Molecular Docking Guided Grid-Independent Descriptor Analysis to Probe the Impact of Water Molecules on Conformational Changes of hERG Inhibitors in Drug Trapping Phenomenon



Abstract
:
1. Introduction
2. Results
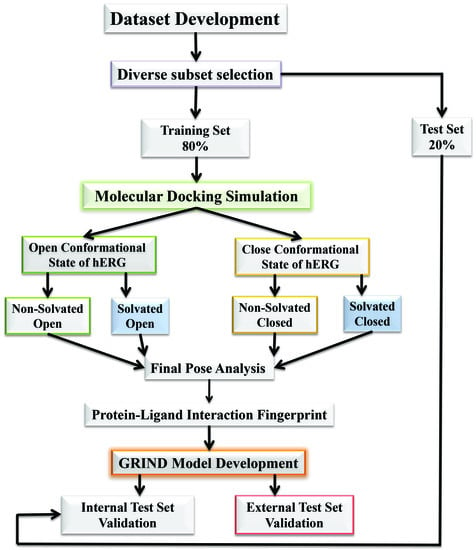
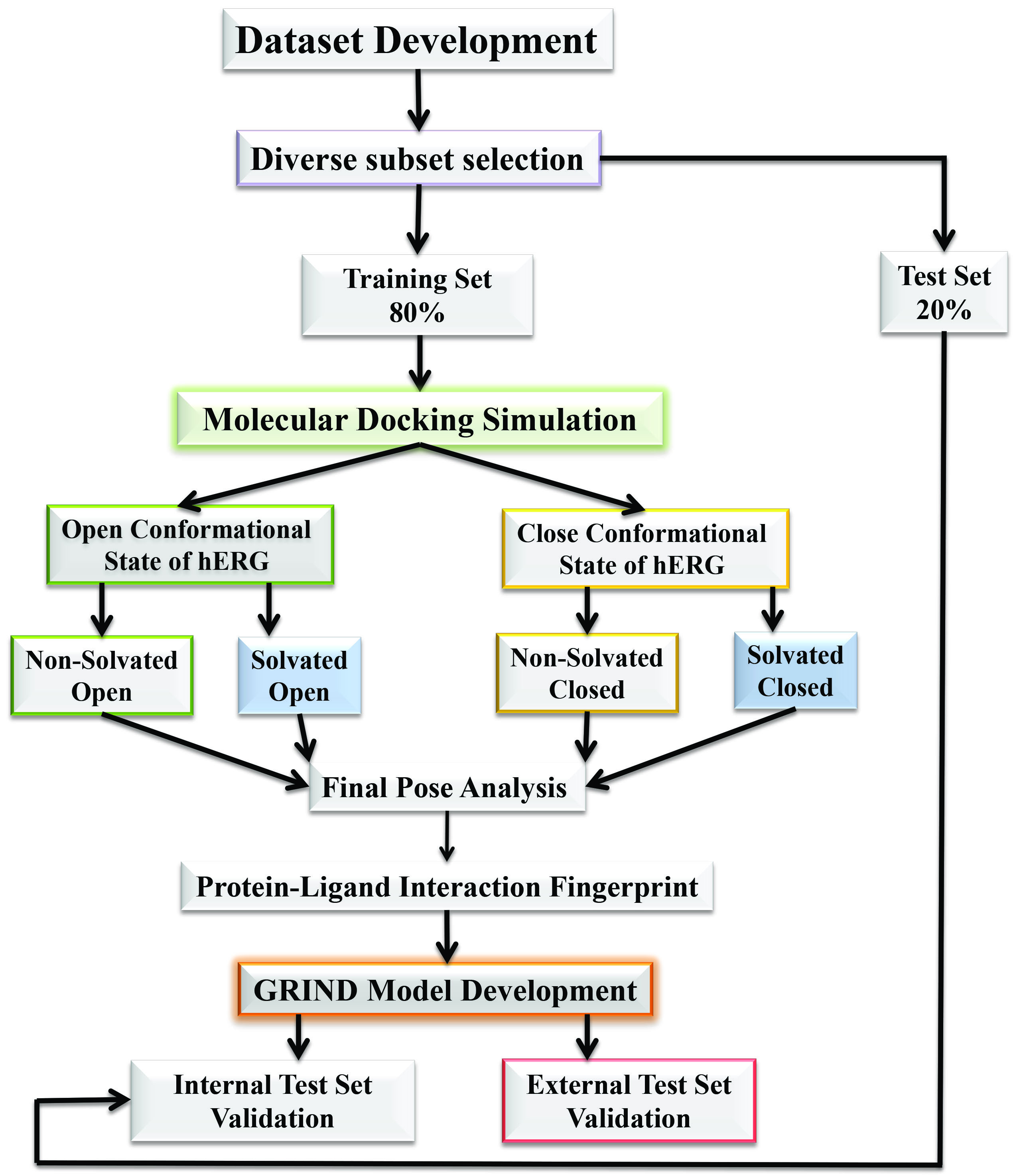
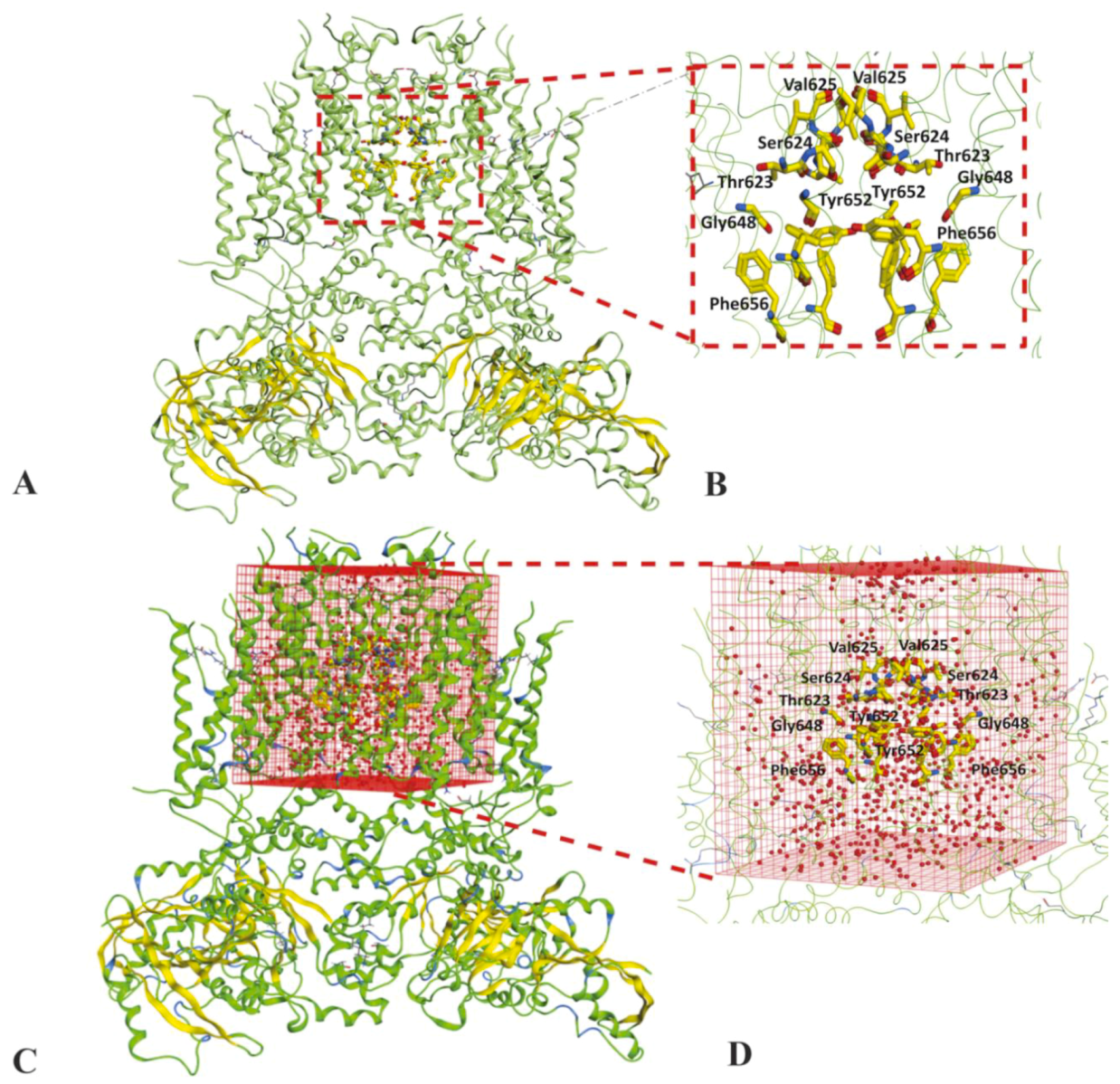
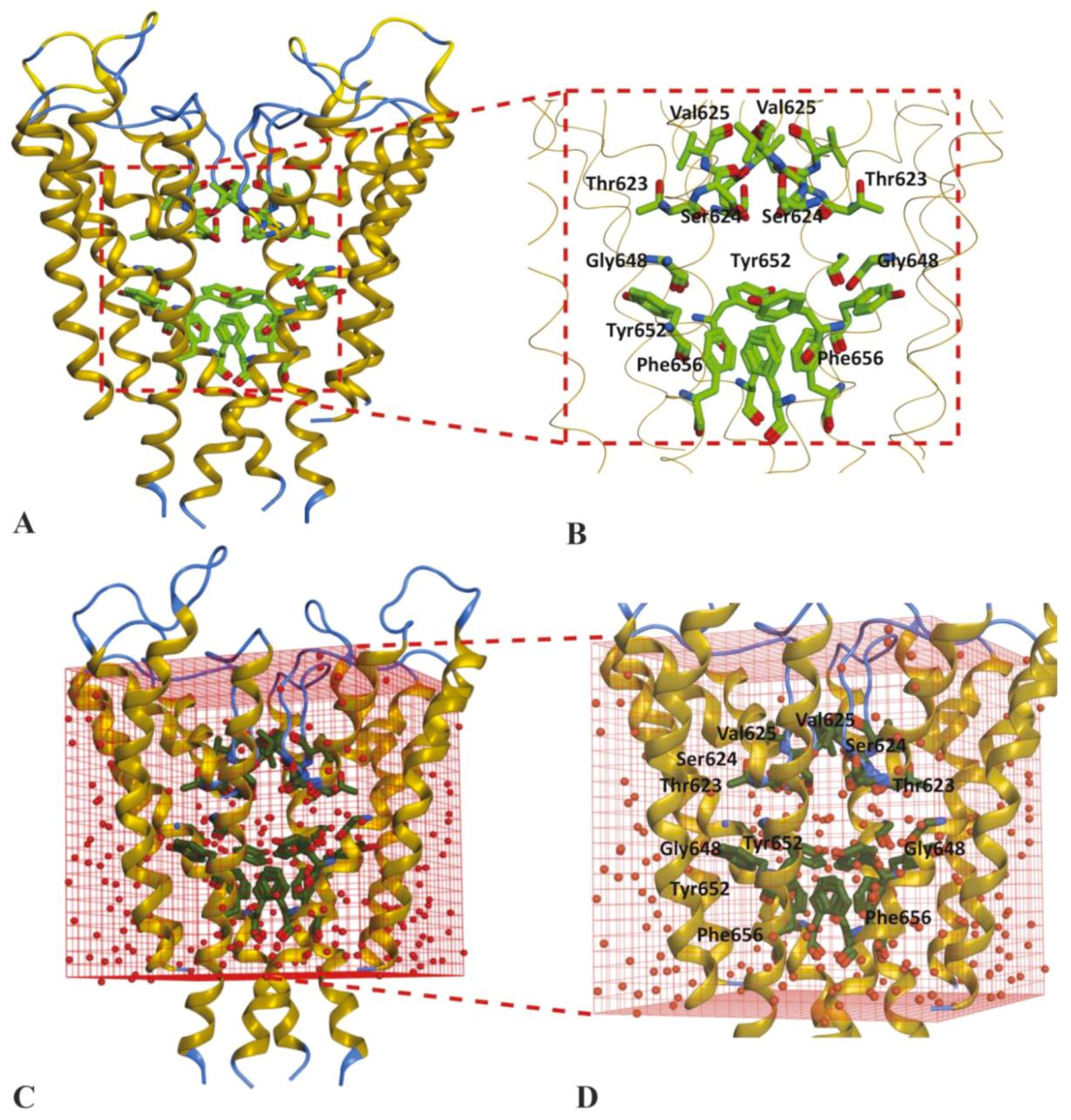
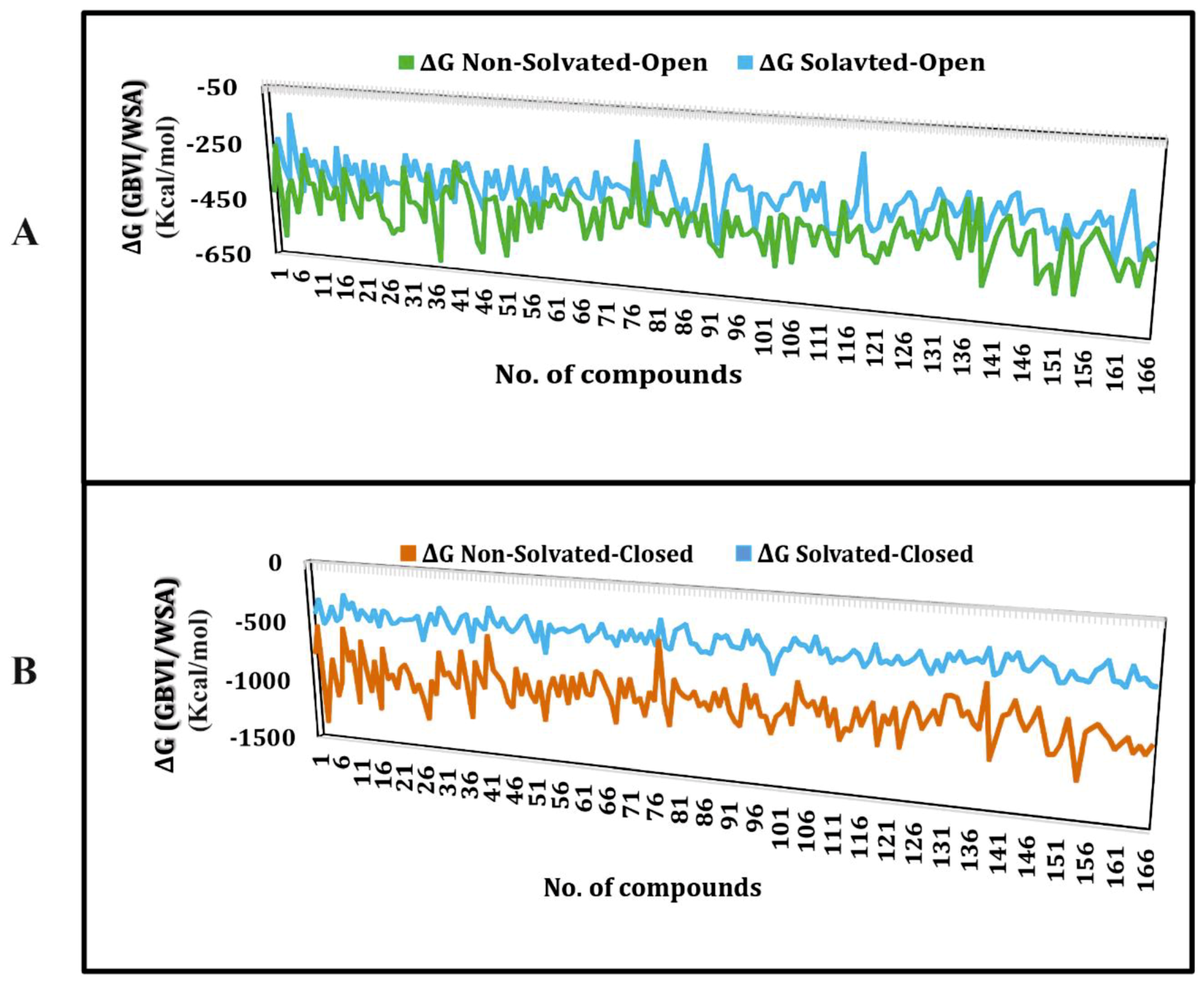
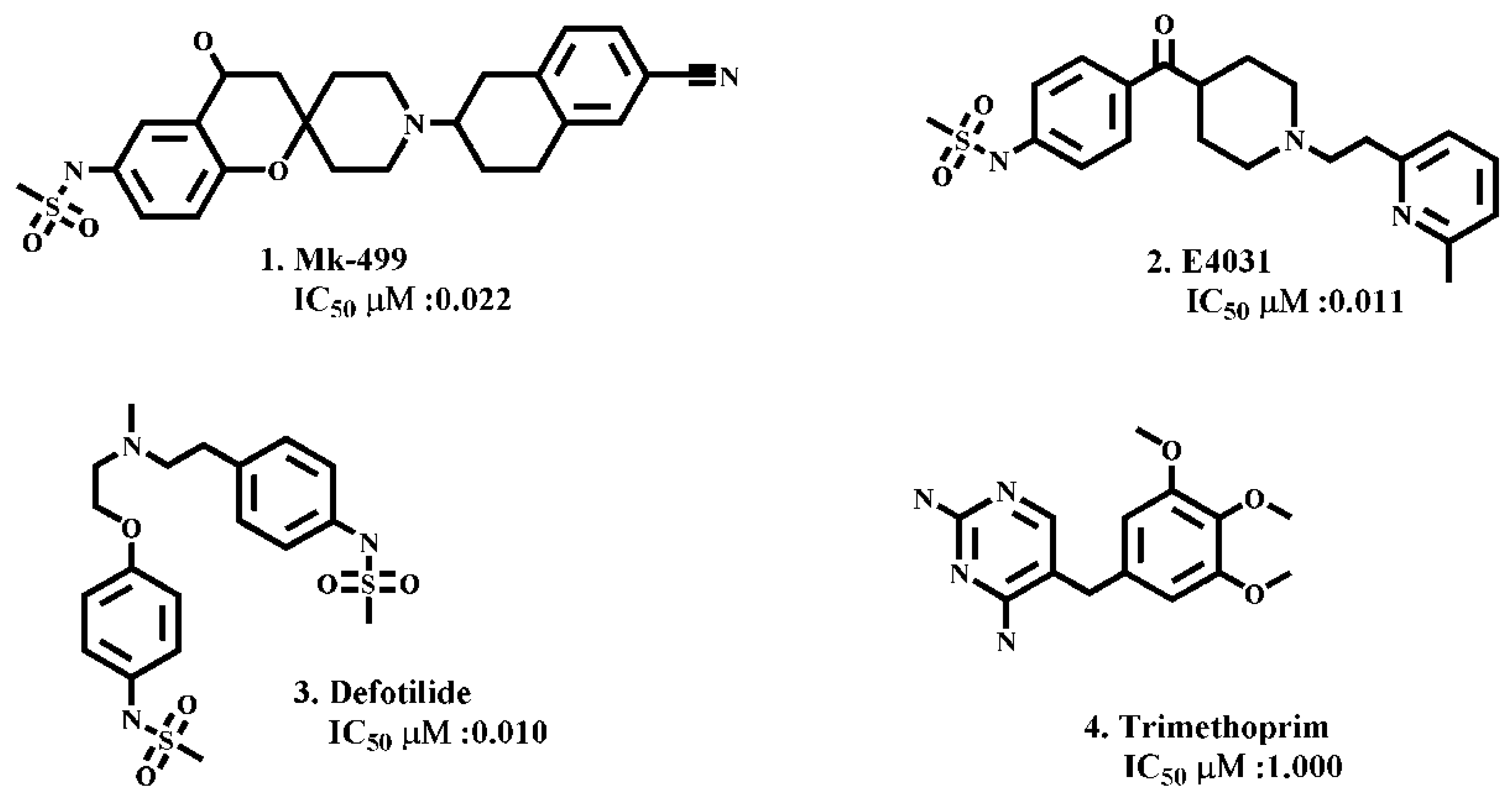
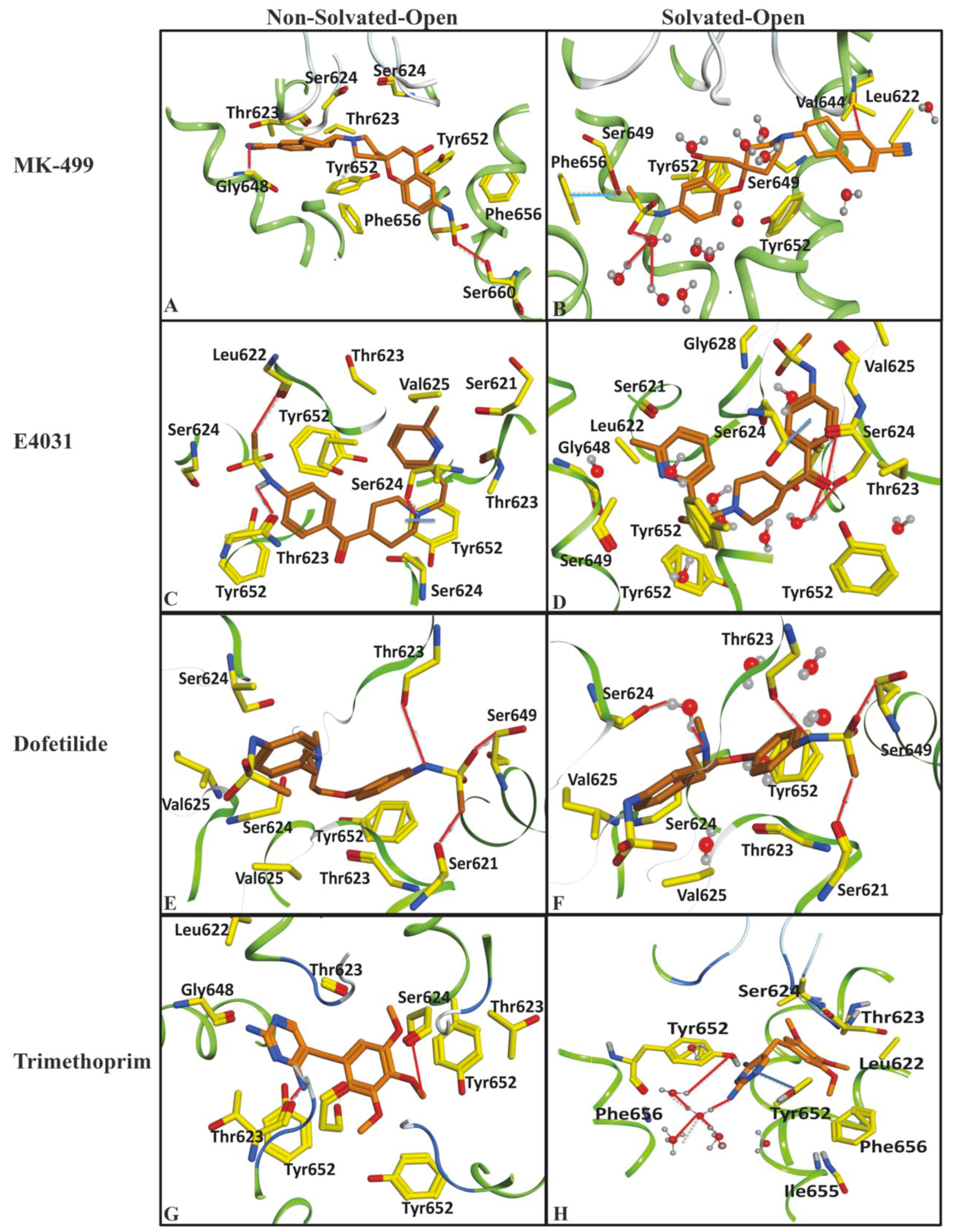
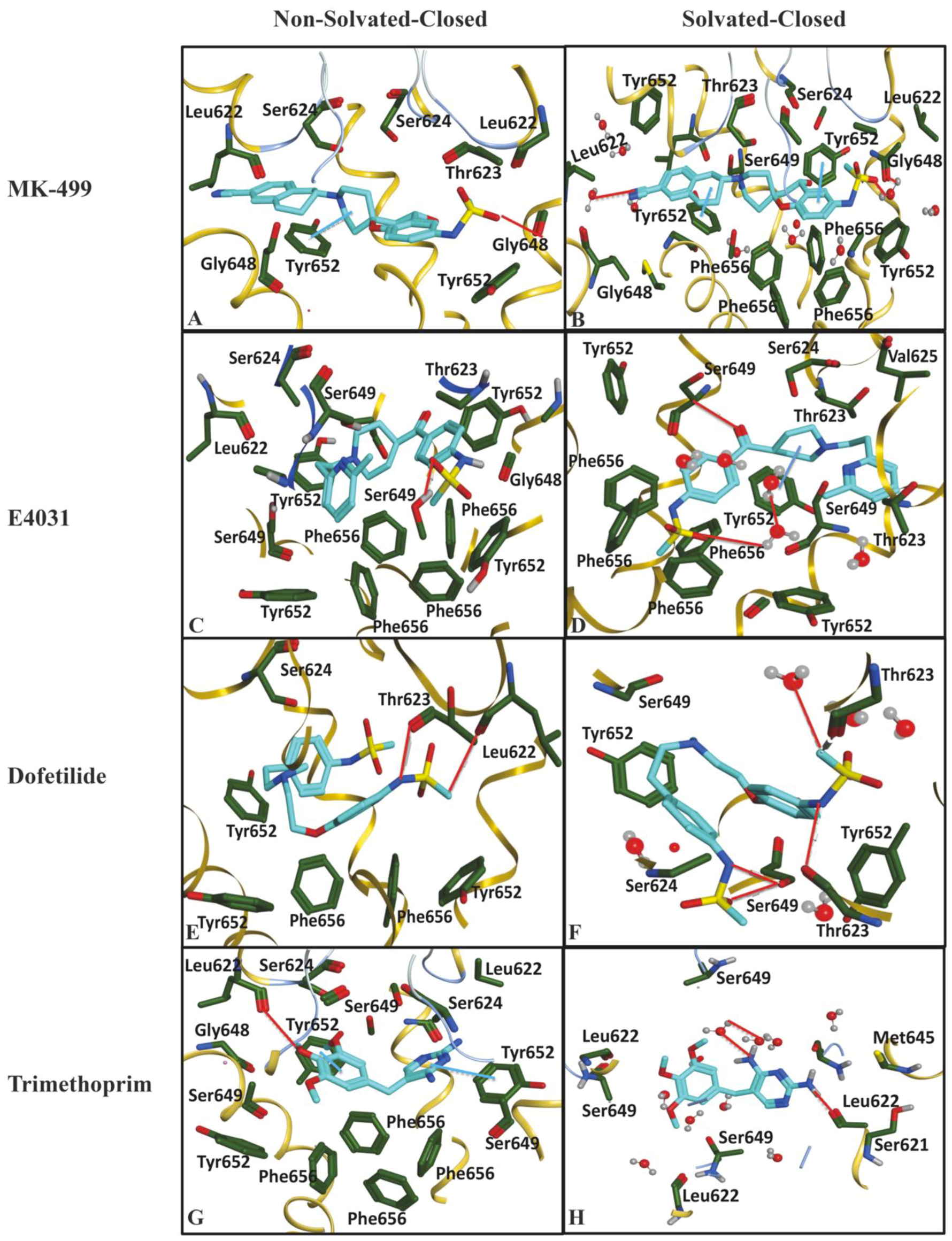
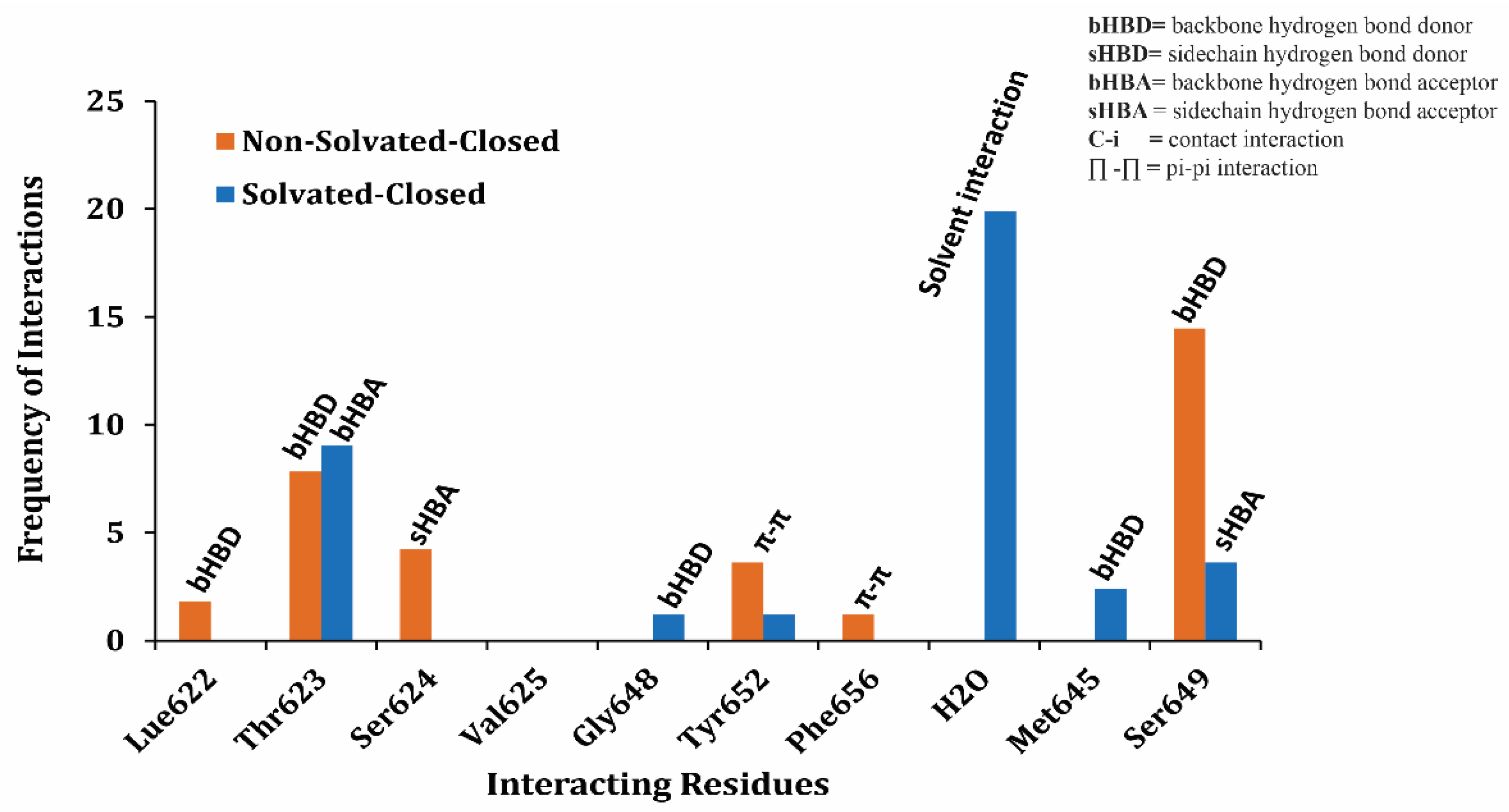
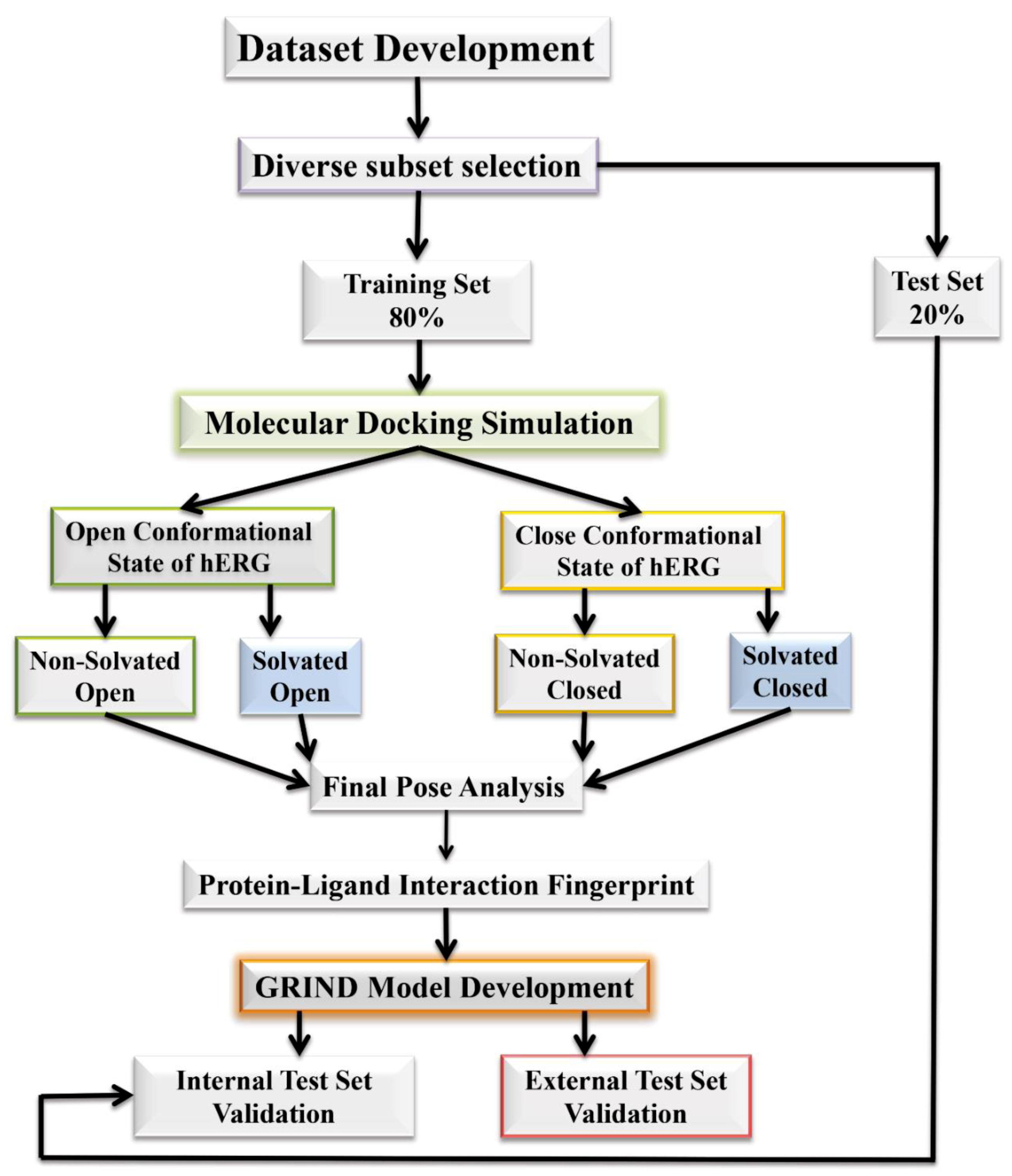
2.1. Molecular Docking Simulations
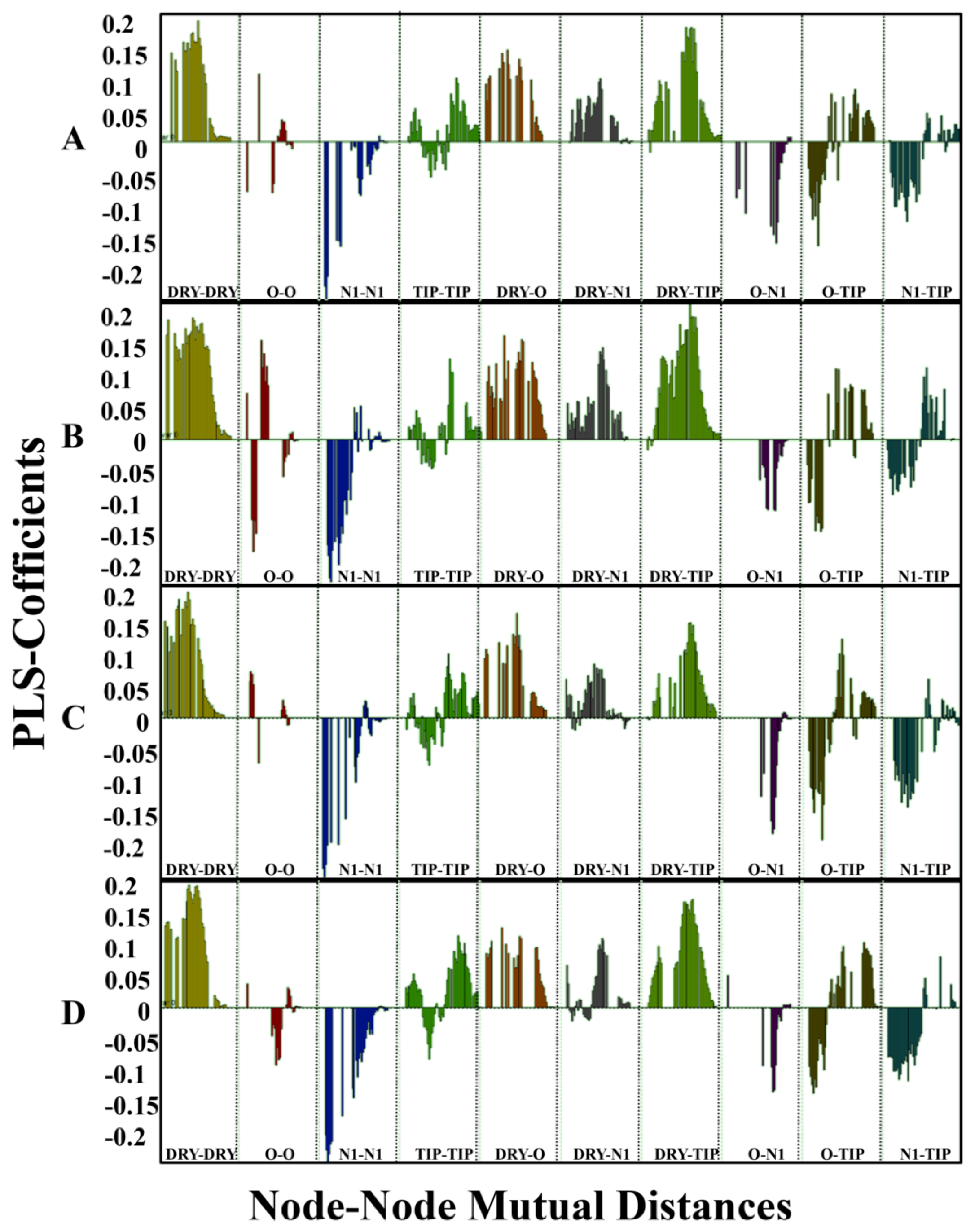
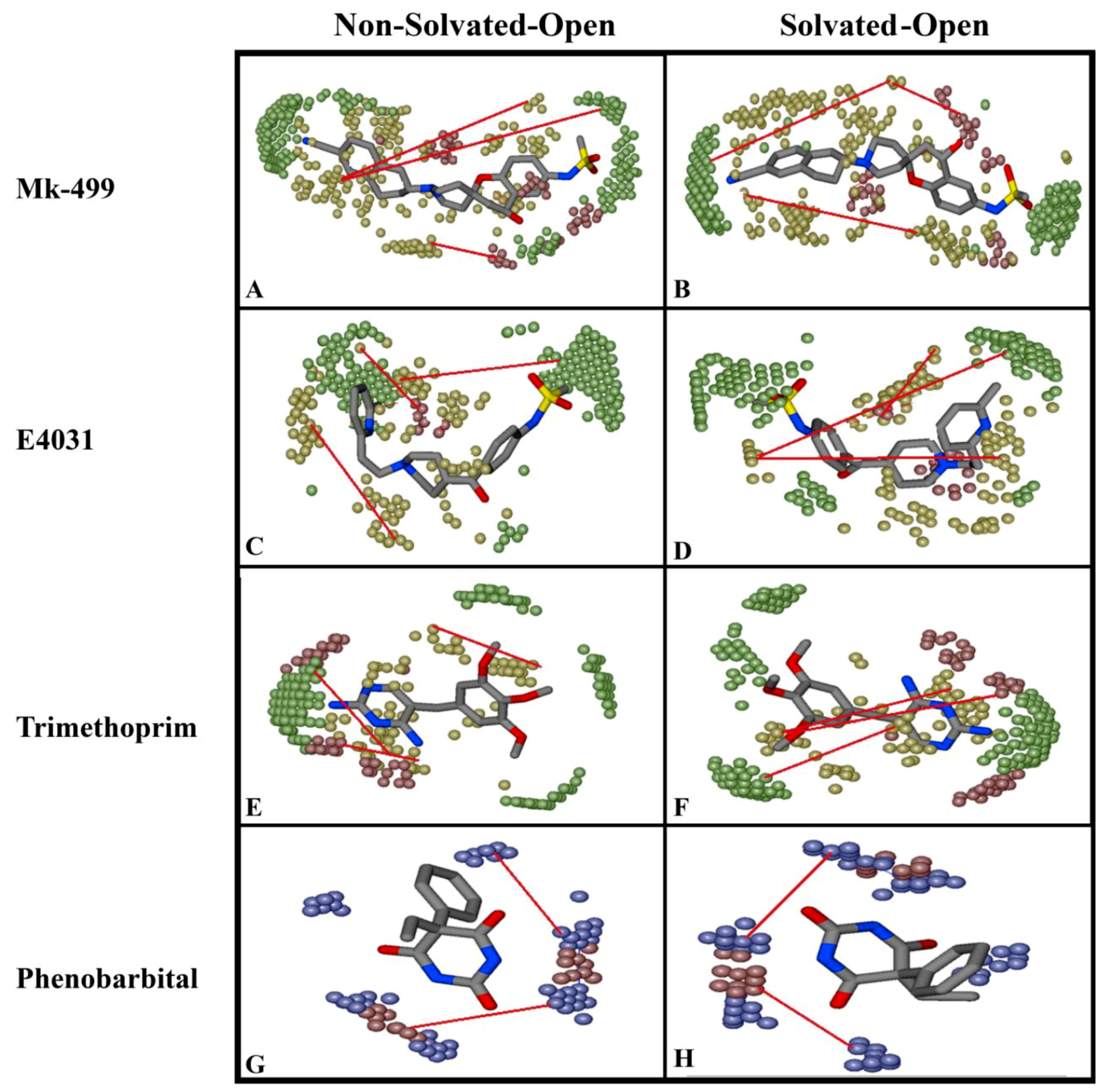
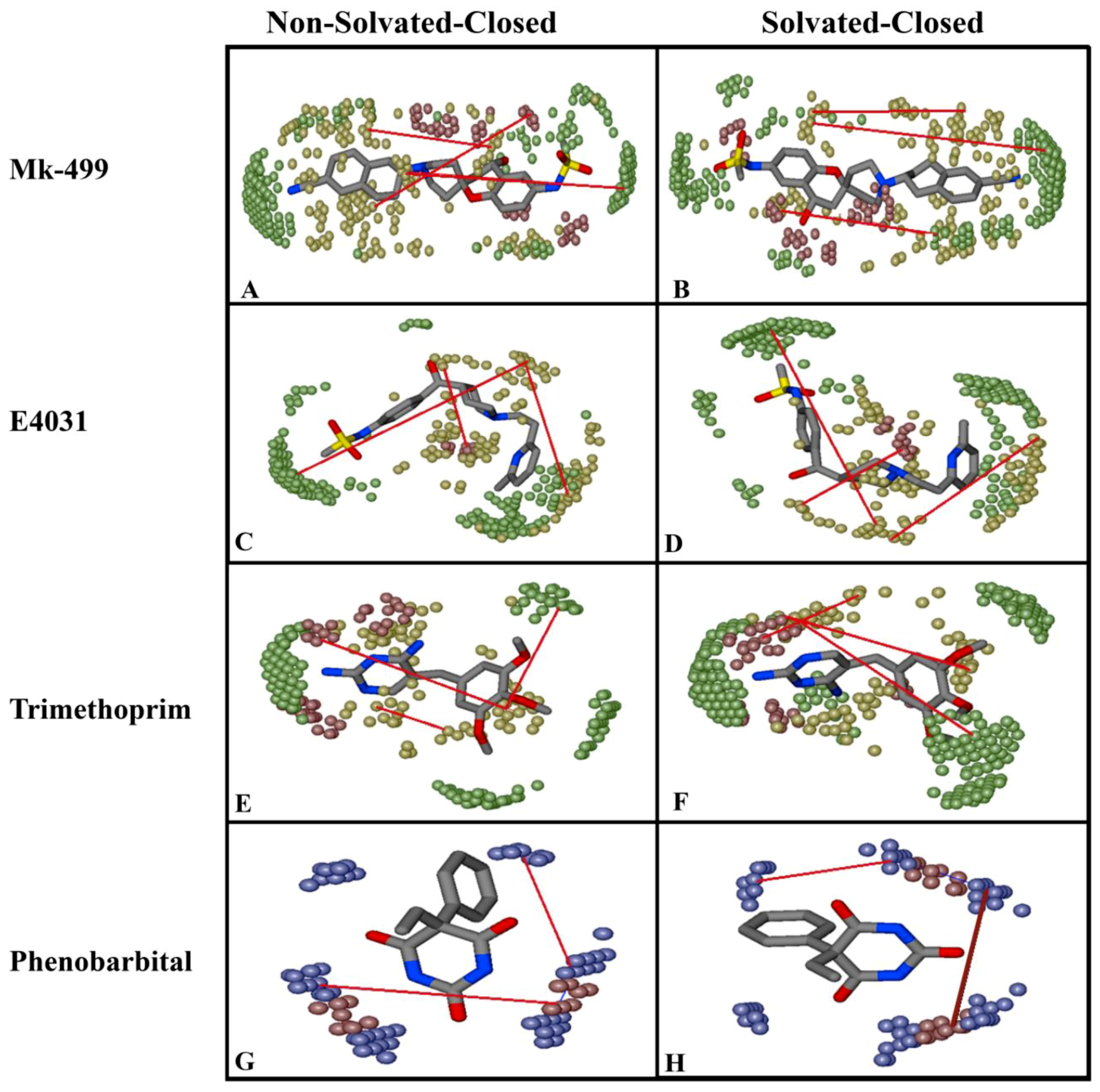
2.2. Grid-Independent Descriptor Analysis
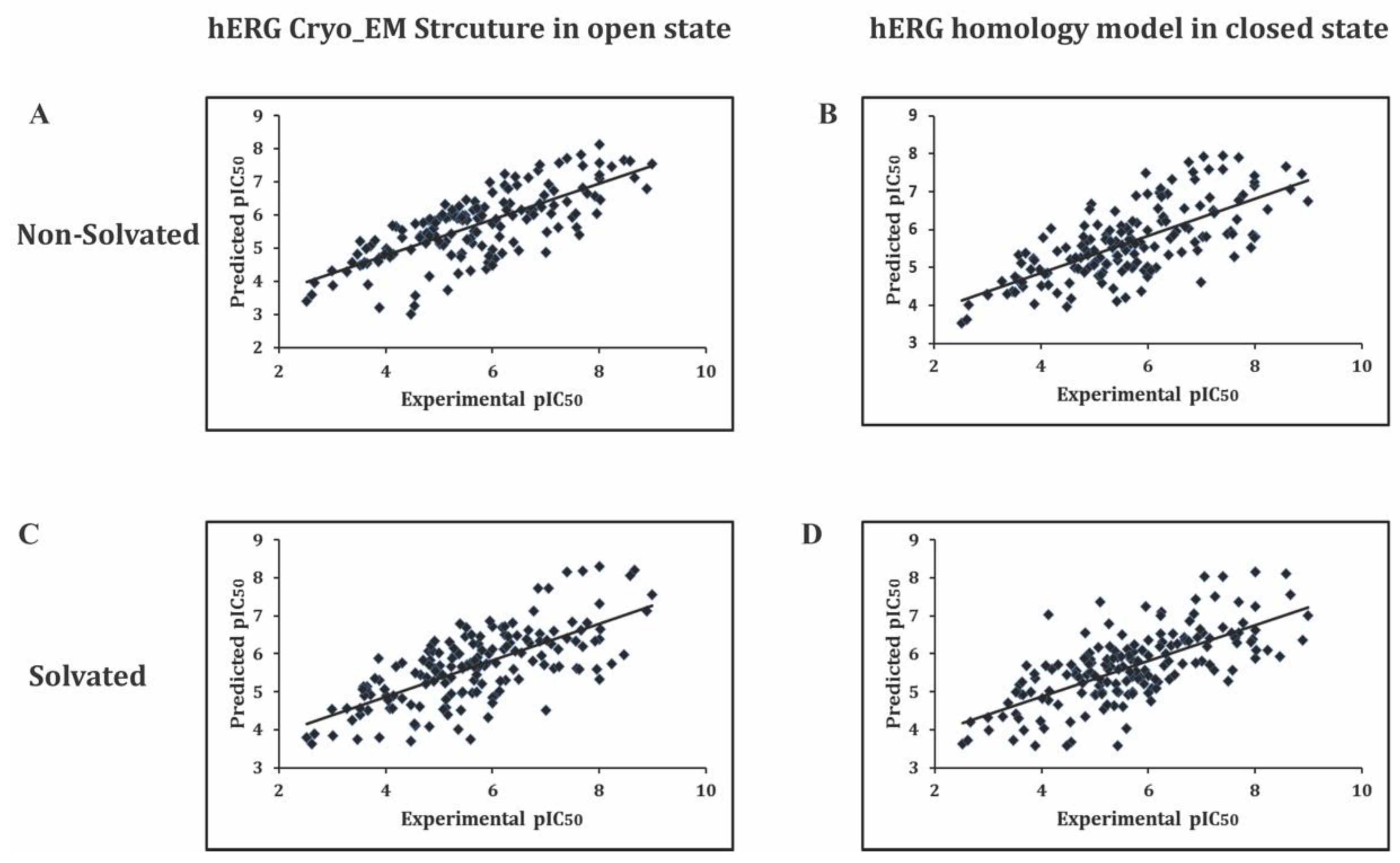
2.3. External Test Set Validation
3. Discussion
4. Materials and Methods
4.1. Molecular Docking Simulations
4.1.1. Structure Preparation
4.1.2. Molecular Dynamic Simulations
4.1.3. Solvation of Binding Cavity

- c: represents the average gain/loss of rotational and translational entropy.
- α, β: are constants which were determined during training (along with c) and are forcefield-dependent. If not using an AMBER forcefield, the parameters will be set by default to the MMFF trained parameters.
- EcoulEcoul: is the coulombic electrostatic term which is calculated using currently loaded charges, using a constant dielectric of 1.
- EsolEsol: is the solvation electrostatic term which is calculated using the GB/VI solvation model. For more information on the GB/VI solvation model.
- Evdw: is the van der Waals contribution to the binding.
- SAweighted: is the surface area, weighted by exposure. This weighting scheme penalizes exposed surface area.
- External H-bond: protein–ligand hydrogen bond energy,
- External vdw: protein–ligand van der Waals (vdw) energy,
- Internal vdw: ligand internal vdw energy,
- Internal torsion: ligand torsional strain energy.
4.1.4. Final Pose Selection and Ligand–Protein Interaction Analysis
4.1.5. Protein–Ligand Interaction Fingerprints
4.2. Grid Independent Descriptors Model Development
Test Set Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| hERG | Human ether a-go-go related gene |
| aLQTS | acquired Long QT syndrome |
| GRIND | GRId-Independent Descriptors |
| QSAR | Quantitative Structure Activity Relationship |
| MIFs | Molecular Interaction Fields |
| PLIF | Protein Ligand Interaction Fingerprints |
| TdPs | Torsade de Pointes |
| ICH | International Conference on Harmonisation |
| MD | Molecular Dynamics |
| GB/VI | Generalized Born Solvation Model |
| PLS | Partial Least Square |
| LOO | Leave One Out |
| SDEP | Standard Deviation of Error Prediction |
| FFD | Fractional Factorial Design |
| CLACC | Consistently Large Auto And Cross-Correlation |
References
- Jurkiewicz, N.K.; Sanguinetti, M.C. Rate-dependent prolongation of cardiac action potentials by a methanesulfonanilide class III antiarrhythmic agent. Specific block of rapidly activating delayed rectifier K+ current by dofetilide. Circ. Res. 1993, 72, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, U.; Schmidt, C.; Netzer, R.; Pongs, O. Effects of fluoroquinolones on HERG currents. Eur. J. Pharmacol. 2000, 406, 341–343. [Google Scholar] [CrossRef]
- Alvarez, P.A.; Pahissa, J. QT alterations in psychopharmacology: Proven candidates and suspects. Curr. Drug Saf. 2010, 5, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Vorperian, V.R.; Gong, Q.; Zhang, S.; January, C.T. Block of HERG potassium channels by the antihistamine astemizole and its metabolites desmethylastemizole and norastemizole. J. Cardiovasc. Electrophysiol. 1999, 10, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Mitcheson, J.S.; Chen, J.; Lin, M.; Culberson, C.; Sanguinetti, M.C. A structural basis for drug-induced long QT syndrome. Proc. Natl. Acad. Sci. USA 2000, 97, 12329–12333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roden, D.M. Drug-induced prolongation of the QT interval. N. Engl. J. Med. 2004, 350, 1013–1022. [Google Scholar] [CrossRef]
- Sanguinetti, M.C.; Jiang, C.; Curran, M.E.; Keating, M.T. A mechanistic link between an inherited and an acquird cardiac arrthytmia: HERG encodes the I Kr potassium channel. Cell 1995, 81, 299–307. [Google Scholar] [CrossRef]
- Raschi, E.; Vasina, V.; Poluzzi, E.; De Ponti, F. The hERG K+ channel: Target and antitarget strategies in drug development. Pharmacol. Res. 2008, 57, 181–195. [Google Scholar] [CrossRef]
- Food and Drug Administration, HHS. International conference on harmonisation; guidance on E14 clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non-antiarrhythmic drugs. Fed. Regist. 2005, 70, 61134–61135. [Google Scholar]
- Food and Drug Administration, HHS. International conference on harmonisation; guidance on S7B nonclinical evaluation of the potential for delayed ventricular repolarization (QT interval prolongation) by human pharmaceuticals. Fed. Regist. 2005, 70, 61133–61134. [Google Scholar]
- Pollard, C.; Gerges, N.A.; Bridgland-Taylor, M.; Easter, A.; Hammond, T.; Valentin, J.P. An introduction to QT interval prolongation and non-clinical approaches to assessing and reducing risk. Br. J. Pharmacol. 2010, 159, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitcheson, J.S. hERG potassium channels and the structural basis of drug-induced arrhythmias. Chem. Res. Toxicol. 2008, 21, 1005–1010. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, P.; Ciantar, M.; Theret, I.; Ducrot, P. Dynamics of hERG closure allow novel insights into hERG blocking by small molecules. J. Chem. Inf. Model. 2014, 54, 2320–2333. [Google Scholar] [CrossRef] [PubMed]
- Durdagi, S.; Deshpande, S.; Duff, H.J.; Noskov, S.Y. Modeling of open, closed, and open-inactivated states of the hERG1 channel: Structural mechanisms of the state-dependent drug binding. J. Chem. Inf. Model. 2012, 52, 2760–2774. [Google Scholar] [CrossRef] [PubMed]
- Farid, R.; Day, T.; Friesner, R.A.; Pearlstein, R.A. New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg. Med. Chem. 2006, 14, 3160–3173. [Google Scholar] [CrossRef] [PubMed]
- Masetti, M.; Cavalli, A.; Recanatini, M. Modeling the hERG potassium channel in a phospholipid bilayer: Molecular dynamics and drug docking studies. J. Comput. Chem. 2008, 29, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Stary, A.; Wacker, S.J.; Boukharta, L.; Zachariae, U.; Karimi-Nejad, Y.; Åqvist, J.; Vriend, G.; de Groot, B.L. Toward a consensus model of the HERG potassium channel. ChemMedChem 2010, 5, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Kayık, G.; Tüzün, N.Ş.; Durdagi, S. Structural investigation of vesnarinone at the pore domains of open and open-inactivated states of hERG1 K+ channel. J. Mol. Graph. Model. 2017, 77, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Barakat, K.H. Development of safe drugs: The hERG challenge. Med. Res. Rev. 2018, 38, 525–555. [Google Scholar] [CrossRef]
- Wang, W.; MacKinnon, R. Cryo-EM Structure of the Open Human Ether-à-go-go-Related K+ Channel hERG. Cell 2017, 169, 422–430. [Google Scholar] [CrossRef]
- Ladbury, J.E. Just add water! The effect of water on the specificity of protein-ligand binding sites and its potential application to drug design. Chem. Biol. 1996, 3, 973–980. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Lazaridis, T. Water at biomolecular binding interfaces. Phys. Chem. Chem. Phys. 2007, 9, 573–581. [Google Scholar] [CrossRef] [PubMed]
- de Beer, S.; Vermeulen, N.P.; Oostenbrink, C. The role of water molecules in computational drug design. Curr. Top. Med. Chem. 2010, 10, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Roux, B. Computation of binding free energy with molecular dynamics and grand canonical Monte Carlo simulations. J. Chem. Phys. 2008, 128, 115103. [Google Scholar] [CrossRef] [PubMed]
- Young, T.; Abel, R.; Kim, B.; Berne, B.J.; Friesner, R.A. Motifs for molecular recognition exploiting hydrophobic enclosure in protein–ligand binding. Proc. Natl. Acad. Sci. USA 2007, 104, 808–813. [Google Scholar] [CrossRef]
- Klebe, G. Virtual ligand screening: Strategies, perspectives and limitations. Drug Discov. Today 2006, 11, 580–594. [Google Scholar] [CrossRef]
- Vitorović-Todorović, M.D.; Cvijetić, I.N.; Juranić, I.O.; Drakulić, B.J. The 3D-QSAR study of 110 diverse, dual binding, acetylcholinesterase inhibitors based on alignment independent descriptors (GRIND-2). The effects of conformation on predictive power and interpretability of the models. J. Mol. Graph. Model. 2012, 38, 194–210. [Google Scholar] [CrossRef]
- Pastor, M.; Cruciani, G.; McLay, I.; Pickett, S.; Clementi, S. GRid-INdependent descriptors (GRIND): A novel class of alignment-independent three-dimensional molecular descriptors. J. Med. Chem. 2000, 43, 3233–3243. [Google Scholar] [CrossRef]
- Munawar, S.; Windley, M.J.; Tse, E.G.; Todd, M.; Hill, A.; Vandenberg, J.; Jabeen, I. Experimentally Validated Pharmacoinformatics Approach to Predict hERG Inhibition Potential of New Chemical Entities. Front. Pharmacol. 2018, 9, 1035. [Google Scholar] [CrossRef]
- MacKerell, A.D., Jr.; Bashford, D.; Bellott, M.; Dunbrack, R.L., Jr.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojciechowski, M.; Lesyng, B. Generalized Born model: Analysis, refinement, and applications to proteins. J. Phys. Chem. B 2004, 108, 18368–18376. [Google Scholar] [CrossRef]
- Biela, A.; Nasief, N.N.; Betz, M.; Heine, A.; Hangauer, D.; Klebe, G. Dissecting the hydrophobic effect on the molecular level: The role of water, enthalpy, and entropy in ligand binding to thermolysin. Angew. Chem. Int. Ed. 2013, 52, 1822–1828. [Google Scholar] [CrossRef] [PubMed]
- Caron, G.; Ermondi, G. Influence of conformation on GRIND-based three-dimensional quantitative structure-activity relationship (3D-QSAR). J. Med. Chem. 2007, 50, 5039–5042. [Google Scholar] [CrossRef]
- Elisseeff, A.; Pontil, M. Leave-one-out error and stability of learning algorithms with applications. Nato Sci. Ser. Sub Ser. III Comput. Syst. Sci. 2003, 190, 111–130. [Google Scholar]
- Durán Alcaide, Á. Development of High-Performance Algorithms for a New Generation of Versatile Molecular Descriptors. The Pentacle Software; Universitat Pompeu Fabra: Barcelona, Spain, 2010. [Google Scholar]
- Baroni, M.; Costantino, G.; Cruciani, G.; Riganelli, D.; Valigi, R.; Clementi, S. Generating Optimal Linear PLS Estimations (GOLPE): An Advanced Chemometric Tool for Handling 3D-QSAR Problems. Quant. Struct. Act. Relatsh. 1993, 12, 9–20. [Google Scholar] [CrossRef]
- Shrestha, S.K.; Kril, L.M.; Green, K.D.; Kwiatkowski, S.; Sviripa, V.M.; Nickell, J.R.; Dwoskin, L.P.; Watt, D.S.; Garneau-Tsodikova, S. Bis (N-amidinohydrazones) and N-(amidino)-N‘-aryl-bishydrazones: New classes of antibacterial/antifungal agents. Bioorg. Med. Chem. 2017, 25, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Higgins, M.A.; Marcin, L.R.; Zusi, F.C.; Gentles, R.; Ding, M.; Pearce, B.C.; Easton, A.; Kostich, W.A.; Seager, M.A.; Bourin, C. Triazolopyridine ethers as potent, orally active mGlu2 positive allosteric modulators for treating schizophrenia. Bioorg. Med. Chem. 2017, 25, 496–513. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Yang, H.Y.; Park, B.G.; Jung, S.Y.; Park, J.-H.; Park, K.D.; Min, S.-J.; Tae, J.; Yang, H.; Cho, S. Discovery of benzimidazole derivatives as modulators of mitochondrial function: A potential treatment for Alzheimer’s disease. Eur. J. Med. Chem. 2017, 125, 1172–1192. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.D.; Fang, H.; King, H.D.; Iwuagwu, C.I.; McDonald, I.M.; Cook, J.; Zusi, F.C.; Mate, R.A.; Knox, R.J.; Post-Munson, D. Development of 4-Heteroarylamino-1‘azaspiro [oxazole-5, 3‘-bicyclo [2.2. 2] octanes] as α7 Nicotinic Receptor Agonists. ACS Med. Chem. Lett. 2016, 8, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Xu, Q.; Guo, S.; Zuo, R.; Hong, Y.; Luo, Y.; Li, Y.; Gong, P.; Liu, Y. Design, synthesis, and structure-activity relationships of novel imidazo [4–c] pyridine derivatives as potent non-nucleoside inhibitors of hepatitis C virus NS5B. Bioorg. Med. Chem. 2018, 26, 2621–2631. [Google Scholar] [CrossRef] [PubMed]
- Williamson, A.E.; Ylioja, P.M.; Robertson, M.N.; Antonova-Koch, Y.; Avery, V.; Baell, J.B.; Batchu, H.; Batra, S.; Burrows, J.N.; Bhattacharyya, S. Open source drug discovery: Highly potent antimalarial compounds derived from the tres cantos arylpyrroles. ACS Cent. Sci. 2016, 2, 687–701. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, J.I.; Perozo, E.; Allen, T.W. Towards a Structural View of Drug Binding to hERG K+ Channels. Trends Pharmacol. Sci. 2017, 38, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Anwar-Mohamed, A.; Barakat, K.H.; Bhat, R.; Noskov, S.Y.; Tyrrell, D.L.; Tuszynski, J.A.; Houghton, M. A human ether-á-go-go-related (hERG) ion channel atomistic model generated by long supercomputer molecular dynamics simulations and its use in predicting drug cardiotoxicity. Toxicol. Lett. 2014, 230, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Ando, H.; Kuno, M.; Shimizu, H.; Muramatsu, I.; Oiki, S. Coupled K+–water flux through the HERG potassium channel measured by an osmotic pulse method. J. Gen. Physiol. 2005, 126, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Huang, R.; Xia, M.; Shahane, S.; Southall, N.; Wang, Y. Prediction of hERG Liability–Using SVM Classification, Bootstrapping and Jackknifing. Mol. Inform. 2017, 36, 1600126. [Google Scholar] [CrossRef] [PubMed]
- Vaz, R.J.; Kang, J.; Luo, Y.; Rampe, D. Molecular determinants of loperamide and N-desmethyl loperamide binding in the hERG cardiac K+ channel. Bioorg. Med. Chem. Lett. 2018, 28, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Aronov, A.M. Predictive in silico modeling for hERG channel blockers. Drug Discov. Today 2005, 10, 149–155. [Google Scholar] [CrossRef]
- Helliwell, M.V.; Zhang, Y.; El Harchi, A.; Du, C.; Hancox, J.C.; Dempsey, C.E. Structural implications of hERG K+ channel block by a high-affinity minimally structured blocker. J. Biol. Chem. 2018, 293, 7040–7057. [Google Scholar] [CrossRef]
- Cavalli, A.; Buonfiglio, R.; Ianni, C.; Masetti, M.; Ceccarini, L.; Caves, R.; Chang, M.W.; Mitcheson, J.S.; Roberti, M.; Recanatini, M. Computational design and discovery of “minimally structured” hERG blockers. J. Med. Chem. 2012, 55, 4010–4014. [Google Scholar] [CrossRef]
- Chen, A.A.; Pappu, R.V. Parameters of monovalent ions in the AMBER-99 forcefield: Assessment of inaccuracies and proposed improvements. J. Phys. Chem. B 2007, 111, 11884–11887. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Vandenberg, J.I.; Perry, M.D.; Perrin, M.J.; Mann, S.A.; Ke, Y.; Hill, A.P. hERG K+ channels: Structure, function, and clinical significance. Physiol. Rev. 2012, 92, 1393–1478. [Google Scholar] [CrossRef] [PubMed]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef] [PubMed]
- Labute, P. The generalized Born/volume integral implicit solvent model: Estimation of the free energy of hydration using London dispersion instead of atomic surface area. J. Comput. Chem. 2008, 29, 1693–1698. [Google Scholar] [CrossRef] [PubMed]
- Durán, A.; Martínez, G.C.; Pastor, M. Development and validation of AMANDA, a new algorithm for selecting highly relevant regions in molecular interaction fields. J. Chem. Inf. Model. 2008, 48, 1813–1823. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | −ΔG of Docked Complexes (kcal/mol) | −ΔG of Docked Complexes (kcal/ mol) | ||
|---|---|---|---|---|
| Non-Solvated-Open | Solvated-Open | Non-Solvated-Closed | Solvated-Closed | |
| MK-499 | −388 | −372 | −949 | −467 |
| E4031 | −487 | −389 | −946 | −447 |
| Dofetilide | −397 | −385 | −861 | −436 |
| Trimethoprim | −363 | −296 | −736 | −338 |
| hERG Channel Docked Conformatio-ns | Complete Variable GBVI/WSA Score | FFD 1ST Cycle GBVI/WSA Score | Test Set 1 Validation | Test Set 2 Validation | Complete Variable GoldScore | FFD 1ST Cycle GoldScore | Test Set 1 Validation | Test Set 2 Validation | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LVa | q2 | r2 | SDEPb | LV | q2 | r2 | SDEP | R2 | R2 | LV | q2 | r2 | SDEP | LV | q2 | r2 | SDEP | R2 | R2 | |
| Non-solvated -open | 1 | 0.43 | 0.55 | 0.10 | 2 | 0.54 | 0.62 | 0.98 | 0.58 | 0.51 | 3 | 0.35 | 0.47 | 1.14 | 2 | 0.43 | 0.52 | 1.07 | 0.48 | 0.41 |
| Solvated -open | 2 | 0.42 | 0.53 | 1.07 | 2 | 0.44 | 0.55 | 1.05 | 0.52 | 0.41 | 2 | 0.34 | 0.47 | 1.14 | 2 | 0.36 | 0.49 | 1.12 | 0.32 | 0.31 |
| Non-solvated-closed | 3 | 0.42 | 0.62 | 1.07 | 2 | 0.49 | 0.58 | 1.00 | 0.48 | 0.46 | 3 | 0.33 | 0.42 | 1.10 | 2 | 0.38 | 0.50 | 1.02 | 0.38 | 0.36 |
| Solvated-closed | 1 | 0.41 | 0.51 | 1.07 | 2 | 0.43 | 0.53 | 1.05 | 0.36 | 0.38 | 2 | 0.31 | 0.38 | 1.12 | 2 | 0.36 | 0.48 | 1.07 | 0.31 | 0.28 |
| Important Feature | Hotspots Indicating | Impact | Open (cryo_EM) Conformations Distance Å | Closed (Homology) Conformations Distance Å | ||
|---|---|---|---|---|---|---|
| Non-Solvated-Open | Solvated-Open | Non-Solvated-Closed | Solvated-Closed | |||
| DRY-DRY | A particular Distance between two hydrophobic moieties | + | 12.0–12.4 | 12.4–12.8 | 10.0–10.4 | 10.8–11.2 |
| DRY-TIP | A particular Distance between hydrophobic moiety and steric hot spot | + | 13.6–14.0 | 14.4–14.8 | 14.8–15.2 | 15.2–15.6 |
| DRY-O | A particular Distance between hydrophobic moiety and hydrogen bond donor feature | + | 7.2–7.6 | 6.8–7.2 | 11.6–12.0 | 12.0–12.4 |
| N1-N1 | A particular Distance between two hydrogen bond acceptor groups | - | 4.8–5.2 | 5.2–5.6 | 4.0–4.4 | 3.2–3.6 |
| O-N1 | A particular Distance between hydrogen bond donor and hydrogen bond acceptor feature | - | 16.0–16.4 | 16.4–16.8 | 15.6–16.0 | 16.0–16.4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munawar, S.; Vandenberg, J.I.; Jabeen, I. Molecular Docking Guided Grid-Independent Descriptor Analysis to Probe the Impact of Water Molecules on Conformational Changes of hERG Inhibitors in Drug Trapping Phenomenon. Int. J. Mol. Sci. 2019, 20, 3385. https://doi.org/10.3390/ijms20143385
Munawar S, Vandenberg JI, Jabeen I. Molecular Docking Guided Grid-Independent Descriptor Analysis to Probe the Impact of Water Molecules on Conformational Changes of hERG Inhibitors in Drug Trapping Phenomenon. International Journal of Molecular Sciences. 2019; 20(14):3385. https://doi.org/10.3390/ijms20143385
Chicago/Turabian StyleMunawar, Saba, Jamie I. Vandenberg, and Ishrat Jabeen. 2019. "Molecular Docking Guided Grid-Independent Descriptor Analysis to Probe the Impact of Water Molecules on Conformational Changes of hERG Inhibitors in Drug Trapping Phenomenon" International Journal of Molecular Sciences 20, no. 14: 3385. https://doi.org/10.3390/ijms20143385