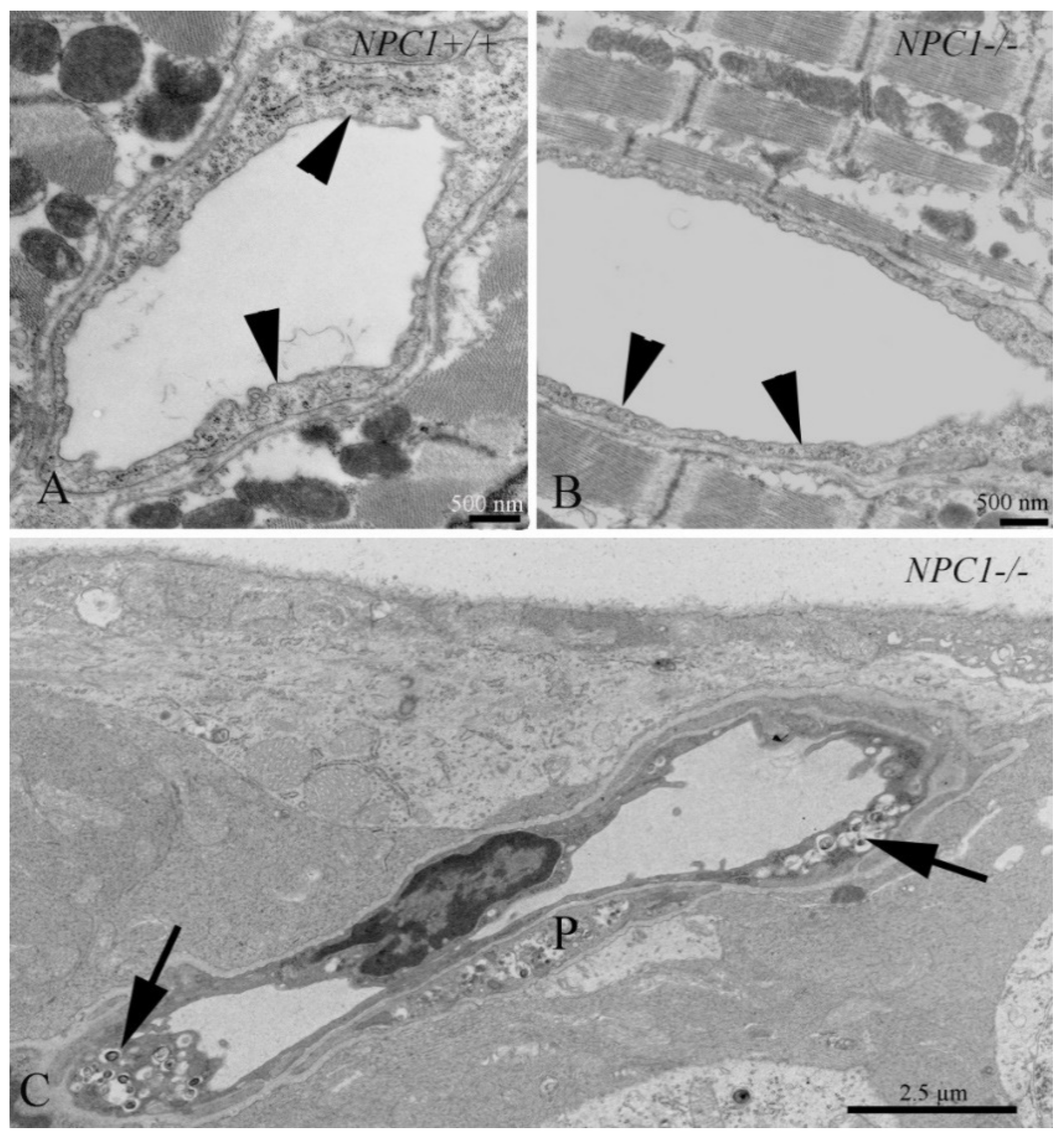
3.10. Cardiovascular System
3.11. Tooth
4. Perspectives
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAV | adeno-associated virus |
| ABC | ATP binding cassette |
| ACMKO | acid sphingomyelinase knock-out |
| acox1 | acyl-coenzyme A oxidase 1 |
| apoE | apolipoprotein E |
| ASMKO | acid sphingomyelinase knock-out |
| CNS | central nervous system |
| ER | endoplasmic reticulum |
| FACS | fluorescence activated cell sorting |
| fatp2 | fatty acid transport protein 2 |
| GI | gastrointestinal |
| HDACi | histone deacetylase inhibitors |
| HPβCD | 2-hydroxypropyl-β-cyclodextrin |
| LDL | low-density lipoprotein |
| LE/LY | late endosome/lysosome |
| lxr | liver X receptor |
| NPC1 | Niemann–Pick disease type C1 |
| PPAR | peroxisome proliferator-activated receptor |
| SphK2 | sphingosine kinase 2 |
| SREBP | sterol regulatory element binding protein |
References
- Naureckiene, S.; Sleat, D.E.; Lackland, H.; Fensom, A.; Vanier, M.T.; Wattiaux, R.; Jadot, M.; Lobel, P. Identification of HE1 as the second gene of Niemann-Pick C disease. Science 2000, 290, 2298–2301. [Google Scholar] [CrossRef]
- Garver, W.S.; Francis, G.A.; Jelinek, D.; Shepherd, G.; Flynn, J.; Castro, G.; Walsh Vockley, C.; Coppock, D.L.; Pettit, K.M.; Heidenreich, R.A.; et al. The national Niemann-Pick C1 disease database: report of clinical features and health problems. Am. J. Med. Genet. A 2007, 143, 1204–1211. [Google Scholar] [CrossRef]
- Garver, W.S.; Jelinek, D.; Oyarzo, J.N.; Flynn, J.; Zuckerman, M.; Krishnan, K.; Chung, B.H.; Heidenreich, R.A. Characterization of liver disease and lipid metabolism in the Niemann-Pick C1 mouse. J. Cell. Biochem. 2007, 101, 498–516. [Google Scholar] [CrossRef]
- Garver, W.S.; Jelinek, D.; Meaney, F.J.; Flynn, J.; Pettit, K.M.; Shepherd, G.; Heidenreich, R.A.; Vockley, C.M.; Castro, G.; Francis, G.A. The national Niemann-Pick Type C1 disease database: correlation of lipid profiles, mutations, and biochemical phenotypes. J. Lipid Res. 2010, 51, 406–415. [Google Scholar] [CrossRef]
- Vanier, M.T.; Millat, G. Niemann-Pick disease type C. Clin. Genet. 2003, 64, 269–281. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, R.; Raas-Rothschild, A.; Reish, O.; Regev, M.; Meiner, V.; Bargal, R.; Sury, V.; Meir, K.; Nadjari, M.; Hermann, G.; et al. The clinical spectrum of fetal Niemann-Pick type C. Am. J. Med. Genet. A 2009, 149, 446–450. [Google Scholar] [CrossRef]
- Yu, X.H.; Jiang, N.; Yao, P.B.; Zheng, X.L.; Cayabyab, F.S.; Tang, C.K. NPC1, intracellular cholesterol trafficking and atherosclerosis. Clin. Chim. Acta. 2014, 429, 69–75. [Google Scholar] [CrossRef]
- Chu, B.B.; Liao, Y.C.; Qi, W.; Xie, C.; Du, X.; Wang, J.; Yang, H.; Miao, H.H.; Li, B.L.; Song, B.L. Cholesterol transport through lysosome-peroxisome membrane contacts. Cell 2015, 161, 291–306. [Google Scholar] [CrossRef]
- Carstea, E.D.; Morris, J.A.; Coleman, K.G.; Loftus, S.K.; Zhang, D.; Cummings, C.; Gu, J.; Rosenfeld, M.A.; Pavan, W.J.; Krizman, D.B.; et al. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science 1997, 277, 228–231. [Google Scholar] [CrossRef]
- Elleder, M.; Jirasek, A.; Smid, F.; Ledvinova, J.; Besley, G.T. Niemann-Pick disease type C. study on the nature of the cerebral storage process. Acta. Neuropathol. 1985, 66, 325–336. [Google Scholar] [CrossRef]
- Tanaka, J.; Nakamura, H.; Miyawaki, S. Cerebellar involvement in murine sphingomyelinosis: a new model of Niemann-Pick disease. J. Neuropathol. Exp. Neurol. 1988, 47, 291–300. [Google Scholar] [CrossRef]
- Sarna, J.R.; Larouche, M.; Marzban, H.; Sillitoe, R.V.; Rancourt, D.E.; Hawkes, R. Patterned purkinje cell degeneration in mouse models of Niemann-Pick type C disease. J. Comp. Neurol. 2003, 456, 279–291. [Google Scholar] [CrossRef]
- Maass, F.; Petersen, J.; Hovakimyan, M.; Schmitt, O.; Witt, M.; Hawlitschka, A.; Lukas, J.; Rolfs, A.; Wree, A. Reduced cerebellar neurodegeneration after combined therapy with cyclodextrin/allopregnanolone and miglustat in NPC1: A mouse model of Niemann-Pick type C1 disease. J. Neurosci. Res. 2015, 93, 433–442. [Google Scholar] [CrossRef]
- Liscum, L. A role for NPC1 and NPC2 in intestinal cholesterol absorption--the hypothesis gutted. Biochem. J. 2007, 408, e1–e3. [Google Scholar] [CrossRef]
- Wassif, C.A.; Cross, J.L.; Iben, J.; Sanchez-Pulido, L.; Cougnoux, A.; Platt, F.M.; Ory, D.S.; Ponting, C.P.; Bailey-Wilson, J.E.; Biesecker, L.G.; et al. High incidence of unrecognized visceral/neurological late-onset Niemann-Pick disease, type C1, predicted by analysis of massively parallel sequencing data sets. Genet. Med. 2016, 18, 41–48. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. A receptor-mediated pathway for cholesterol homeostasis. Science 1986, 232, 34–47. [Google Scholar] [CrossRef]
- Liu, B.; Xie, C.; Richardson, J.A.; Turley, S.D.; Dietschy, J.M. Receptor-mediated and bulk-phase endocytosis cause macrophage and cholesterol accumulation in Niemann-Pick C disease. J. Lipid Res. 2007, 48, 1710–1723. [Google Scholar] [CrossRef] [Green Version]
- Maxfield, F.R.; Menon, A.K. Intracellular sterol transport and distribution. Curr. Opin. Cell. Biol. 2006, 18, 379–385. [Google Scholar] [CrossRef]
- Chang, T.Y.; Chang, C.C.; Ohgami, N.; Yamauchi, Y. Cholesterol sensing, trafficking, and esterification. Annu. Rev. Cell. Dev. Biol. 2006, 22, 129–157. [Google Scholar] [CrossRef]
- Vance, J.E. Lipid imbalance in the neurological disorder, Niemann-Pick C disease. FEBS Lett. 2006, 580, 5518–5524. [Google Scholar] [CrossRef] [Green Version]
- Yu, L. The structure and function of Niemann-Pick C1-like 1 protein. Curr. Opin. Lipidol. 2008, 19, 263–269. [Google Scholar] [CrossRef]
- Scott, C.; Ioannou, Y.A. The NPC1 protein: structure implies function. Biochim. Biophys. Acta. 2004, 1685, 8–13. [Google Scholar] [CrossRef]
- Liscum, L.; Sturley, S.L. Intracellular trafficking of Niemann-Pick C proteins 1 and 2: obligate components of subcellular lipid transport. Biochim. Biophys. Acta. 2004, 1685, 22–27. [Google Scholar] [CrossRef]
- Xu, S.; Benoff, B.; Liou, H.L.; Lobel, P.; Stock, A.M. Structural basis of sterol binding by NPC2, a lysosomal protein deficient in Niemann-Pick type C2 disease. J. Biol. Chem. 2007, 282, 23525–23531. [Google Scholar] [CrossRef]
- Cheruku, S.R.; Xu, Z.; Dutia, R.; Lobel, P.; Storch, J. Mechanism of cholesterol transfer from the Niemann-Pick type C2 protein to model membranes supports a role in lysosomal cholesterol transport. J. Biol. Chem. 2006, 281, 31594–31604. [Google Scholar] [CrossRef]
- Peake, K.B.; Vance, J.E. Defective cholesterol trafficking in Niemann-Pick C-deficient cells. FEBS Lett. 2010, 584, 2731–2739. [Google Scholar] [CrossRef] [Green Version]
- Reddy, J.V.; Ganley, I.G.; Pfeffer, S.R. Clues to neuro-degeneration in Niemann-Pick type C disease from global gene expression profiling. PLoS ONE 2006, 1, e19. [Google Scholar] [CrossRef]
- Abe, K.; Sakai, N. Patient with Niemann-Pick disease type C: over 20 years’ follow-up. BMJ Case Rep. 2017, 2017. [Google Scholar] [CrossRef]
- Kelly, D.A.; Portmann, B.; Mowat, A.P.; Sherlock, S.; Lake, B.D. Niemann-Pick disease type C: diagnosis and outcome in children, with particular reference to liver disease. J. Pediatr. 1993, 123, 242–247. [Google Scholar] [CrossRef]
- Gumus, E.; Haliloglu, G.; Karhan, A.N.; Demir, H.; Gurakan, F.; Topcu, M.; Yuce, A. Niemann-Pick disease type C in the newborn period: a single-center experience. Eur. J. Pediatr. 2017, 176, 1669–1676. [Google Scholar] [CrossRef]
- Rodrigues, A.F.; Gray, R.G.; Preece, M.A.; Brown, R.; Hill, F.G.; Baumann, U.; McKiernan, P.J. The usefulness of bone marrow aspiration in the diagnosis of Niemann-Pick disease type C in infantile liver disease. Arch. Dis. Child. 2006, 91, 841–844. [Google Scholar] [CrossRef] [Green Version]
- Porter, F.D.; Scherrer, D.E.; Lanier, M.H.; Langmade, S.J.; Molugu, V.; Gale, S.E.; Olzeski, D.; Sidhu, R.; Dietzen, D.J.; Fu, R.; et al. Cholesterol oxidation products are sensitive and specific blood-based biomarkers for Niemann-Pick C1 disease. Sci. Transl. Med. 2010, 2, 56ra81. [Google Scholar] [CrossRef]
- McKay Bounford, K.; Gissen, P. Genetic and laboratory diagnostic approach in Niemann Pick disease type C. J. Neurol. 2014, 261, S569–S575. [Google Scholar] [CrossRef]
- Jiang, X.; Ory, D.S. Towards a new diagnostic standard for Niemann-Pick C disease. EBioMedicine 2016, 4, 18–19. [Google Scholar] [CrossRef]
- Vanier, M.T.; Gissen, P.; Bauer, P.; Coll, M.J.; Burlina, A.; Hendriksz, C.J.; Latour, P.; Goizet, C.; Welford, R.W.; Marquardt, T.; et al. Diagnostic tests for Niemann-Pick disease type C (NP-C): A critical review. Mol. Genet. Metab. 2016, 118, 244–254. [Google Scholar] [CrossRef]
- Meyer, A.; Gläser, A.; Bräuer, A.U.; Wree, A.; Strotmann, J.; Rolfs, A.; Witt, M. Olfactory performance as an indicator for protective treatment effects in an animal model of neurodegeneration. Front. Integr. Neurosci. 2018, 12, 35. [Google Scholar] [CrossRef]
- Patterson, M.C.; Vecchio, D.; Prady, H.; Abel, L.; Wraith, J.E. Miglustat for treatment of Niemann-Pick C disease: a randomised controlled study. Lancet Neurol. 2007, 6, 765–772. [Google Scholar] [CrossRef]
- Platt, F.M.; Jeyakumar, M. Substrate reduction therapy. Acta. Paediatr. Suppl. 2008, 97, 88–93. [Google Scholar] [CrossRef]
- Ribas, G.S.; Pires, R.; Coelho, J.C.; Rodrigues, D.; Mescka, C.P.; Vanzin, C.S.; Biancini, G.B.; Negretto, G.; Wayhs, C.A.; Wajner, M.; et al. Oxidative stress in Niemann-Pick type C patients: a protective role of N-butyl-deoxynojirimycin therapy. Int. J. Dev. Neurosci. 2012, 30, 439–444. [Google Scholar] [CrossRef]
- Wraith, J.E.; Vecchio, D.; Jacklin, E.; Abel, L.; Chadha-Boreham, H.; Luzy, C.; Giorgino, R.; Patterson, M.C. Miglustat in adult and juvenile patients with Niemann-Pick disease type C: long-term data from a clinical trial. Mol. Genet. Metab. 2010, 99, 351–357. [Google Scholar] [CrossRef]
- Erickson, R.P.; Fiorenza, M.T. A hopeful therapy for Niemann-Pick C diseases. Lancet 2017, 390, 1720–1721. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, M.; Togawa, M.; Hirabaru, K.; Mochinaga, S.; Narita, A.; Adachi, M.; Egashira, M.; Irie, T.; Ohno, K. Effects of cyclodextrin in two patients with Niemann-Pick Type C disease. Mol. Genet. Metab. 2013, 108, 76–81. [Google Scholar] [CrossRef]
- Liu, B.; Ramirez, C.M.; Miller, A.M.; Repa, J.J.; Turley, S.D.; Dietschy, J.M. Cyclodextrin overcomes the transport defect in nearly every organ of NPC1 mice leading to excretion of sequestered cholesterol as bile acid. J. Lipid Res. 2010, 51, 933–944. [Google Scholar] [CrossRef] [Green Version]
- Ebner, L.; Glaser, A.; Brauer, A.; Witt, M.; Wree, A.; Rolfs, A.; Frank, M.; Vollmar, B.; Kuhla, A. Evaluation of two liver treatment strategies in a mouse model of Niemann-Pick-Disease Type C1. Int. J. Mol. Sci. 2018, 19, 972. [Google Scholar] [CrossRef]
- Crumling, M.A.; Liu, L.; Thomas, P.V.; Benson, J.; Kanicki, A.; Kabara, L.; Halsey, K.; Dolan, D.; Duncan, R.K. Hearing loss and hair cell death in mice given the cholesterol-chelating agent hydroxypropyl-beta-cyclodextrin. PLoS ONE 2012, 7, e53280. [Google Scholar] [CrossRef]
- Vite, C.H.; Bagel, J.H.; Swain, G.P.; Prociuk, M.; Sikora, T.U.; Stein, V.M.; O’Donnell, P.; Ruane, T.; Ward, S.; Crooks, A.; et al. Intracisternal cyclodextrin prevents cerebellar dysfunction and Purkinje cell death in feline Niemann-Pick type C1 disease. Sci. Transl. Med. 2015, 7, 276ra26. [Google Scholar] [CrossRef]
- Ory, D.S.; Ottinger, E.A.; Farhat, N.Y.; King, K.A.; Jiang, X.; Weissfeld, L.; Berry-Kravis, E.; Davidson, C.D.; Bianconi, S.; Keener, L.A.; et al. Intrathecal 2-hydroxypropyl-beta-cyclodextrin decreases neurological disease progression in Niemann-Pick disease, type C1: a non-randomised, open-label, phase 1–2 trial. Lancet 2017, 390, 1758–1768. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Study of VTS-270 (2-hydroxypropyl-β-cyclodextrin) to Treat Niemann-Pick Type C1 (NPC1) Disease. Available online: https://clinicaltrials.gov/ct2/show/study/NCT02534844 (accessed on 4 September 2019).
- Wadman, M. Update: Twins who were face of controversial rare disease treatment have died. Science 2019. [Google Scholar] [CrossRef]
- Puskas, I. HPBCD Treatment for Niemann-Pick C: Mallinckrodt Communicated Results of Clinical Trial. Available online: https://cyclodextrinnews.com/2018/11/13/hpbcd-treatment-for-niemann-pick-type-c-performed-no-differently-than-placebo/ (accessed on 4 September 2019).
- Davidson, C.D.; Ali, N.F.; Micsenyi, M.C.; Stephney, G.; Renault, S.; Dobrenis, K.; Ory, D.S.; Vanier, M.T.; Walkley, S.U. Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS ONE 2009, 4, e6951. [Google Scholar] [CrossRef]
- Davidson, C.D.; Fishman, Y.I.; Puskas, I.; Szeman, J.; Sohajda, T.; McCauliff, L.A.; Sikora, J.; Storch, J.; Vanier, M.T.; Szente, L.; et al. Efficacy and ototoxicity of different cyclodextrins in Niemann-Pick C disease. Ann. Clin. Transl. Neurol. 2016, 3, 366–380. [Google Scholar] [CrossRef]
- Hovakimyan, M.; Maass, F.; Petersen, J.; Holzmann, C.; Witt, M.; Lukas, J.; Frech, M.J.; Hubner, R.; Rolfs, A.; Wree, A. Combined therapy with cyclodextrin/allopregnanolone and miglustat improves motor but not cognitive functions in Niemann-Pick Type C1 mice. Neuroscience 2013, 252, 201–211. [Google Scholar] [CrossRef]
- Repa, J.J.; Li, H.; Frank-Cannon, T.C.; Valasek, M.A.; Turley, S.D.; Tansey, M.G.; Dietschy, J.M. Liver X receptor activation enhances cholesterol loss from the brain, decreases neuroinflammation, and increases survival of the NPC1 mouse. J. Neurosci. 2007, 27, 14470–14480. [Google Scholar] [CrossRef]
- Maceyka, M.; Milstien, S.; Spiegel, S. The potential of histone deacetylase inhibitors in Niemann—Pick type C disease. FEBS J. 2013, 280, 6367–6372. [Google Scholar] [CrossRef]
- Munkacsi, A.B.; Chen, F.W.; Brinkman, M.A.; Higaki, K.; Gutierrez, G.D.; Chaudhari, J.; Layer, J.V.; Tong, A.; Bard, M.; Boone, C.; et al. An “exacerbate-reverse” strategy in yeast identifies histone deacetylase inhibition as a correction for cholesterol and sphingolipid transport defects in human Niemann-Pick type C disease. J. Biol. Chem. 2011, 286, 23842–23851. [Google Scholar] [CrossRef]
- Pipalia, N.H.; Cosner, C.C.; Huang, A.; Chatterjee, A.; Bourbon, P.; Farley, N.; Helquist, P.; Wiest, O.; Maxfield, F.R. Histone deacetylase inhibitor treatment dramatically reduces cholesterol accumulation in Niemann-Pick type C1 mutant human fibroblasts. Proc. Natl. Acad. Sci. USA 2011, 108, 5620–5625. [Google Scholar] [CrossRef] [Green Version]
- Helquist, P.; Maxfield, F.R.; Wiech, N.L.; Wiest, O. Treatment of Niemann--pick type C disease by histone deacetylase inhibitors. Neurotherapeutics 2013, 10, 688–697. [Google Scholar] [CrossRef]
- De Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef]
- Newton, J.; Hait, N.C.; Maceyka, M.; Colaco, A.; Maczis, M.; Wassif, C.A.; Cougnoux, A.; Porter, F.D.; Milstien, S.; Platt, N.; et al. FTY720/fingolimod increases NPC1 and NPC2 expression and reduces cholesterol and sphingolipid accumulation in Niemann-Pick type C mutant fibroblasts. FASEB J. 2017, 31, 1719–1730. [Google Scholar] [CrossRef]
- Hait, N.C.; Avni, D.; Yamada, A.; Nagahashi, M.; Aoyagi, T.; Aoki, H.; Dumur, C.I.; Zelenko, Z.; Gallagher, E.J.; Leroith, D.; et al. The phosphorylated prodrug FTY720 is a histone deacetylase inhibitor that reactivates ERalpha expression and enhances hormonal therapy for breast cancer. Oncogenesis 2015, 4, e156. [Google Scholar] [CrossRef]
- Kirkegaard, T.; Roth, A.G.; Petersen, N.H.; Mahalka, A.K.; Olsen, O.D.; Moilanen, I.; Zylicz, A.; Knudsen, J.; Sandhoff, K.; Arenz, C.; et al. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature 2010, 463, 549–553. [Google Scholar] [CrossRef]
- Fog, C.K.; Kirkegaard, T. Animal models for Niemann-Pick type C: implications for drug discovery & development. Expert Opin. Drug Discov. 2019, 14, 499–509. [Google Scholar] [Green Version]
- Evans, W.R.H.; Nicoli, E.R.; Wang, R.Y.; Movsesyan, N.; Platt, F.M. Case report: Ursodeoxycholic acid treatment in Niemann-Pick disease type C; clinical experience in four cases. Wellcome Open Res. 2017, 2, 75. [Google Scholar] [CrossRef]
- Nicoli, E.R.; Al Eisa, N.; Cluzeau, C.V.; Wassif, C.A.; Gray, J.; Burkert, K.R.; Smith, D.A.; Morris, L.; Cologna, S.M.; Peer, C.J.; et al. Defective cytochrome P450-catalysed drug metabolism in Niemann-Pick type C disease. PLoS ONE 2016, 11, e0152007. [Google Scholar] [CrossRef]
- Bremova, T.; Malinova, V.; Amraoui, Y.; Mengel, E.; Reinke, J.; Kolnikova, M.; Strupp, M. Acetyl-dl-leucine in Niemann-Pick type C: A case series. Neurology 2015, 85, 1368–1375. [Google Scholar] [CrossRef]
- Xie, C.; Gong, X.M.; Luo, J.; Li, B.L.; Song, B.L. AAV9-NPC1 significantly ameliorates Purkinje cell death and behavioral abnormalities in mouse NPC disease. J. Lipid Res. 2017, 58, 512–518. [Google Scholar] [CrossRef] [Green Version]
- Hughes, M.P.; Smith, D.A.; Morris, L.; Fletcher, C.; Colaco, A.; Huebecker, M.; Tordo, J.; Palomar, N.; Massaro, G.; Henckaerts, E.; et al. AAV9 intracerebroventricular gene therapy improves lifespan, locomotor function and pathology in a mouse model of Niemann-Pick type C1 disease. Hum. Mol. Genet. 2018, 27, 3079–3098. [Google Scholar] [CrossRef]
- Chandler, R.J.; Williams, I.M.; Gibson, A.L.; Davidson, C.D.; Incao, A.A.; Hubbard, B.T.; Porter, F.D.; Pavan, W.J.; Venditti, C.P. Systemic AAV9 gene therapy improves the lifespan of mice with Niemann-Pick disease, type C1. Hum. Mol. Genet. 2017, 26, 52–64. [Google Scholar] [CrossRef]
- Schlegel, V.; Thieme, M.; Holzmann, C.; Witt, M.; Grittner, U.; Rolfs, A.; Wree, A. Pharmacologic treatment assigned for Niemann Pick type C1 disease partly changes behavioral traits in wild-type mice. Int. J. Mol. Sci. 2016, 17, 1866. [Google Scholar] [CrossRef]
- Williams, I.M.; Wallom, K.L.; Smith, D.A.; Al Eisa, N.; Smith, C.; Platt, F.M. Improved neuroprotection using miglustat, curcumin and ibuprofen as a triple combination therapy in Niemann-Pick disease type C1 mice. Neurobiol. Dis. 2014, 67, 9–17. [Google Scholar] [CrossRef]
- King, K.A.; Gordon-Salant, S.; Yanjanin, N.; Zalewski, C.; Houser, A.; Porter, F.D.; Brewer, C.C. Auditory phenotype of Niemann-Pick disease, type C1. Ear. Hear. 2014, 35, 110–117. [Google Scholar] [CrossRef]
- King, K.A.; Gordon-Salant, S.; Pawlowski, K.S.; Taylor, A.M.; Griffith, A.J.; Houser, A.; Kurima, K.; Wassif, C.A.; Wright, C.G.; Porter, F.D.; et al. Hearing loss is an early consequence of Npc1 gene deletion in the mouse model of Niemann-Pick disease, type C. J. Assoc. Re. Otolaryngol. 2014, 15, 529–541. [Google Scholar] [CrossRef]
- Ward, S.; O’Donnell, P.; Fernandez, S.; Vite, C.H. 2-hydroxypropyl-beta-cyclodextrin raises hearing threshold in normal cats and in cats with Niemann-Pick type C disease. Pediatr. Res. 2010, 68, 52–56. [Google Scholar] [CrossRef]
- Takahashi, S.; Homma, K.; Zhou, Y.; Nishimura, S.; Duan, C.; Chen, J.; Ahmad, A.; Cheatham, M.A.; Zheng, J. Susceptibility of outer hair cells to cholesterol chelator 2-hydroxypropyl-beta-cyclodextrine is prestin-dependent. Sci. Rep. 2016, 6, 21973. [Google Scholar] [CrossRef]
- Iodice, R.; Dubbioso, R.; Topa, A.; Ruggiero, L.; Pisciotta, C.; Esposito, M.; Tozza, S.; Santoro, L.; Manganelli, F. Electrophysiological characterization of adult-onset Niemann-Pick type C disease. J. Neurol. Sci. 2015, 348, 262–265. [Google Scholar] [CrossRef]
- Claudepierre, T.; Paques, M.; Simonutti, M.; Buard, I.; Sahel, J.; Maue, R.A.; Picaud, S.; Pfrieger, F.W. Lack of Niemann-Pick type C1 induces age-related degeneration in the mouse retina. Mol. Cell. Neurosci. 2010, 43, 164–176. [Google Scholar] [CrossRef]
- Yan, X.; Ma, L.; Hovakimyan, M.; Lukas, J.; Wree, A.; Frank, M.; Guthoff, R.; Rolfs, A.; Witt, M.; Luo, J. Defects in the retina of Niemann-pick type C 1 mutant mice. BMC Neurosci. 2014, 15, 126. [Google Scholar] [CrossRef]
- Palladino, G.; Loizzo, S.; Fortuna, A.; Canterini, S.; Palombi, F.; Erickson, R.P.; Mangia, F.; Fiorenza, M.T. Visual evoked potentials of Niemann-Pick type C1 mice reveal an impairment of the visual pathway that is rescued by 2-hydroxypropyl-ss-cyclodextrin. Orphanet. J. Rare Dis. 2015, 10, 133. [Google Scholar] [CrossRef]
- Abel, L.A.; Walterfang, M.; Fietz, M.; Bowman, E.A.; Velakoulis, D. Saccades in adult Niemann-Pick disease type C reflect frontal, brainstem, and biochemical deficits. Neurology 2009, 72, 1083–1086. [Google Scholar] [CrossRef]
- Hovakimyan, M.; Meyer, A.; Lukas, J.; Luo, J.; Gudziol, V.; Hummel, T.; Rolfs, A.; Wree, A.; Witt, M. Olfactory deficits in Niemann-Pick type C1 (NPC1) disease. PLoS ONE 2013, 8, e82216. [Google Scholar] [CrossRef]
- Seo, Y.; Kim, H.S.; Shin, Y.; Kang, I.; Choi, S.W.; Yu, K.R.; Seo, K.W.; Kang, K.S. Excessive microglial activation aggravates olfactory dysfunction by impeding the survival of newborn neurons in the olfactory bulb of Niemann-Pick disease type C1 mice. Biochim. Biophys. Acta. 2014, 1842, 2193–2203. [Google Scholar] [CrossRef]
- Meyer, A.; Wree, A.; Günther, R.; Holzmann, C.; Schmitt, O.; Rolfs, A.; Witt, M. Increased regenerative capacity of the olfactory epithelium in Niemann-Pick disease type C1. Int. J. Mol. Sci. 2017, 18, 777. [Google Scholar] [CrossRef]
- Bagel, J.H.; Sikora, T.U.; Prociuk, M.; Pesayco, J.P.; Mizisin, A.P.; Shelton, G.D.; Vite, C.H. Electrodiagnostic testing and histopathologic changes confirm peripheral nervous system myelin abnormalities in the feline model of niemann-pick disease type C. J. Neuropathol. Exp. Neurol. 2013, 72, 256–262. [Google Scholar] [CrossRef]
- Marmiroli, P.; Rodriguez-Menendez, V.; Rigamonti, L.; Tonoli, E.; Rigolio, R.; Cavaletti, G.; Tredici, G.; Vercelli, A. Neuropathological changes in the peripheral nervous system and spinal cord in a transgenic mouse model of Niemann-Pick disease type A. Clin. Neuropathol. 2009, 28, 263–274. [Google Scholar]
- Donohue, C.; Marion, S.; Erickson, R.P. Expression of Npc1 in glial cells corrects sterility in Npc1(−/−) mice. J. Appl. Genet. 2009, 50, 385–390. [Google Scholar] [CrossRef]
- Gevry, N.Y.; Lopes, F.L.; Ledoux, S.; Murphy, B.D. Aberrant intracellular cholesterol transport disrupts pituitary and ovarian function. Molecul. Endocrinol. 2004, 18, 1778–1786. [Google Scholar] [CrossRef]
- Akpovi, C.D.; Murphy, B.D.; Erickson, R.P.; Pelletier, R.M. Dysregulation of testicular cholesterol metabolism following spontaneous mutation of the niemann-pick c1 gene in mice. Biol. Reprod. 2014, 91, 42. [Google Scholar] [CrossRef]
- Roff, C.F.; Strauss, J.F., 3rd; Goldin, E.; Jaffe, H.; Patterson, M.C.; Agritellis, G.C.; Hibbs, A.M.; Garfield, M.; Brady, R.O.; Pentchev, P.G. The murine Niemann-Pick type C lesion affects testosterone production. Endocrinology 1993, 133, 2913–2923. [Google Scholar] [CrossRef]
- Fan, J.; Akabane, H.; Graham, S.N.; Richardson, L.L.; Zhu, G.Z. Sperm defects in mice lacking a functional Niemann-Pick C1 protein. Mol. Reprod. Dev. 2006, 73, 1284–1291. [Google Scholar] [CrossRef]
- Elleder, M.; Šmíd, F. Adrenal changes in Niemann-Pick disease: Differences between sphingomyelinase deficiency and type C. Acta. Histochemica. 1985, 76, 163–176. [Google Scholar] [CrossRef]
- Kapur, R.; Donohue, C.; Jelinek, D.; Erickson, R.P. Amelioration of enteric neuropathology in a mouse model of Niemann-Pick C by Npc1 expression in enteric glia. J. Neurosci. Res. 2009, 87, 2994–3001. [Google Scholar] [CrossRef] [Green Version]
- Cougnoux, A.; Movassaghi, M.; Picache, J.A.; Iben, J.R.; Navid, F.; Salman, A.; Martin, K.; Farhat, N.Y.; Cluzeau, C.; Tseng, W.C.; et al. Gastrointestinal tract pathology in a BALB/c Niemann-Pick disease type C1 null mouse model. Dig. Dis. Sci. 2018, 63, 870–880. [Google Scholar] [CrossRef]
- Cavounidis, A.; Uhlig, H.H. Crohn’s disease in Niemann-Pick disease type C1: Caught in the cross-Fire of host-microbial interactions. Dig. Dis. Sci. 2018, 63, 811–813. [Google Scholar] [CrossRef]
- Schwerd, T.; Pandey, S.; Yang, H.T.; Bagola, K.; Jameson, E.; Jung, J.; Lachmann, R.H.; Shah, N.; Patel, S.Y.; Booth, C.; et al. Impaired antibacterial autophagy links granulomatous intestinal inflammation in Niemann-Pick disease type C1 and XIAP deficiency with NOD2 variants in Crohn’s disease. Gut 2017, 66, 1060–1073. [Google Scholar] [CrossRef]
- Steven, L.C.; Driver, C.P. Niemann-pick disease type C and Crohn’s disease. Scott. Med. J. 2005, 50, 80–81. [Google Scholar] [CrossRef]
- Hammel, I.; Alroy, J. The effect of lysosomal storage diseases on secretory cells: An ultrastructural study of pancreas as an example. J. Submicrosc. Cytol. Pathol. 1995, 27, 143–160. [Google Scholar]
- Staretz-Chacham, O.; Aviram, M.; Morag, I.; Goldbart, A.; Hershkovitz, E. Pulmonary involvement in Niemann-Pick C type 1. Eur. J. Pediatr. 2018. [Google Scholar] [CrossRef]
- Roszell, B.R.; Tao, J.Q.; Yu, K.J.; Gao, L.; Huang, S.; Ning, Y.; Feinstein, S.I.; Vite, C.H.; Bates, S.R. Pulmonary abnormalities in animal models due to Niemann-Pick type C1 (NPC1) or C2 (NPC2) disease. PLoS ONE 2013, 8, e67084. [Google Scholar] [CrossRef]
- Erickson, R.P.; Deutsch, G.; Patil, R. A pilot study of direct delivery of hydroxypropyl-beta-cyclodextrin to the lung by the nasal route in a mouse model of Niemann-Pick C1 disease: motor performance is unaltered and lung disease is worsened. J. Appl. Genet. 2018, 59, 187–191. [Google Scholar] [CrossRef]
- Capron, T.; Trigui, Y.; Gautier, C.; Puech, B.; Chanez, P.; Reynaud-Gaubert, M. Respiratory impairment in Niemann-Pick B disease: Two case reports and review for the pulmonologist. Respir. Med. Res. 2019, 76, 13–18. [Google Scholar] [CrossRef]
- Kuemmel, T.A.; Thiele, J.; Schroeder, R.; Stoffel, W. Pathology of visceral organs and bone marrow in an acid sphingomyelinase deficient knock-out mouse line, mimicking human Niemann-Pick disease type A. A light and electron microscopic study. Pathol. Res. Pract. 1997, 193, 663–671. [Google Scholar] [CrossRef]
- Pitha, J.; Gerloczy, A.; Olivi, A. Parenteral hydroxypropyl cyclodextrins: intravenous and intracerebral administration of lipophiles. J. Pharm. Sci. 1994, 83, 833–837. [Google Scholar] [CrossRef]
- Reif, S.; Spirer, Z.; Messer, G.; Baratz, M.; Bembi, B.; Bujanover, Y. Severe failure to thrive and liver dysfunction as the main manifestations of a new variant of Niemann-Pick disease. Clin. Pediatr. (Phila) 1994, 33, 628–630. [Google Scholar] [CrossRef]
- Putterman, C.; Zelingher, J.; Shouval, D. Liver failure and the sea-blue histiocyte/adult Niemann-Pick disease. Case report and review of the literature. J. Clin. Gastroenterol. 1992, 15, 146–149. [Google Scholar] [CrossRef]
- Rutledge, J.C. Progressive neonatal liver failure due to type C Niemann-Pick isease. Pediatr. Pathol. 1989, 9, 779–784. [Google Scholar] [CrossRef]
- Dumontel, C.; Girod, C.; Dijoud, F.; Dumez, Y.; Vanier, M.T. Fetal Niemann-Pick disease type C: ultrastructural and lipid findings in liver and spleen. Virchows Arch. A Pathol. Anat. Histopathol. 1993, 422, 253–259. [Google Scholar] [CrossRef]
- Yerushalmi, B.; Sokol, R.J.; Narkewicz, M.R.; Smith, D.; Ashmead, J.W.; Wenger, D.A. Niemann-pick disease type C in neonatal cholestasis at a North American Center. J. Pediatr. Gastroenterol. Nutr. 2002, 35, 44–50. [Google Scholar] [CrossRef]
- Beltroy, E.P.; Richardson, J.A.; Horton, J.D.; Turley, S.D.; Dietschy, J.M. Cholesterol accumulation and liver cell death in mice with Niemann-Pick type C disease. Hepatology 2005, 42, 886–893. [Google Scholar] [CrossRef]
- Rimkunas, V.M.; Graham, M.J.; Crooke, R.M.; Liscum, L. TNF-(alpha) plays a role in hepatocyte apoptosis in Niemann-Pick type C liver disease. J. Lipid Res. 2009, 50, 327–333. [Google Scholar] [CrossRef]
- Sayre, N.L.; Rimkunas, V.M.; Graham, M.J.; Crooke, R.M.; Liscum, L. Recovery from liver disease in a Niemann-Pick type C mouse model. J. Lipid Res. 2010, 51, 2372–2383. [Google Scholar] [CrossRef] [Green Version]
- Jelinek, D.; Castillo, J.J.; Garver, W.S. The C57BL/6J Niemann-Pick C1 mouse model with decreased gene dosage has impaired glucose tolerance independent of body weight. Gene 2013, 527, 65–70. [Google Scholar] [CrossRef]
- Li, A.C.; Glass, C.K. PPAR- and LXR-dependent pathways controlling lipid metabolism and the development of atherosclerosis. J. Lipid Res. 2004, 45, 2161–2173. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Yamada, Y.; Ishitsuka, Y.; Matsuo, M.; Shiraishi, K.; Wada, K.; Uchio, Y.; Kondo, Y.; Takeo, T.; Nakagata, N.; et al. Efficacy of 2-hydroxypropyl-beta-cyclodextrin in Niemann-Pick disease type C model mice and its pharmacokinetic analysis in a patient with the disease. Biol. Pharm. Bull. 2015, 38, 844–851. [Google Scholar] [CrossRef]
- Rigante, D.; Cipolla, C.; Basile, U.; Gulli, F.; Savastano, M.C. Overview of immune abnormalities in lysosomal storage disorders. Immunol. Lett. 2017, 188, 79–85. [Google Scholar] [CrossRef]
- Nesslauer, A.M.; Gläser, A.; Gräler, M.; Engelmann, R.; Müller-Hilke, B.; Frank, M.; Burstein, C.; Rolfs, A.; Neidhardt, J.; Wree, A.; et al. A therapy with miglustat, 2-hydroxypropyl-ss-cyclodextrin and allopregnanolone restores splenic cholesterol homeostasis in Niemann-pick disease type C1. Lipids Health Dis. 2019, 18, 146. [Google Scholar] [CrossRef]
- Angheloiu, G.O.; Haka, A.S.; Georgakoudi, I.; Arendt, J.; Muller, M.G.; Scepanovic, O.R.; Evanko, S.P.; Wight, T.N.; Mukherjee, P.; Waldeck, D.H.; et al. Detection of coronary atherosclerotic plaques with superficial proteoglycans and foam cells using real-time intrinsic fluorescence spectroscopy. Atherosclerosis 2011, 215, 96–102. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, K.D.; Gordon, D.; Deeb, S.; Ferguson, M.; Chait, A. Lipoprotein lipase is synthesized by macrophage-derived foam cells in human coronary atherosclerotic plaques. J. Clin. Invest. 1992, 89, 1544–1550. [Google Scholar] [CrossRef]
- Dubland, J.A.; Francis, G.A. So much cholesterol: the unrecognized importance of smooth muscle cells in atherosclerotic foam cell formation. Curr. Opin. Lipidol. 2016, 27, 155–161. [Google Scholar] [CrossRef]
- Coisne, C.; Hallier-Vanuxeem, D.; Boucau, M.C.; Hachani, J.; Tilloy, S.; Bricout, H.; Monflier, E.; Wils, D.; Serpelloni, M.; Parissaux, X.; et al. Beta-cyclodextrins decrease cholesterol release and ABC-associated transporter expression in smooth muscle cells and aortic endothelial cells. Front. Physiol. 2016, 7, 185. [Google Scholar] [CrossRef]
- Montecucco, F.; Lenglet, S.; Carbone, F.; Boero, S.; Pelli, G.; Burger, F.; Roth, A.; Bertolotto, M.; Nencioni, A.; Cea, M.; et al. Treatment with KLEPTOSE(R) CRYSMEB reduces mouse atherogenesis by impacting on lipid profile and Th1 lymphocyte response. Vascul. Pharmacol. 2015, 72, 197–208. [Google Scholar] [CrossRef]
- Zimmer, S.; Grebe, A.; Bakke, S.S.; Bode, N.; Halvorsen, B.; Ulas, T.; Skjelland, M.; De Nardo, D.; Labzin, L.I.; Kerksiek, A.; et al. Cyclodextrin promotes atherosclerosis regression via macrophage reprogramming. Sci. Transl. Med. 2016, 8, 333ra50. [Google Scholar] [CrossRef]
- Mesquita-Guimaraes, K.S.; De Rossi, A.; Freitas, A.C.; Nelson-Filho, P.; da Silva, R.A.; de Queiroz, A.M. Changes in caries risk and activity of a 9-year-old patient with niemann-pick disease type C. Case Rep. Dent. 2015, 2015, 571098. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bräuer, A.U.; Kuhla, A.; Holzmann, C.; Wree, A.; Witt, M. Current Challenges in Understanding the Cellular and Molecular Mechanisms in Niemann–Pick Disease Type C1. Int. J. Mol. Sci. 2019, 20, 4392. https://doi.org/10.3390/ijms20184392
Bräuer AU, Kuhla A, Holzmann C, Wree A, Witt M. Current Challenges in Understanding the Cellular and Molecular Mechanisms in Niemann–Pick Disease Type C1. International Journal of Molecular Sciences. 2019; 20(18):4392. https://doi.org/10.3390/ijms20184392
Chicago/Turabian StyleBräuer, Anja U., Angela Kuhla, Carsten Holzmann, Andreas Wree, and Martin Witt. 2019. "Current Challenges in Understanding the Cellular and Molecular Mechanisms in Niemann–Pick Disease Type C1" International Journal of Molecular Sciences 20, no. 18: 4392. https://doi.org/10.3390/ijms20184392