IGF1R Is a Potential New Therapeutic Target for HGNET-BCOR Brain Tumor Patients

 , and
, and 
Abstract
:1. Introduction
2. Results
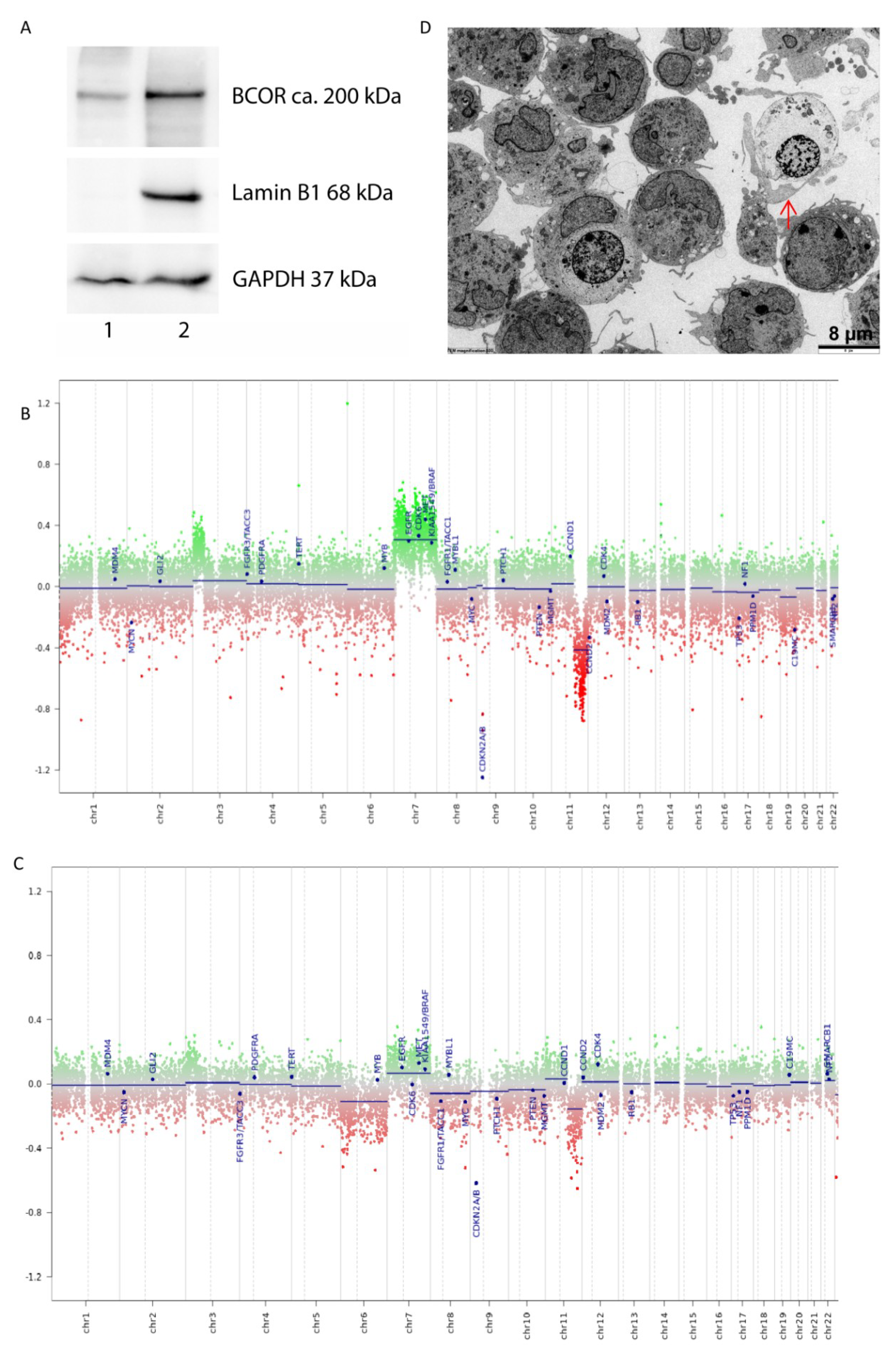
2.1. Characterization of the HGNET-BCOR in Vitro Model PhKh1
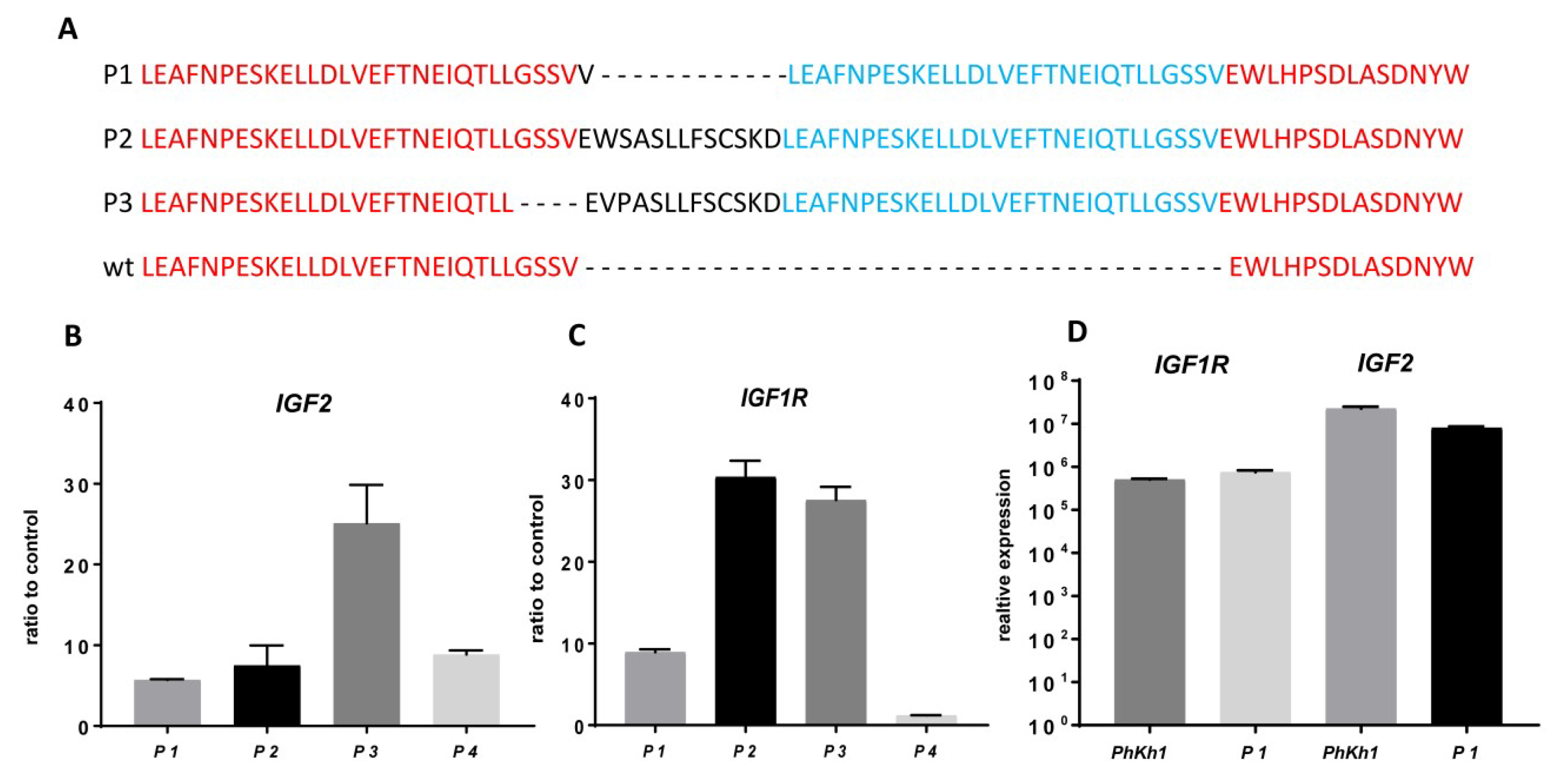
2.2. IGF2 and IGF1R are Highly Expressed in HGNET-BCOR
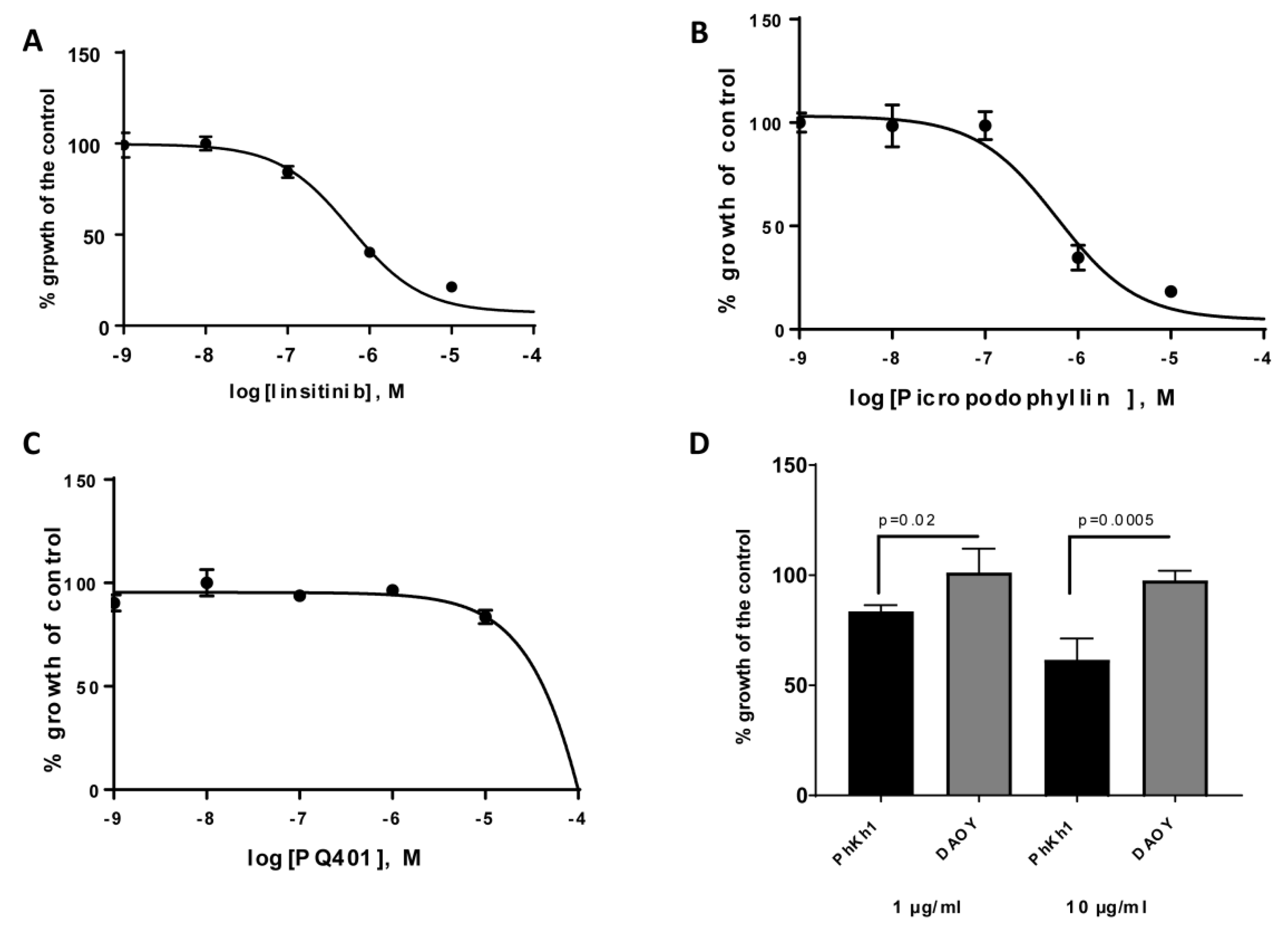
2.3. HGNET-BCOR is Responsive to Actinomycin D, Vinca Alkaloids, and IGF1R Inhibitors
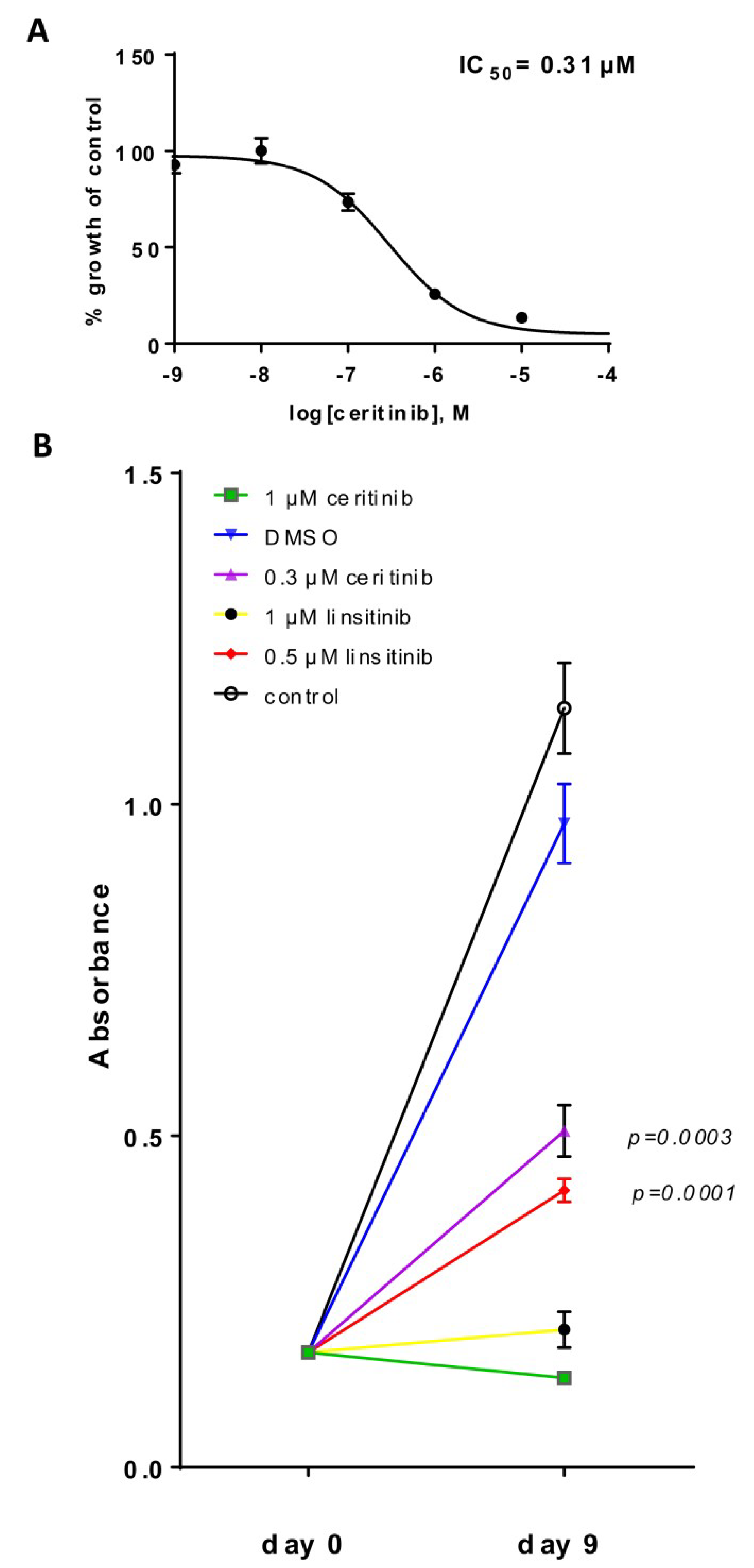
2.4. HGNET-BCOR is Responsive to Ceritinib
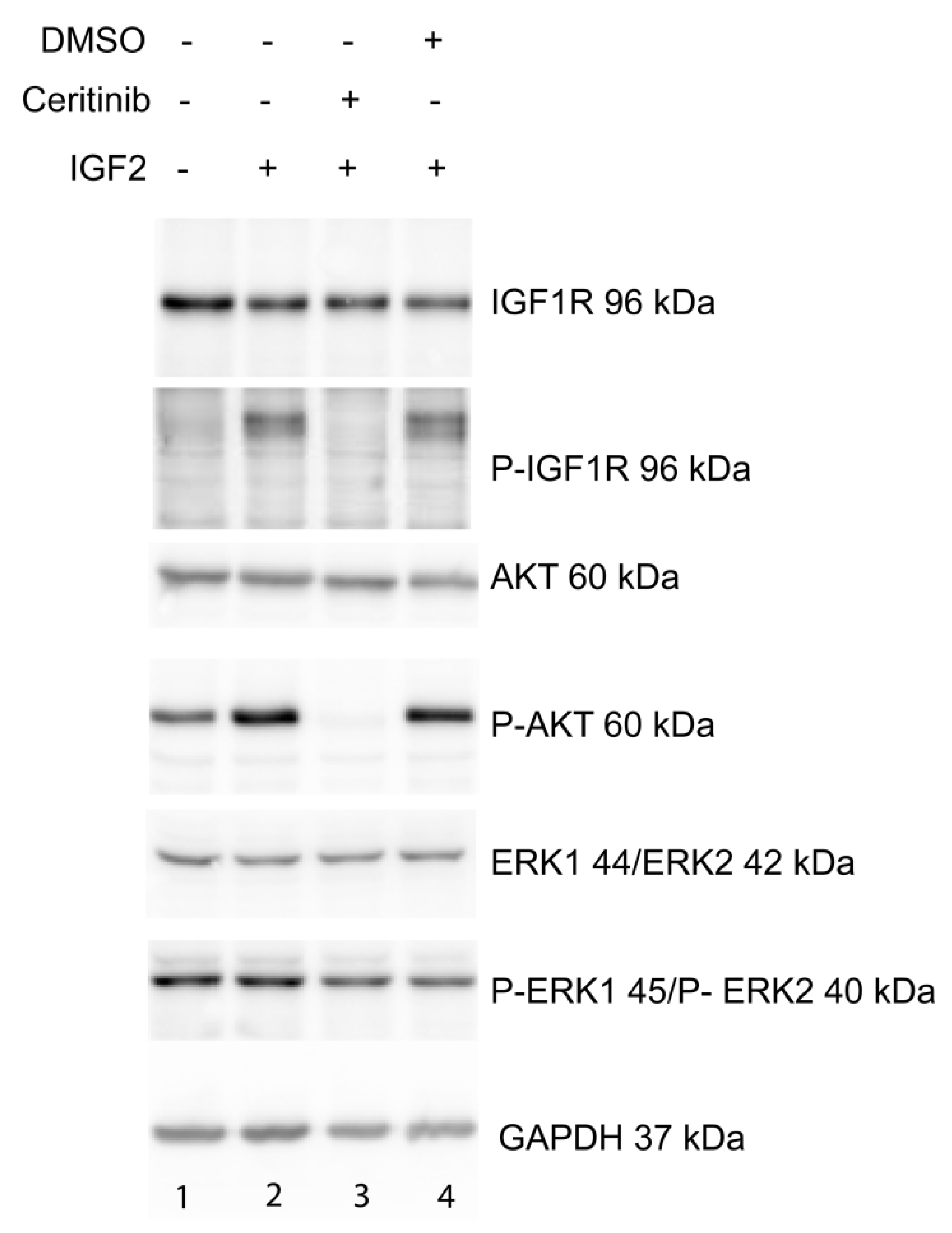
2.5. Ceritinib Acts Via the IGF1R/AKT Pathway
2.6. Figures





3. Discussion
4. Materials and Methods
4.1. Patient (P) Tissue Samples
4.2. Cells
4.3. Nucleic Acid Extraction
4.4. RT-PCR and qRT-PCR
4.5. DNA Sequencing
4.6. In Vitro Drug Screening
4.7. Phosphorylation Assay
4.8. Preparation of Subcellular Compartments and Lysis of Tumor Tissue
4.9. Western Blot Analysis
4.10. Transmission Electron Microscopy (TEM)
4.11. DNA Methylation Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABCB1 | ATP-Binding Cassette Subfamily B Member 1 |
| ABCG2 | ATP-Binding Cassette Subfamily G Member 2 |
| AKT | Protein kinase B |
| ALK | Anaplastic Lymphoma Kinase |
| BCOR | BCL-6 Co-Repressor |
| BCRP | Breast Cancer Resistance Protein |
| CNS-PN | Primitive neuroectodermal tumors of the central nervous system |
| ERK | Extracellular signal-regulated Kinase |
| HGNET | High-grade neuroepithelial Tumor |
| IGF | Insulin-like growth factor |
| IGF1R | Insulin-like growth factor receptor |
| ITD | Internal Tandem Duplication |
| MB | Medulloblastoma |
| MEK | Mitogen/Extracellular signal-regulated Kinase |
| mTOR | Mechanistic Target Of Rapamycin |
| NSCLC | Non-small cell lung cancer |
| P-GP | P-Glycoprotein |
| PI3K | phosphatidyl-inositol-3 kinase |
| qRT-PCR | quantitative Real-Time Polymerase Chain Reaction |
| RAF | Rapidly Accelerated Fibrosarcoma Kinase |
| RAS | Rat Sarcoma Oncogene |
| ROS1 | c-ros oncogene 1 |
| SHH | Sonic Hedgehog |
| URCSI | Soft tissue undifferentiated round cell sarcoma of infancy |
References
- Sturm, D.; Orr, B.A.; Toprak, U.H.; Hovestadt, V.; Jones, D.T.; Capper, D.; Sill, M.; Buchhalter, I.; Northcott, P.A.; Leis, I.; et al. New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 2016, 164, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W.; et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Appay, R.; Macagno, N.; Padovani, L.; Korshunov, A.; Kool, M.; Andre, N.; Scavarda, D.; Pietsch, T.; Figarella-Branger, D. HGNET-BCOR Tumors of the Cerebellum: Clinicopathologic and Molecular Characterization of 3 Cases. Am. J. Surg. Pathol. 2017, 41, 1254–1260. [Google Scholar] [CrossRef] [PubMed]
- Kirkman, M.A.; Pickles, J.C.; Fairchild, A.R.; Avery, A.; Pietsch, T.; Jacques, T.S.; Aquilina, K. Early Wound Site Seeding in a Patient with Central Nervous System High-Grade Neuroepithelial Tumor with BCOR Alteration. World Neurosurg. 2018, 116, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Paret, C.; Russo, A.; Otto, H.; Mayer, A.; Zahnreich, S.; Wagner, W.; Samuel, D.; Scharnhorst, D.; Solomon, D.A.; Dhall, G.; et al. Personalized therapy: CNS HGNET-BCOR responsiveness to arsenic trioxide combined with radiotherapy. Oncotarget 2017, 8, 114210–114225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, Y.; Nobusawa, S.; Nakata, S.; Nakada, M.; Arakawa, Y.; Mineharu, Y.; Sugita, Y.; Yoshioka, T.; Araki, A.; Sato, Y.; et al. CNS high-grade neuroepithelial tumor with BCOR internal tandem duplication: A comparison with its counterparts in the kidney and soft tissue. Brain Pathol. 2018, 28, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kumar, V.; Zorman, B.; Fang, E.; Haines, K.M.; Doddapaneni, H.; Hampton, O.A.; White, S.; Bavle, A.A.; Patel, N.R.; et al. Recurrent internal tandem duplications of BCOR in clear cell sarcoma of the kidney. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef]
- Kao, Y.C.; Sung, Y.S.; Zhang, L.; Huang, S.C.; Argani, P.; Chung, C.T.; Graf, N.S.; Wright, D.C.; Kellie, S.J.; Agaram, N.P.; et al. Recurrent BCOR Internal Tandem Duplication and YWHAE-NUTM2B Fusions in Soft Tissue Undifferentiated Round Cell Sarcoma of Infancy: Overlapping Genetic Features With Clear Cell Sarcoma of Kidney. Am. J. Surg. Pathol. 2016, 40, 1009–1020. [Google Scholar] [CrossRef]
- Kim, S.Y.; Toretsky, J.A.; Scher, D.; Helman, L.J. The role of IGF-1R in pediatric malignancies. Oncologist 2009, 14, 83–91. [Google Scholar] [CrossRef]
- Simpson, A.; Petnga, W.; Macaulay, V.M.; Weyer-Czernilofsky, U.; Bogenrieder, T. Insulin-Like Growth Factor (IGF) Pathway Targeting in Cancer: Role of the IGF Axis and Opportunities for Future Combination Studies. Target. Oncol. 2017, 12, 571–597. [Google Scholar] [CrossRef] [Green Version]
- Marsilje, T.H.; Pei, W.; Chen, B.; Lu, W.; Uno, T.; Jin, Y.; Jiang, T.; Kim, S.; Li, N.; Warmuth, M.; et al. Synthesis, structure-activity relationships, and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulf onyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J. Med. Chem. 2013, 56, 5675–5690. [Google Scholar] [CrossRef] [PubMed]
- Paret, C.; Theruvath, J.; Russo, A.; Kron, B.; El Malki, K.; Lehmann, N.; Wingerter, A.; Neu, M.A.; Gerhold-Ay, A.; Wagner, W.; et al. Activation of the basal cell carcinoma pathway in a patient with CNS HGNET-BCOR diagnosis: Consequences for personalized targeted therapy. Oncotarget 2016, 7, 83378–83391. [Google Scholar] [CrossRef] [PubMed]
- Huynh, K.D.; Fischle, W.; Verdin, E.; Bardwell, V.J. BCoR, a novel corepressor involved in BCL-6 repression. Genes Dev. 2000, 14, 1810–1823. [Google Scholar] [PubMed]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Honda, S.; Arai, Y.; Haruta, M.; Sasaki, F.; Ohira, M.; Yamaoka, H.; Horie, H.; Nakagawara, A.; Hiyama, E.; Todo, S.; et al. Loss of imprinting of IGF2 correlates with hypermethylation of the H19 differentially methylated region in hepatoblastoma. Br. J. Cancer 2008, 99, 1891–1899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulvihill, M.J.; Cooke, A.; Rosenfeld-Franklin, M.; Buck, E.; Foreman, K.; Landfair, D.; O’Connor, M.; Pirritt, C.; Sun, Y.; Yao, Y.; et al. Discovery of OSI-906: A selective and orally efficacious dual inhibitor of the IGF-1 receptor and insulin receptor. Future Med. Chem. 2009, 1, 1153–1171. [Google Scholar] [CrossRef] [PubMed]
- Gable, K.L.; Maddux, B.A.; Penaranda, C.; Zavodovskaya, M.; Campbell, M.J.; Lobo, M.; Robinson, L.; Schow, S.; Kerner, J.A.; Goldfine, I.D.; et al. Diarylureas are small-molecule inhibitors of insulin-like growth factor I receptor signaling and breast cancer cell growth. Mol. Cancer Ther. 2006, 5, 1079–1086. [Google Scholar] [CrossRef] [Green Version]
- Girnita, A.; Girnita, L.; del Prete, F.; Bartolazzi, A.; Larsson, O.; Axelson, M. Cyclolignans as inhibitors of the insulin-like growth factor-1 receptor and malignant cell growth. Cancer Res. 2004, 64, 236–242. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Bachar Raveh, T.; Barakat, M.T.; Lee, E.Y.; Scott, M.P. Insulin-like growth factor 2 is required for progression to advanced medulloblastoma in patched1 heterozygous mice. Cancer Res. 2008, 68, 8788–8795. [Google Scholar] [CrossRef]
- Hartmann, W.; Koch, A.; Brune, H.; Waha, A.; Schuller, U.; Dani, I.; Denkhaus, D.; Langmann, W.; Bode, U.; Wiestler, O.D.; et al. Insulin-like growth factor II is involved in the proliferation control of medulloblastoma and its cerebellar precursor cells. Am. J. Pathol. 2005, 166, 1153–1162. [Google Scholar] [CrossRef]
- Poondru, S.; Chaves, J.; Yuen, G.; Parker, B.; Conklin, E.; Singh, M.; Nagata, M.; Gill, S. Mass balance, pharmacokinetics, and metabolism of linsitinib in cancer patients. Cancer Chemother. Pharmacol. 2016, 77, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Kim, D.W.; Mehra, R.; Tan, D.S.; Felip, E.; Chow, L.Q.; Camidge, D.R.; Vansteenkiste, J.; Sharma, S.; De Pas, T.; et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 370, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Miller, W.T. Role of the activation loop tyrosines in regulation of the insulin-like growth factor I receptor-tyrosine kinase. J. Biol. Chem. 2006, 281, 23785–23791. [Google Scholar] [CrossRef] [PubMed]
- Girnita, L.; Worrall, C.; Takahashi, S.; Seregard, S.; Girnita, A. Something old, something new and something borrowed: Emerging paradigm of insulin-like growth factor type 1 receptor (IGF-1R) signaling regulation. Cell. Mol. Life Sci. 2014, 71, 2403–2427. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Huysentruyt, L.C. On the origin of cancer metastasis. Crit. Rev. Oncog. 2013, 18, 43–73. [Google Scholar] [CrossRef] [PubMed]
- Fais, S. Cannibalism: A way to feed on metastatic tumors. Cancer Lett. 2007, 258, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Bouffet, E.; Jakacki, R.; Goldman, S.; Hargrave, D.; Hawkins, C.; Shroff, M.; Hukin, J.; Bartels, U.; Foreman, N.; Kellie, S.; et al. Phase II study of weekly vinblastine in recurrent or refractory pediatric low-grade glioma. J. Clin. Oncol. 2012, 30, 1358–1363. [Google Scholar] [CrossRef] [PubMed]
- Houghton, P.J.; Morton, C.L.; Gorlick, R.; Kolb, E.A.; Keir, S.T.; Reynolds, C.P.; Kang, M.H.; Maris, J.M.; Wu, J.; Smith, M.A.; et al. Initial testing of a monoclonal antibody (IMC-A12) against IGF-1R by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 2010, 54, 921–926. [Google Scholar] [CrossRef]
- Zhu, Z.; Chai, Y. Crizotinib resistance overcome by ceritinib in an ALK-positive non-small cell lung cancer patient with brain metastases: A case report. Medicine (Baltim.) 2017, 96, e8652. [Google Scholar] [CrossRef]
- Kort, A.; Sparidans, R.W.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Brain accumulation of the EML4-ALK inhibitor ceritinib is restricted by P-glycoprotein (P-GP/ABCB1) and breast cancer resistance protein (BCRP/ABCG2). Pharmacol. Res. 2015, 102, 200–207. [Google Scholar] [CrossRef]
- van Erp, A.E.M.; Hillebrandt-Roeffen, M.H.S.; van Houdt, L.; Fleuren, E.D.G.; van der Graaf, W.T.A.; Versleijen-Jonkers, Y.M.H. Targeting Anaplastic Lymphoma Kinase (ALK) in Rhabdomyosarcoma (RMS) with the Second-Generation ALK Inhibitor Ceritinib. Target. Oncol. 2017, 12, 815–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Chen, J.; Karner, C.M.; Long, F. Hedgehog signaling activates a positive feedback mechanism involving insulin-like growth factors to induce osteoblast differentiation. Proc. Natl. Acad. Sci. USA 2015, 112, 4678–4683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodzic, D.; Frey, B.; Marechal, D.; Scarcez, T.; Grooteclaes, M.; Winkler, R. Cloning of breakpoints in and downstream the IGF2 gene that are associated with overexpression of IGF2 transcripts in colorectal tumours. Oncogene 1999, 18, 4710–4717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regairaz, M.; Munier, F.; Sartelet, H.; Castaing, M.; Marty, V.; Renauleaud, C.; Doux, C.; Delbe, J.; Courty, J.; Fabre, M.; et al. Mutation-Independent Activation of the Anaplastic Lymphoma Kinase in Neuroblastoma. Am. J. Pathol. 2016, 186, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Holden, P.; Horton, W.A. Crude subcellular fractionation of cultured mammalian cell lines. BMC Res. Notes 2009, 2, 243. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | IC50 nM (±SD) | Class | Mechanism of Action | Entity |
|---|---|---|---|---|
| Actinomycin D (n = 3) | <1 | Antibiotic | Intercalation into DNA | Sarcoma |
| Vinblastine (n = 2) | <1 | Vinca alkaloid | Binds tubulin | Sarcoma |
| Vincristine (n = 3) | 6.4 (±4) | Vinca alkaloid | Binds tubulin | Sarcoma, Brain tumors |
| Doxorubicin (n = 4) | 89.3 (±65) | anthracycline | Intercalation into DNA | Sarcoma |
| Ceritinib (n = 5) | 277 (±99) | Kinase inhibitor | ALK, ROS1, IGF1R inhibitor | Lung Cancer |
| Linsitinib (n = 3) | 516.7 (±89) | Kinase inhibitor | IGF1R Inhibitor | Not yet FDA-approved |
| Picropodophyllin (n = 3) | 475.1 (±111) | Kinase inhibitor | IGF1R Inhibitor | Not yet FDA-approved |
| Etoposide (n = 3) | 5808 (±795) | Derivative of podophyllotoxin | Forms complex with topoisomerase II | Sarcoma |
| Cisplatin (n = 3) | 6976 (± 3100) | Platin derivative | Binds to DNA | Sarcoma, Brain tumors |
| Carboplatin (n = 3) | >10,000 | Platin derivative | Binds to DNA | Brain tumors |
| PQ401 (n = 3) | >10,000 | Kinase inhibitor | IGF1R Inhibitor | Not yet FDA-approved |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vewinger, N.; Huprich, S.; Seidmann, L.; Russo, A.; Alt, F.; Bender, H.; Sommer, C.; Samuel, D.; Lehmann, N.; Backes, N.; et al. IGF1R Is a Potential New Therapeutic Target for HGNET-BCOR Brain Tumor Patients. Int. J. Mol. Sci. 2019, 20, 3027. https://doi.org/10.3390/ijms20123027
Vewinger N, Huprich S, Seidmann L, Russo A, Alt F, Bender H, Sommer C, Samuel D, Lehmann N, Backes N, et al. IGF1R Is a Potential New Therapeutic Target for HGNET-BCOR Brain Tumor Patients. International Journal of Molecular Sciences. 2019; 20(12):3027. https://doi.org/10.3390/ijms20123027
Chicago/Turabian StyleVewinger, Nadine, Sabrina Huprich, Larissa Seidmann, Alexandra Russo, Francesca Alt, Hannah Bender, Clemens Sommer, David Samuel, Nadine Lehmann, Nora Backes, and et al. 2019. "IGF1R Is a Potential New Therapeutic Target for HGNET-BCOR Brain Tumor Patients" International Journal of Molecular Sciences 20, no. 12: 3027. https://doi.org/10.3390/ijms20123027