The Clinical Potential of Oligonucleotide Therapeutics against Pancreatic Cancer

 , ,
, ,
Abstract
:1. Introduction
2. History of Oligonucleotide Therapeutics: From Pioneer Works to Current Diversity
3. Antisense Oligonucleotides
4. SiRNA
5. MicroRNA
6. Aptamers
7. Decoys
8. Clustered, Regularly Interspaced, Short Palindromic Repeats
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ANXA1 | Annexin A1 |
| CCKBR | cholecystokinin B receptor |
| C/EBPα | CCAAT/enhancer-binding protein-α |
| CHST15 | carbohydrate sulfotransferase15 |
| CRISPR | clustered regularly interspaced short palindromic repeats |
| CRISPR/Cas9 | CRISPR/CRISPR-Associated Protein 9 |
| CS-E | chondroitin sulfate-E |
| CypB | cyclophilin B |
| DNA | deoxyribonucleic acid |
| EGFR | epidermal growth factor receptor |
| EUS | endoscopic ultrasonography |
| EUS-FNI | endoscopic ultrasonography guided fine-needle injection |
| GMP | good manufacturing practice |
| HIV-1 | human immunodeficiency virus-1 |
| HMGA1 | high mobility group AT-hook 1 |
| Hsp27 | Heat shock protein 27 |
| IL-8 | interlukein-8 |
| MAZ | myc-associated zinc-finger |
| miRNA | micro RNA |
| MMAE | monomethyl auristatin E |
| MMAF | monomethyl auristatin F |
| NHE | nuclear-hypersensitive element |
| NIH | National Institutes of Health |
| OS | overall survival |
| PAUF | pancreatic adenocarcinoma upregulated factor |
| PD-L1 | programmed cell-death 1 ligand 1 |
| P12FR2 | 2’-fluoropyrimidine modified RNA aptamer |
| RNA | ribonucleic acid |
| RNAi | RNA interference |
| RSV | respiratory syncytial virus |
| SELEX | Systematic Evolution of Ligands by Exponential Enrichment |
| siRNA | small interfering RNA |
| STAT | signal transducer and activator of transcription |
| TGF-β | transforming growth factor-beta |
| XIAP | X-linked inhibitor of apoptosis |
References
- Scherman, D.; Rousseau, A.; Bigey, P.; Escriou, V. Genetic pharmacology: Progresses in siRNA delivery and therapeutic applications. Gene Ther. 2017, 24, 151. [Google Scholar] [CrossRef] [PubMed]
- Aier, I.; Semwal, R.; Sharma, A.; Varadwaj, P.K. A systematic assessment of statistics, risk factors, and underlying features involved in pancreatic cancer. Cancer Epidemiol. 2019, 58, 104–110. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. WJG 2018, 24, 4846–4861. [Google Scholar] [CrossRef] [PubMed]
- Takakura, K.; Shibazaki, Y.; Yoneyama, H.; Fujii, M.; Hashiguchi, T.; Ito, Z.; Kajihara, M.; Misawa, T.; Homma, S.; Ohkusa, T.; et al. Inhibition of Cell Proliferation and Growth of Pancreatic Cancer by Silencing of Carbohydrate Sulfotransferase 15 In Vitro and in a Xenograft Model. PLoS ONE 2015, 10, e0142981. [Google Scholar] [CrossRef] [PubMed]
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 280–284. [Google Scholar] [CrossRef]
- Rapaport, E.; Zamecnik, P.C. Increased incorporation of adenosine into adenine nucleotide pools in serum-deprived mammalian cells. Proc. Natl. Acad. Sci. USA 1978, 75, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Matsukura, M.; Zon, G.; Shinozuka, K.; Robert-Guroff, M.; Shimada, T.; Stein, C.A.; Mitsuya, H.; Wong-Staal, F.; Cohen, J.S.; Broder, S. Regulation of viral expression of human immunodeficiency virus in vitro by an antisense phosphorothioate oligodeoxynucleotide against rev (art/trs) in chronically infected cells. Proc. Natl. Acad. Sci. USA 1989, 86, 4244–4248. [Google Scholar] [CrossRef]
- Cullen, B.R.; Greene, W.C. Regulatory pathways governing HIV-1 replication. Cell 1989, 58, 423–426. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- DeVincenzo, J.; Cehelsky, J.E.; Alvarez, R.; Elbashir, S.; Harborth, J.; Toudjarska, I.; Nechev, L.; Murugaiah, V.; Van Vliet, A.; Vaishnaw, A.K.; et al. Evaluation of the safety, tolerability and pharmacokinetics of ALN-RSV01, a novel RNAi antiviral therapeutic directed against respiratory syncytial virus (RSV). Antivir. Res. 2008, 77, 225–231. [Google Scholar] [CrossRef]
- Almeida, M.I.; Reis, R.M.; Calin, G.A. MicroRNA history: Discovery, recent applications, and next frontiers. Mutat. Res. 2011, 717, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohn, D.B.; Bauer, G.; Rice, C.R.; Rothschild, J.C.; Carbonaro, D.A.; Valdez, P.; Hao, Q.; Zhou, C.; Bahner, I.; Kearns, K.; et al. A clinical trial of retroviral-mediated transfer of a rev-responsive element decoy gene into CD34(+) cells from the bone marrow of human immunodeficiency virus-1-infected children. Blood 1999, 94, 368–371. [Google Scholar] [PubMed]
- MacLeod, A.R.; Crooke, S.T. RNA Therapeutics in Oncology: Advances, Challenges, and Future Directions. J. Clin. Pharmacol. 2017, 57 (Suppl. 10), S43–S59. [Google Scholar] [CrossRef] [PubMed]
- Kole, R.; Krainer, A.R.; Altman, S. RNA therapeutics: Beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012, 11, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Monia, B.P.; Johnston, J.F.; Ecker, D.J.; Zounes, M.A.; Lima, W.F.; Freier, S.M. Selective inhibition of mutant Ha-RAS mRNA expression by antisense oligonucleotides. J. Biol. Chem. 1992, 267, 19954–19962. [Google Scholar] [PubMed]
- Cunningham, C.C.; Holmlund, J.T.; Geary, R.S.; Kwoh, T.J.; Dorr, A.; Johnston, J.F.; Monia, B.; Nemunaitis, J. A Phase I trial of H-ras antisense oligonucleotide ISIS 2503 administered as a continuous intravenous infusion in patients with advanced carcinoma. Cancer 2001, 92, 1265–1271. [Google Scholar] [CrossRef]
- Adjei, A.A.; Dy, G.K.; Erlichman, C.; Reid, J.M.; Sloan, J.A.; Pitot, H.C.; Alberts, S.R.; Goldberg, R.M.; Hanson, L.J.; Atherton, P.J.; et al. A phase I trial of ISIS 2503, an antisense inhibitor of H-ras, in combination with gemcitabine in patients with advanced cancer. Clin. Cancer Res. 2003, 9, 115–123. [Google Scholar] [PubMed]
- Alberts, S.R.; Schroeder, M.; Erlichman, C.; Steen, P.D.; Foster, N.R.; Moore, D.F., Jr.; Rowland, K.M., Jr.; Nair, S.; Tschetter, L.K.; Fitch, T.R. Gemcitabine and ISIS-2503 for patients with locally advanced or metastatic pancreatic adenocarcinoma: A North Central Cancer Treatment Group phase II trial. J. Clin. Oncol. 2004, 22, 4944–4950. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Friess, H.; Yamanaka, Y.; Buchler, M.; Ebert, M.; Beger, H.G.; Gold, L.I.; Korc, M. Enhanced expression of transforming growth factor beta isoforms in pancreatic cancer correlates with decreased survival. Gastroenterology 1993, 105, 1846–1856. [Google Scholar] [CrossRef]
- von Bernstorff, W.; Voss, M.; Freichel, S.; Schmid, A.; Vogel, I.; Johnk, C.; Henne-Bruns, D.; Kremer, B.; Kalthoff, H. Systemic and local immunosuppression in pancreatic cancer patients. Clin. Cancer Res. 2001, 7 (Suppl. 3), 925s–932s. [Google Scholar] [PubMed]
- Schlingensiepen, K.H.; Schlingensiepen, R.; Steinbrecher, A.; Hau, P.; Bogdahn, U.; Fischer-Blass, B.; Jachimczak, P. Targeted tumor therapy with the TGF-beta 2 antisense compound AP 12009. Cytokine Growth Factor Rev. 2006, 17, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Jaschinski, F.; Rothhammer, T.; Jachimczak, P.; Seitz, C.; Schneider, A.; Schlingensiepen, K.H. The antisense oligonucleotide trabedersen (AP 12009) for the targeted inhibition of TGF-beta2. Curr. Pharm. Biotechnol. 2011, 12, 2203–2213. [Google Scholar] [CrossRef] [PubMed]
- Schlingensiepen, K.H.; Jaschinski, F.; Lang, S.A.; Moser, C.; Geissler, E.K.; Schlitt, H.J.; Kielmanowicz, M.; Schneider, A. Transforming growth factor-beta 2 gene silencing with trabedersen (AP 12009) in pancreatic cancer. Cancer Sci. 2011, 102, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- D’Cruz, O.J.; Qazi, S.; Hwang, L.; Ng, K.; Trieu, V. Impact of targeting transforming growth factor beta-2 with antisense OT-101 on the cytokine and chemokine profile in patients with advanced pancreatic cancer. Oncotargets Ther. 2018, 11, 2779–2796. [Google Scholar] [CrossRef] [PubMed]
- Furqan, M.; Akinleye, A.; Mukhi, N.; Mittal, V.; Chen, Y.; Liu, D. STAT inhibitors for cancer therapy. J. Hematol. Oncol. 2013, 6, 90. [Google Scholar] [CrossRef]
- Hong, D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; Zhou, T.; Schmidt, J.; Jo, M.; et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015, 7, 314ra185. [Google Scholar] [CrossRef]
- Reilley, M.J.; McCoon, P.; Cook, C.; Lyne, P.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; et al. STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pretreated lymphoma: Results of a phase 1b trial. J. Immunother. Cancer 2018, 6, 119. [Google Scholar] [CrossRef]
- Odate, S.; Veschi, V.; Yan, S.; Lam, N.; Woessner, R.; Thiele, C.J. Inhibition of STAT3 with the Generation 2.5 Antisense Oligonucleotide, AZD9150, Decreases Neuroblastoma Tumorigenicity and Increases Chemosensitivity. Clin. Cancer Res. 2017, 23, 1771–1784. [Google Scholar] [CrossRef]
- Stewart, R.; Morrow, M.; Hammond, S.A.; Mulgrew, K.; Marcus, D.; Poon, E.; Watkins, A.; Mullins, S.; Chodorge, M.; Andrews, J.; et al. Identification and Characterization of MEDI4736, an Antagonistic Anti-PD-L1 Monoclonal Antibody. Cancer Immunol. Res. 2015, 3, 1052–1062. [Google Scholar] [CrossRef]
- Cheung, H.H.; LaCasse, E.C.; Korneluk, R.G. X-linked inhibitor of apoptosis antagonism: Strategies in cancer treatment. Clin. Cancer Res. 2006, 12, 3238–3242. [Google Scholar] [CrossRef] [PubMed]
- Lacasse, E.C.; Kandimalla, E.R.; Winocour, P.; Sullivan, T.; Agrawal, S.; Gillard, J.W.; Durkin, J. Application of XIAP antisense to cancer and other proliferative disorders: Development of AEG35156/GEM640. Ann. N. Y. Acad. Sci. 2005, 1058, 215–234. [Google Scholar] [CrossRef] [PubMed]
- LaCasse, E.C.; Cherton-Horvat, G.G.; Hewitt, K.E.; Jerome, L.J.; Morris, S.J.; Kandimalla, E.R.; Yu, D.; Wang, H.; Wang, W.; Zhang, R.; et al. Preclinical characterization of AEG35156/GEM 640, a second-generation antisense oligonucleotide targeting X-linked inhibitor of apoptosis. Clin. Cancer Res. 2006, 12, 5231–5241. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, D.; Chalasani, P.; Rensvold, D.; Kurtin, S.; Pretzinger, C.; Jolivet, J.; Ramanathan, R.K.; Von Hoff, D.D.; Weiss, G.J. Phase I trial of AEG35156 an antisense oligonucleotide to XIAP plus gemcitabine in patients with metastatic pancreatic ductal adenocarcinoma. Am. J. Clin. Oncol. 2013, 36, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Melle, C.; Ernst, G.; Escher, N.; Hartmann, D.; Schimmel, B.; Bleul, A.; Thieme, H.; Kaufmann, R.; Felix, K.; Friess, H.M.; et al. Protein profiling of microdissected pancreas carcinoma and identification of HSP27 as a potential serum marker. Clin. Chem. 2007, 53, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Mori-Iwamoto, S.; Kuramitsu, Y.; Ryozawa, S.; Mikuria, K.; Fujimoto, M.; Maehara, S.; Maehara, Y.; Okita, K.; Nakamura, K.; Sakaida, I. Proteomics finding heat shock protein 27 as a biomarker for resistance of pancreatic cancer cells to gemcitabine. Int. J. Oncol. 2007, 31, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Baylot, V.; Andrieu, C.; Katsogiannou, M.; Taieb, D.; Garcia, S.; Giusiano, S.; Acunzo, J.; Iovanna, J.; Gleave, M.; Garrido, C.; et al. OGX-427 inhibits tumor progression and enhances gemcitabine chemotherapy in pancreatic cancer. Cell Death Dis. 2011, 2, e221. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.H.; Murphy, P.B.; Peyton, J.D.; Shipley, D.L.; Al-Hazzouri, A.; Rodriguez, F.A.; Womack, M.S.; Xiong, H.Q.; Waterhouse, D.M.; Tempero, M.A.; et al. A Randomized, Double-Blinded, Phase II Trial of Gemcitabine and Nab-Paclitaxel Plus Apatorsen or Placebo in Patients with Metastatic Pancreatic Cancer: The RAINIER Trial. Oncologist 2017, 22, 1427–e129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuckerman, J.E.; Davis, M.E. Clinical experiences with systemically administered siRNA-based therapeutics in cancer. Nat. Rev. Drug Discov. 2015, 14, 843–856. [Google Scholar] [CrossRef]
- Kim, D.H.; Rossi, J.J. Strategies for silencing human disease using RNA interference. Nat. Rev. Genet. 2007, 8, 173–184. [Google Scholar] [CrossRef]
- Chang, H. RNAi-mediated knockdown of target genes: A promising strategy for pancreatic cancer research. Cancer Gene Ther. 2007, 14, 677–685. [Google Scholar] [CrossRef]
- Leenders, F.; Mopert, K.; Schmiedeknecht, A.; Santel, A.; Czauderna, F.; Aleku, M.; Penschuck, S.; Dames, S.; Sternberger, M.; Rohl, T.; et al. PKN3 is required for malignant prostate cell growth downstream of activated PI 3-kinase. EMBO J. 2004, 23, 3303–3313. [Google Scholar] [CrossRef] [Green Version]
- Aleku, M.; Schulz, P.; Keil, O.; Santel, A.; Schaeper, U.; Dieckhoff, B.; Janke, O.; Endruschat, J.; Durieux, B.; Roder, N.; et al. Atu027, a liposomal small interfering RNA formulation targeting protein kinase N3, inhibits cancer progression. Cancer Res. 2008, 68, 9788–9798. [Google Scholar] [CrossRef] [PubMed]
- Schultheis, B.; Strumberg, D.; Santel, A.; Vank, C.; Gebhardt, F.; Keil, O.; Lange, C.; Giese, K.; Kaufmann, J.; Khan, M.; et al. First-in-human phase I study of the liposomal RNA interference therapeutic Atu027 in patients with advanced solid tumors. J. Clin. Oncol. 2014, 32, 4141–4148. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.A.; Pasca di Magliano, M. Kras as a key oncogene and therapeutic target in pancreatic cancer. Front. Physiol. 2013, 4, 407. [Google Scholar] [CrossRef]
- Singh, A.; Greninger, P.; Rhodes, D.; Koopman, L.; Violette, S.; Bardeesy, N.; Settleman, J. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell 2009, 15, 489–500. [Google Scholar] [CrossRef]
- Rejiba, S.; Wack, S.; Aprahamian, M.; Hajri, A. K-ras oncogene silencing strategy reduces tumor growth and enhances gemcitabine chemotherapy efficacy for pancreatic cancer treatment. Cancer Sci. 2007, 98, 1128–1136. [Google Scholar] [CrossRef]
- Zorde Khvalevsky, E.; Gabai, R.; Rachmut, I.H.; Horwitz, E.; Brunschwig, Z.; Orbach, A.; Shemi, A.; Golan, T.; Domb, A.J.; Yavin, E.; et al. Mutant KRAS is a druggable target for pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20723–20728. [Google Scholar] [CrossRef] [Green Version]
- Eser, S.; Schnieke, A.; Schneider, G.; Saur, D. Oncogenic KRAS signalling in pancreatic cancer. Br. J. Cancer 2014, 111, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, Y.; Zhang, L.; Huang, L. Nanoparticle-delivered transforming growth factor-beta siRNA enhances vaccination against advanced melanoma by modifying tumor microenvironment. ACS Nano 2014, 8, 3636–3645. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Transport of molecules, particles, and cells in solid tumors. Annu. Rev. Biomed. Eng. 1999, 1, 241–263. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Khvalevsky, E.Z.; Hubert, A.; Gabai, R.M.; Hen, N.; Segal, A.; Domb, A.; Harari, G.; David, E.B.; Raskin, S.; et al. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget 2015, 6, 24560–24570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramot, Y.; Rotkopf, S.; Gabai, R.M.; Zorde Khvalevsky, E.; Muravnik, S.; Marzoli, G.A.; Domb, A.J.; Shemi, A.; Nyska, A. Preclinical Safety Evaluation in Rats of a Polymeric Matrix Containing an siRNA Drug Used as a Local and Prolonged Delivery System for Pancreatic Cancer Therapy. Toxicol. Pathol. 2016, 44, 856–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Mendt, M.; Kamerkar, S.; Sugimoto, H.; McAndrews, K.M.; Wu, C.C.; Gagea, M.; Yang, S.; Blanko, E.V.R.; Peng, Q.; Ma, X.; et al. Generation and testing of clinical-grade exosomes for pancreatic cancer. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Valkenburg, K.C.; de Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef]
- Neesse, A.; Krug, S.; Gress, T.M.; Tuveson, D.A.; Michl, P. Emerging concepts in pancreatic cancer medicine: Targeting the tumor stroma. Oncotargets Ther. 2013, 7, 33–43. [Google Scholar] [CrossRef]
- Fuster, M.M.; Esko, J.D. The sweet and sour of cancer: Glycans as novel therapeutic targets. Nat. Rev. Cancer 2005, 5, 526–542. [Google Scholar] [CrossRef]
- Sugahara, K.N.; Hirata, T.; Tanaka, T.; Ogino, S.; Takeda, M.; Terasawa, H.; Shimada, I.; Tamura, J.; ten Dam, G.B.; van Kuppevelt, T.H.; et al. Chondroitin sulfate E fragments enhance CD44 cleavage and CD44-dependent motility in tumor cells. Cancer Res. 2008, 68, 7191–7199. [Google Scholar] [CrossRef] [PubMed]
- Mizumoto, S.; Sugahara, K. Glycosaminoglycans are functional ligands for receptor for advanced glycation end-products in tumors. FEBS J. 2013, 280, 2462–2470. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.; Mikami, T.; Uyama, T.; Mizuguchi, S.; Nomura, K.; Kitagawa, H. Recent advances in the structural biology of chondroitin sulfate and dermatan sulfate. Curr. Opin. Struct. Biol. 2003, 13, 612–620. [Google Scholar] [CrossRef] [PubMed]
- Ito, Z.; Takakura, K.; Suka, M.; Kanai, T.; Saito, R.; Fujioka, S.; Kajihara, M.; Yanagisawa, H.; Misawa, T.; Akiba, T.; et al. Prognostic impact of carbohydrate sulfotransferase 15 in patients with pancreatic ductal adenocarcinoma. Oncol. Lett. 2017, 13, 4799–4805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, M.; Matsukawa, M.; Fujii, Y.; Matsuda, Y.; Arai, T.; Ochiai, Y.; Itoi, T.; Yahagi, N. Effects of EUS-guided intratumoral injection of oligonucleotide STNM01 on tumor growth, histology, and overall survival in patients with unresectable pancreatic cancer. Gastrointest. Endosc. 2018, 87, 1126–1131. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.U.; Prieto-Vila, M.; Kohama, I.; Ochiya, T. Development of miRNA-based therapeutic approaches for cancer patients. Cancer Sci. 2019, 110, 1140. [Google Scholar] [CrossRef] [PubMed]
- Gurbuz, N.; Ozpolat, B. MicroRNA-based Targeted Therapeutics in Pancreatic Cancer. Anticancer Res. 2019, 39, 529–532. [Google Scholar] [CrossRef] [Green Version]
- Mollaei, H.; Safaralizadeh, R.; Rostami, Z. MicroRNA replacement therapy in cancer. J. Cell. Physiol. 2019, 234, 12369–12384. [Google Scholar] [CrossRef]
- Ors-Kumoglu, G.; Gulce-Iz, S.; Biray-Avci, C. Therapeutic microRNAs in human cancer. Cytotechnology 2019, 71, 411–425. [Google Scholar] [CrossRef]
- Slotwinski, R.; Lech, G.; Slotwinska, S.M. MicroRNAs in pancreatic cancer diagnosis and therapy. Cent. Eur. J. Immunol. 2018, 43, 314–324. [Google Scholar] [CrossRef] [Green Version]
- Tesfaye, A.A.; Azmi, A.S.; Philip, P.A. miRNA and Gene Expression in Pancreatic Ductal Adenocarcinoma. Am. J. Pathol. 2019, 189, 58–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baradaran, B.; Shahbazi, R.; Khordadmehr, M. Dysregulation of key microRNAs in pancreatic cancer development. Biomed. Pharmacother. Biomed. Pharmacother. 2019, 109, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S. MicroRNAs as Therapeutic Agents: The Future of the Battle against Cancer. Curr. Top. Med. Chem. 2018, 18, 2544–2554. [Google Scholar] [CrossRef] [PubMed]
- Kwok, G.T.; Zhao, J.T.; Weiss, J.; Mugridge, N.; Brahmbhatt, H.; MacDiarmid, J.A.; Robinson, B.G.; Sidhu, S.B. Translational applications of microRNAs in cancer, and therapeutic implications. Non-Coding RNA Res. 2017, 2, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Ghasabi, M.; Mansoori, B.; Mohammadi, A.; Duijf, P.H.; Shomali, N.; Shirafkan, N.; Mokhtarzadeh, A.; Baradaran, B. MicroRNAs in cancer drug resistance: Basic evidence and clinical applications. J. Cell. Physiol. 2019, 234, 2152–2168. [Google Scholar] [CrossRef] [PubMed]
- Dragomir, M.; Mafra, A.C.P.; Dias, S.M.G.; Vasilescu, C.; Calin, G.A. Using microRNA Networks to Understand Cancer. Int. J. Mol. Sci. 2018, 19, 1871. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Fesler, A.; Wang, H.; Ju, J. microRNA based prognostic biomarkers in pancreatic Cancer. Biomark. Res. 2018, 6, 18. [Google Scholar] [CrossRef]
- Sun, X.; Zhou, X.; Zhang, Y.; Zhu, X.; Liu, H. Systematic Review and Meta-Analysis of Diagnostic Accuracy of miRNAs in Patients with Pancreatic Cancer. Dis. Markers 2018, 2018, 6292396. [Google Scholar] [CrossRef]
- Kian, R.; Moradi, S.; Ghorbian, S. Role of components of microRNA machinery in carcinogenesis. Exp. Oncol. 2018, 40, 2–9. [Google Scholar] [CrossRef]
- Hosseinahli, N.; Aghapour, M.; Duijf, P.H.G.; Baradaran, B. Treating cancer with microRNA replacement therapy: A literature review. J. Cell. Physiol. 2018, 233, 5574–5588. [Google Scholar] [CrossRef]
- Vannini, I.; Fanini, F.; Fabbri, M. Emerging roles of microRNAs in cancer. Curr. Opin. Genet. Dev. 2018, 48, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Petrovic, N.; Ergun, S. miRNAs as Potential Treatment Targets and Treatment Options in Cancer. Mol. Diagn. Ther. 2018, 22, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.D.; Chang, S. Development of Novel Therapeutic Agents by Inhibition of Oncogenic MicroRNAs. Int. J. Mol. Sci. 2017, 19, 65. [Google Scholar] [CrossRef]
- Qadir, M.I.; Faheem, A. miRNA: A Diagnostic and Therapeutic Tool for Pancreatic Cancer. Crit. Rev. Eukaryot. Gene Expr. 2017, 27, 197–204. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Shirjang, S.; Baradaran, B. MicroRNAs in the Diagnosis and Treatment of Cancer. Immunol. Investig. 2017, 46, 880–897. [Google Scholar] [CrossRef]
- Drusco, A.; Croce, C.M. MicroRNAs and Cancer: A Long Story for Short RNAs. Adv. Cancer Res. 2017, 135, 1–24. [Google Scholar]
- Schultz, N.A.; Dehlendorff, C.; Jensen, B.V.; Bjerregaard, J.K.; Nielsen, K.R.; Bojesen, S.E.; Calatayud, D.; Nielsen, S.E.; Yilmaz, M.; Hollander, N.H.; et al. MicroRNA biomarkers in whole blood for detection of pancreatic cancer. JAMA 2014, 311, 392–404. [Google Scholar] [CrossRef]
- Su, Q.; Zhu, E.C.; Qu, Y.L.; Wang, D.Y.; Qu, W.W.; Zhang, C.G.; Wu, T.; Gao, Z.H. Serum level of co-expressed hub miRNAs as diagnostic and prognostic biomarkers for pancreatic ductal adenocarcinoma. J. Cancer 2018, 9, 3991–3999. [Google Scholar] [CrossRef] [PubMed]
- Deng, T.; Yuan, Y.; Zhang, C.; Zhang, C.; Yao, W.; Wang, C.; Liu, R.; Ba, Y. Identification of Circulating MiR-25 as a Potential Biomarker for Pancreatic Cancer Diagnosis. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2016, 39, 1716–1722. [Google Scholar] [CrossRef]
- Liu, R.; Chen, X.; Du, Y.; Yao, W.; Shen, L.; Wang, C.; Hu, Z.; Zhuang, R.; Ning, G.; Zhang, C.; et al. Serum microRNA expression profile as a biomarker in the diagnosis and prognosis of pancreatic cancer. Clin. Chem. 2012, 58, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Mayer, G. The chemical biology of aptamers. Angew. Chem. 2009, 48, 2672–2689. [Google Scholar] [CrossRef] [PubMed]
- Ireson, C.R.; Kelland, L.R. Discovery and development of anticancer aptamers. Mol. Cancer Ther. 2006, 5, 2957–2962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djordjevic, M. SELEX experiments: New prospects, applications and data analysis in inferring regulatory pathways. Biomol. Eng. 2007, 24, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Sung, H.J.; Kim, S.; Kim, E.O.; Lee, J.W.; Moon, J.Y.; Choi, K.; Jung, J.E.; Lee, Y.; Koh, S.S.; et al. An RNA aptamer that specifically binds pancreatic adenocarcinoma up-regulated factor inhibits migration and growth of pancreatic cancer cells. Cancer Lett. 2011, 313, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.A.; Lee, Y.; Jung, D.E.; Park, K.H.; Park, J.Y.; Gang, J.; Jeon, S.B.; Park, E.C.; Kim, Y.G.; Lee, B.; et al. Pancreatic adenocarcinoma up-regulated factor (PAUF), a novel up-regulated secretory protein in pancreatic ductal adenocarcinoma. Cancer Sci. 2009, 100, 828–836. [Google Scholar] [CrossRef]
- Ray, P.; Rialon-Guevara, K.L.; Veras, E.; Sullenger, B.A.; White, R.R. Comparing human pancreatic cell secretomes by in vitro aptamer selection identifies cyclophilin B as a candidate pancreatic cancer biomarker. J. Clin. Investig. 2012, 122, 1734–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, P.; Sullenger, B.A.; White, R.R. Further characterization of the target of a potential aptamer biomarker for pancreatic cancer: Cyclophilin B and its posttranslational modifications. Nucleic Acid Ther. 2013, 23, 435–442. [Google Scholar] [CrossRef]
- Xiong, H.Q.; Rosenberg, A.; LoBuglio, A.; Schmidt, W.; Wolff, R.A.; Deutsch, J.; Needle, M.; Abbruzzese, J.L. Cetuximab, a monoclonal antibody targeting the epidermal growth factor receptor, in combination with gemcitabine for advanced pancreatic cancer: A multicenter phase II Trial. J. Clin. Oncol. 2004, 22, 2610–2616. [Google Scholar] [CrossRef]
- Ray, P.; Cheek, M.A.; Sharaf, M.L.; Li, N.; Ellington, A.D.; Sullenger, B.A.; Shaw, B.R.; White, R.R. Aptamer-mediated delivery of chemotherapy to pancreatic cancer cells. Nucleic Acid Ther. 2012, 22, 295–305. [Google Scholar] [CrossRef]
- Zhang, J.; Li, S.; Liu, F.; Zhou, L.; Shao, N.; Zhao, X. SELEX aptamer used as a probe to detect circulating tumor cells in peripheral blood of pancreatic cancer patients. PLoS ONE 2015, 10, e0121920. [Google Scholar] [CrossRef]
- Champanhac, C.; Teng, I.T.; Cansiz, S.; Zhang, L.; Wu, X.; Zhoa, Z.; Fu, T.; Tan, W. Development of a panel of DNA Aptamers with High Affinity for Pancreatic Ductal Adenocarcinoma. Sci. Rep. 2015, 5, 16788. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhao, Z.; Bai, H.; Fu, T.; Yang, C.; Hu, X.; Liu, Q.; Champanhac, C.; Teng, I.T.; Ye, M.; et al. DNA Aptamer Selected against Pancreatic Ductal Adenocarcinoma for in vivo Imaging and Clinical Tissue Recognition. Theranostics 2015, 5, 985–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dua, P.; Kim, S.; Lee, D.K. ALPPL2 Aptamer-Mediated Targeted Delivery of 5-Fluoro-2′-Deoxyuridine to Pancreatic Cancer. Nucleic Acid Ther. 2015, 25, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Huang, K.W.; Reebye, V.; Mintz, P.; Tien, Y.W.; Lai, H.S.; Saetrom, P.; Reccia, I.; Swiderski, P.; Armstrong, B.; et al. Targeted Delivery of C/EBPalpha-saRNA by Pancreatic Ductal Adenocarcinoma-specific RNA Aptamers Inhibits Tumor Growth In Vivo. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 1106–1116. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Huang, K.W.; Reebye, V.; Spalding, D.; Przytycka, T.M.; Wang, Y.; Swiderski, P.; Li, L.; Armstrong, B.; Reccia, I.; et al. Aptamer-Drug Conjugates of Active Metabolites of Nucleoside Analogs and Cytotoxic Agents Inhibit Pancreatic Tumor Cell Growth. Mol. Ther. Nucleic Acids 2017, 6, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Rossi, J.J. Treatment of Pancreatic Cancer by Aptamer Conjugated C/EBPalpha-saRNA. Adv. Exp. Med. Biol. 2017, 983, 173–188. [Google Scholar] [PubMed]
- Kratschmer, C.; Levy, M. Targeted Delivery of Auristatin-Modified Toxins to Pancreatic Cancer Using Aptamers. Mol. Ther. Nucleic Acids 2018, 10, 227–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetze, J.P.; Nielsen, F.C.; Burcharth, F.; Rehfeld, J.F. Closing the gastrin loop in pancreatic carcinoma: Coexpression of gastrin and its receptor in solid human pancreatic adenocarcinoma. Cancer 2000, 88, 2487–2494. [Google Scholar] [CrossRef]
- Clawson, G.A.; Abraham, T.; Pan, W.; Tang, X.; Linton, S.S.; McGovern, C.O.; Loc, W.S.; Smith, J.P.; Butler, P.J.; Kester, M.; et al. A Cholecystokinin B Receptor-Specific DNA Aptamer for Targeting Pancreatic Ductal Adenocarcinoma. Nucleic Acid Ther. 2017, 27, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.J.; Lee, H.S.; Jung, D.E.; Kim, J.M.; Song, S.Y. The DNA aptamer binds stemness-enriched cancer cells in pancreatic cancer. J. Mol. Recognit. JMR 2017, 30, e2591. [Google Scholar] [CrossRef]
- Park, J.Y.; Cho, Y.L.; Chae, J.R.; Moon, S.H.; Cho, W.G.; Choi, Y.J.; Lee, S.J.; Kang, W.J. Gemcitabine-Incorporated G-Quadruplex Aptamer for Targeted Drug Delivery into Pancreas Cancer. Mol. Ther. Nucleic Acids 2018, 12, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Cogoi, S.; Zorzet, S.; Rapozzi, V.; Geci, I.; Pedersen, E.B.; Xodo, L.E. MAZ-binding G4-decoy with locked nucleic acid and twisted intercalating nucleic acid modifications suppresses KRAS in pancreatic cancer cells and delays tumor growth in mice. Nucleic Acids Res. 2013, 41, 4049–4064. [Google Scholar] [CrossRef] [PubMed]
- Cogoi, S.; Jakobsen, U.; Pedersen, E.B.; Vogel, S.; Xodo, L.E. Lipid-modified G4-decoy oligonucleotide anchored to nanoparticles: Delivery and bioactivity in pancreatic cancer cells. Sci. Rep. 2016, 6, 38468. [Google Scholar] [CrossRef] [PubMed]
- Liau, S.S.; Whang, E. HMGA1 is a molecular determinant of chemoresistance to gemcitabine in pancreatic adenocarcinoma. Clin. Cancer Res. 2008, 14, 1470–1477. [Google Scholar] [CrossRef] [PubMed]
- Hassan, F.; Ni, S.; Arnett, T.C.; McKell, M.C.; Kennedy, M.A. Adenovirus-Mediated Delivery of Decoy Hyper Binding Sites Targeting Oncogenic HMGA1 Reduces Pancreatic and Liver Cancer Cell Viability. Mol. Ther. Oncolytics 2018, 8, 52–61. [Google Scholar] [CrossRef] [Green Version]
- Knott, G.J.; Doudna, J.A. CRISPR-Cas guides the future of genetic engineering. Science 2018, 361, 866–869. [Google Scholar] [CrossRef] [Green Version]
- Vorvis, C.; Hatziapostolou, M.; Mahurkar-Joshi, S.; Koutsioumpa, M.; Williams, J.; Donahue, T.R.; Poultsides, G.A.; Eibl, G.; Iliopoulos, D. Transcriptomic and CRISPR/Cas9 technologies reveal FOXA2 as a tumor suppressor gene in pancreatic cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G1124–G1137. [Google Scholar] [CrossRef]
- Belvedere, R.; Saggese, P.; Pessolano, E.; Memoli, D.; Bizzarro, V.; Rizzo, F.; Parente, L.; Weisz, A.; Petrella, A. miR-196a Is Able to Restore the Aggressive Phenotype of Annexin A1 Knock-Out in Pancreatic Cancer Cells by CRISPR/Cas9 Genome Editing. Int. J. Mol. Sci. 2018, 19, 1967. [Google Scholar] [CrossRef]
- Steinhart, Z.; Pavlovic, Z.; Chandrashekhar, M.; Hart, T.; Wang, X.; Zhang, X.; Robitaille, M.; Brown, K.R.; Jaksani, S.; Overmeer, R.; et al. Genome-wide CRISPR screens reveal a Wnt-FZD5 signaling circuit as a druggable vulnerability of RNF43-mutant pancreatic tumors. Nat. Med. 2017, 23, 60–68. [Google Scholar] [CrossRef]
- Szlachta, K.; Kuscu, C.; Tufan, T.; Adair, S.J.; Shang, S.; Michaels, A.D.; Mullen, M.G.; Fischer, N.L.; Yang, J.; Liu, L.; et al. CRISPR knockout screening identifies combinatorial drug targets in pancreatic cancer and models cellular drug response. Nat. Commun. 2018, 9, 4275. [Google Scholar] [CrossRef]
- Bakke, J.; Wright, W.C.; Zamora, A.E.; Oladimeji, P.; Crawford, J.C.; Brewer, C.T.; Autry, R.J.; Evans, W.E.; Thomas, P.G.; Chen, T. Genome-wide CRISPR screen reveals PSMA6 to be an essential gene in pancreatic cancer cells. BMC Cancer 2019, 19, 253. [Google Scholar] [CrossRef] [PubMed]



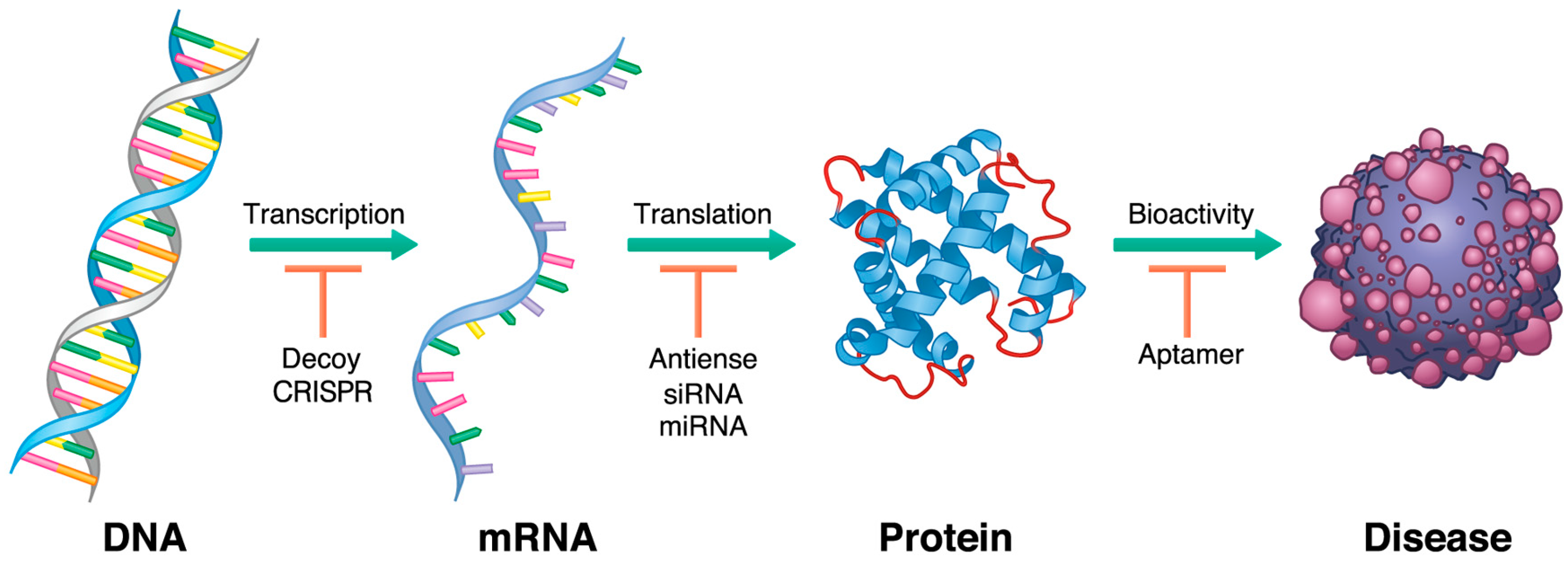{kind=link}
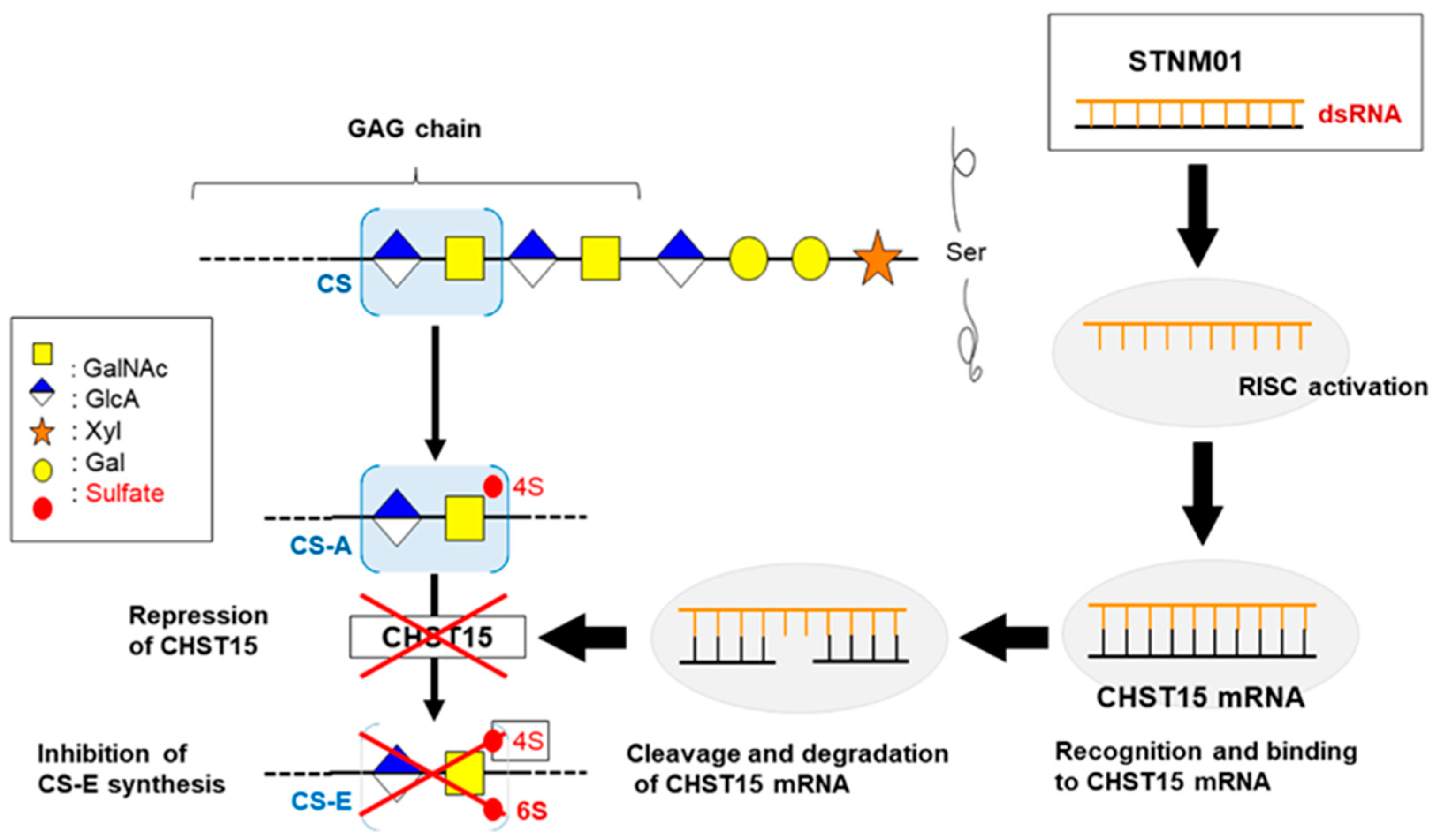{kind=link}
| Trial Identifer | The Name of Tested Oligonucleotide | Target Molecule | Category of Agent | Enrollment | Organizing Location | Study Phase |
|---|---|---|---|---|---|---|
| NCT00005594 | ISIS 2503 | Hras | Antisense | 48 | United States | Phase 2 |
| NCT00844064 | AP 12009/OT-101 | TGF-β2 | Antisense | 37 | Germany | Phase 1/2 |
| NCT02983578 | AZD9150 | STAT3 | Antisense | 75 (estimated) | United States | Phase 2 |
| NCT00557596 | AEG35156 | XIAP | Antisense | 14 | United States | Phase 1 |
| NCT01844817 | OGX-427/apatorsen | Hsp27 | Antisense | 132 | United States | Phase 2 |
| NCT01808638 | Atu027 | PKN3 | siRNA | 29 | Germany | Phase 1/2 |
| NCT01188785 | siG12D LODER | KrasG12D | siRNA | 15 | United States | Phase 1 |
| NCT01676259 | siG12D LODER | KrasG12D | siRNA | 80 (estimated) | United States | Phase 2 |
| NCT03608631 | iExosomes | KrasG12D | siRNA | 28 (estimated) | United States | Phase 1 |
| NCT03432624 | Detection Kit | MiR-25 | miRNA | 750 (estimated) | China |
| Antisense | siRNA | miRNA | Aptamer | Decoy | CRISPR | |
|---|---|---|---|---|---|---|
| Structure | ssDNA/RNA | dsRNA | dsRNA, shRNA | ssDNA/RNA | dsDNA | sgRNA |
| Target | mRNA, miRNA | mRNA | mRNA | Protein | Protein | dsDNA |
| Pre-mRNA | (transcription factor) | |||||
| Active site | Intracellular | Intracellular | Intracellular | Extracellular | Intracellular | Intracellular |
| Action | mRNA decay | miRNA | Functional | Transcriptional | Adaptive | |
| Mechanism | Splicing inhibition | mRNA decay | Complement | Inhibition | Inhibition | Immunity |
| miRNA inhibition |
| No. | Target Agent | How to Work? | Ref. |
|---|---|---|---|
| 1 | P12FR2 | Oncosuppressor | [94] |
| 2 | C/EBPα-saRNA | Oncosuppressor | [106] |
| 3 | cyclophilin B | Biomarker | [96,97] |
| 4 | circulating tumor cells | Biomarker | [100] |
| 5 | EGFR | Targeted delivery | [99] |
| 6 | ALPPL2 | Targeted delivery | [103] |
| 7 | ApDCs P19 | Targeted delivery | [105] |
| 8 | Auristatin-Modified Toxins | Targeted delivery | [107] |
| 9 | CCKBR | Targeted delivery | [109] |
| 10 | APTA-12 | Targeted delivery | [111] |
| 11 | XQ-2d | Detector | [102] |
| 12 | cancer stem cells | Detector | [110] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takakura, K.; Kawamura, A.; Torisu, Y.; Koido, S.; Yahagi, N.; Saruta, M. The Clinical Potential of Oligonucleotide Therapeutics against Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 3331. https://doi.org/10.3390/ijms20133331
Takakura K, Kawamura A, Torisu Y, Koido S, Yahagi N, Saruta M. The Clinical Potential of Oligonucleotide Therapeutics against Pancreatic Cancer. International Journal of Molecular Sciences. 2019; 20(13):3331. https://doi.org/10.3390/ijms20133331
Chicago/Turabian StyleTakakura, Kazuki, Atsushi Kawamura, Yuichi Torisu, Shigeo Koido, Naohisa Yahagi, and Masayuki Saruta. 2019. "The Clinical Potential of Oligonucleotide Therapeutics against Pancreatic Cancer" International Journal of Molecular Sciences 20, no. 13: 3331. https://doi.org/10.3390/ijms20133331