The Role of Telomerase and Telomeres in Interstitial Lung Diseases: From Molecules to Clinical Implications

Abstract
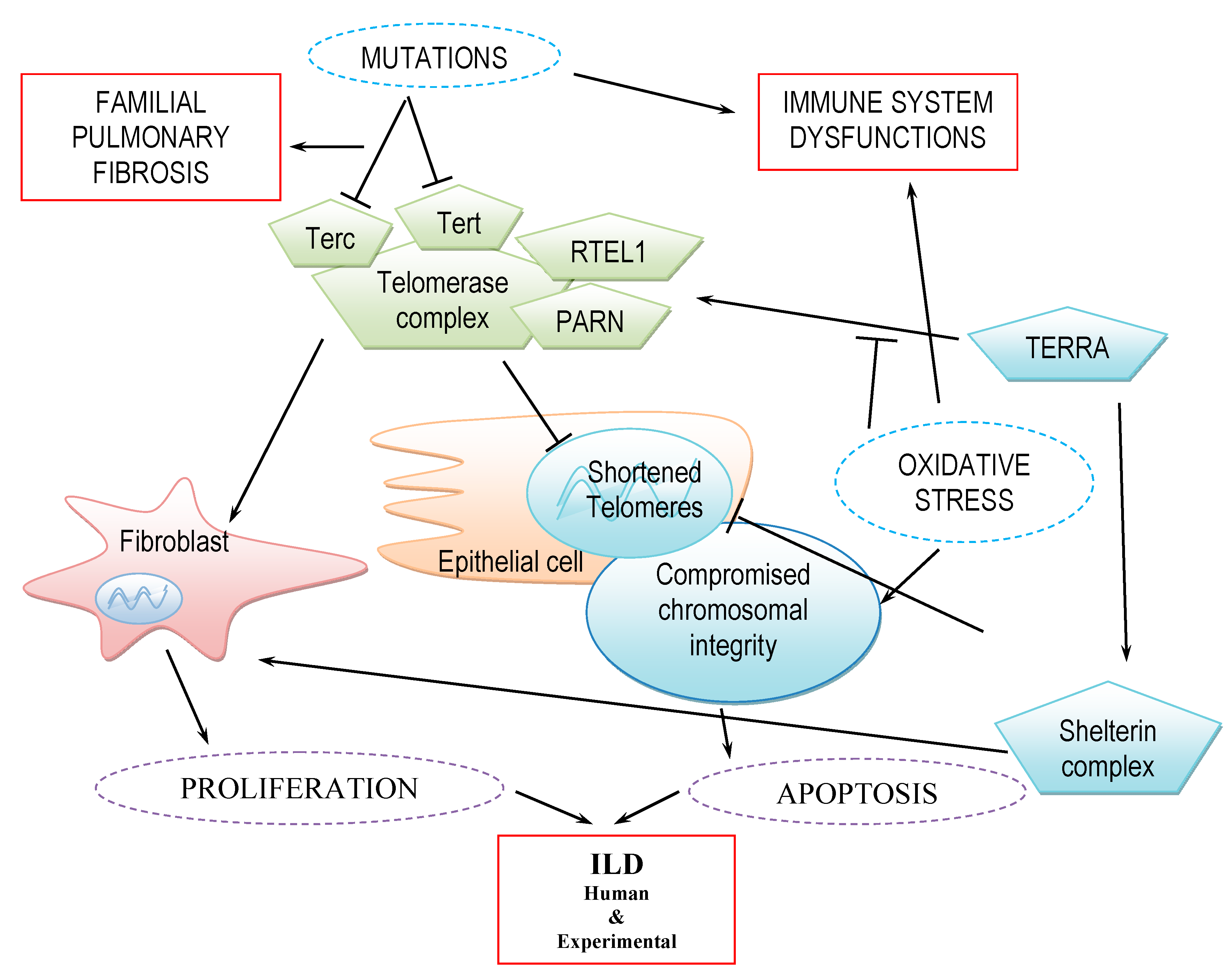
:1. Introduction
2. Experimental ILDs
2.1. Telomerase-Deficiency and Spontaneous Development of Experimental ILD
2.2. Stress-Induced ILDs in the Context of Deficient Telomerase Function
2.3. Experimental ILDs in the Context of Irregular Telomere Maintenance
2.4. ILD Development and Telomere Length in Fibroblast vs. Epithelial Cells
3. Telomere and Telomerase in Human ILDs
4. Clinical Implications
5. Perspectives for Further Studies
6. Summary and Unsolved Problems
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ILD | Interstitial lung disease |
| IPF | Idiopathic pulmonary fibrosis |
| UIP | Usual interstitial pneumonia |
| FPF | Familial pulmonary fibrosis |
| lncRNA | Long non-coding RNA |
| NSIP | Nonspecific intestinal pneumonitis |
References
- Prasad, K.N.; Wu, M.; Bondy, S.C. Telomere shortening during aging: Attenuation by antioxidants and anti-inflammatory agents. Mech. Ageing Dev. 2017, 164, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Bettin, N.; Oss Pegorar, C.; Cusanelli, E. The Emerging Roles of TERRA in Telomere Maintenance and Genome Stability. Cells 2019, 8, 246. [Google Scholar] [CrossRef]
- Cusanelli, E.; Chartrand, P. Telomeric noncoding RNA: Telomeric repeat-containing RNA in telomere biology. Wiley Interdiscip. Rev. RNA 2014, 5, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Snetselaar, R.; van Moorsel, C.H.M.; Kazemier, K.M.; van der Vis, J.J.; Zanen, P.; van Oosterhout, M.F.M.; Grutters, J.C. Telomere length in interstitial lung diseases. Chest 2015, 148, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef] [PubMed]
- Kropski, J.A.; Blackwell, T.S.; Loyd, J.E. The genetic basis of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1717–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonough, J.E.; Martens, D.S.; Tanabe, N.; Ahangari, F.; Verleden, S.E.; Maes, K.; Verleden, G.M.; Kaminski, N.; Hogg, J.C.; Nawrot, T.S.; et al. A role for telomere length and chromosomal damage in idiopathic pulmonary fibrosis. Respir. Res. 2018, 19, 132. [Google Scholar] [CrossRef] [PubMed]
- Vece, T.J.; Schecter, M.G.; Gatti, R.A.; Tunuguntla, R.; Garcia, C.K.; Langston, C.; Dishop, M.K.; Moore, R.H.; Fan, L.L. Rapid and progressive pulmonary fibrosis in 2 families with DNA repair deficiencies of undetermined etiology. J. Pediatr. 2012, 160, 700–702.e3. [Google Scholar] [CrossRef] [PubMed]
- Kage, H.; Borok, Z. EMT and interstitial lung disease: A mysterious relationship. Curr. Opin. Pulm. Med. 2012, 18, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Salton, F.; Volpe, M.C.; Confalonieri, M. Epithelial−Mesenchymal Transition in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Medicina (Kaunas) 2019, 55, 83. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A.; Barrera, L.; Estrada, A.; Watson, S.R.; Wilson, K.; Aziz, N.; Kaminski, N.; Zlotnik, A. Gene expression profiles distinguish idiopathic pulmonary fibrosis from hypersensitivity pneumonitis. Am. J. Respir. Crit. Care Med. 2006, 173, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Horimasu, Y.; Ishikawa, N.; Taniwaki, M.; Yamaguchi, K.; Hamai, K.; Iwamoto, H.; Ohshimo, S.; Hamada, H.; Hattori, N.; Okada, M.; et al. Gene expression profiling of idiopathic interstitial pneumonias (IIPs): Identification of potential diagnostic markers and therapeutic targets. BMC Med. Genet. 2017, 18, 88. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, P.; Rossi, G.; Cavazza, A. Pathogenesis of idiopathic pulmonary fibrosis and its clinical implications. Expert Rev. Clin. Immunol. 2014, 10, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- King, T.E.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Blasco, M.A. Mice with bad ends: Mouse models for the study of telomeres and telomerase in cancer and aging. EMBO J. 2005, 24, 1095–1103. [Google Scholar] [CrossRef]
- Degryse, A.L.; Xu, X.C.; Newman, J.L.; Mitchell, D.B.; Tanjore, H.; Polosukhin, V.V.; Jones, B.R.; McMahon, F.B.; Gleaves, L.A.; Phillips, J.A.; et al. Telomerase deficiency does not alter bleomycin-induced fibrosis in mice. Exp. Lung Res. 2012, 38, 124–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Reddy, R.; Barsky, L.; Scholes, J.; Chen, H.; Shi, W.; Driscoll, B. Lung alveolar integrity is compromised by telomere shortening in telomerase-null mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 296, L57–L70. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Ullenbruch, M.; Young Choi, Y.; Yu, H.; Ding, L.; Xaubet, A.; Pereda, J.; Feghali-Bostwick, C.A.; Bitterman, P.B.; Henke, C.A.; et al. Telomerase and telomere length in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2013, 49, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Blasco, M.A.; Lee, H.W.; Hande, M.P.; Samper, E.; Lansdorp, P.M.; DePinho, R.A.; Greider, C.W. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 1997, 91, 25–34. [Google Scholar] [CrossRef]
- Saretzki, G. Telomeres, Telomerase and Ageing. Subcell. Biochem. 2018, 90, 221–308. [Google Scholar] [PubMed]
- Nishina, H.; Wada, T.; Katada, T. Physiological roles of SAPK/JNK signaling pathway. J. Biochem. 2004, 136, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Povedano, J.M.; Martinez, P.; Serrano, R.; Tejera, Á.; Gómez-López, G.; Bobadilla, M.; Flores, J.M.; Bosch, F.; Blasco, M.A. Therapeutic effects of telomerase in mice with pulmonary fibrosis induced by damage to the lungs and short telomeres. eLife 2018, 7, e31299. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-Y.; Shi, Y.; Liu, Y.; Pan, X.-H.; Zhang, K.-X. Telomere shortening activates TGF-β/Smads signaling in lungs and enhances both lipopolysaccharide and bleomycin-induced pulmonary fibrosis. Acta Pharmacol. Sin. 2018, 39, 1735–1745. [Google Scholar] [CrossRef] [PubMed]
- Alder, J.K.; Guo, N.; Kembou, F.; Parry, E.M.; Anderson, C.J.; Gorgy, A.I.; Walsh, M.F.; Sussan, T.; Biswal, S.; Mitzner, W.; et al. Telomere length is a determinant of emphysema susceptibility. Am. J. Respir. Crit. Care Med. 2011, 184, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, D.P.; Bitar, M.; Jacobs, F.M.J.; Barry, G. Long Non-Coding RNAs in Neuronal Aging. Noncoding RNA 2018, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Sfeir, A.; de Lange, T. Removal of shelterin reveals the telomere end-protection problem. Science 2012, 336, 593–597. [Google Scholar] [CrossRef] [PubMed]
- Rai, R.; Chen, Y.; Lei, M.; Chang, S. TRF2-RAP1 is required to protect telomeres from engaging in homologous recombination-mediated deletions and fusions. Nat. Commun. 2016, 7, 10881. [Google Scholar] [CrossRef] [Green Version]
- Rice, C.; Skordalakes, E. Structure and function of the telomeric CST complex. Comput. Struct. Biotechnol. J. 2016, 14, 161–167. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.-Y.; Redon, S.; Lingner, J. The human CST complex is a terminator of telomerase activity. Nature 2012, 488, 540–544. [Google Scholar] [CrossRef]
- Shoeb, M.; Mustafa, G.M.; Joseph, P.; Umbright, C.; Kodali, V.; Roach, K.A.; Meighan, T.; Roberts, J.R.; Erdely, A.; Antonini, J.M. Initiation of Pulmonary Fibrosis after Silica Inhalation in Rats is linked with Dysfunctional Shelterin Complex and DNA Damage Response. Sci. Rep. 2019, 9, 471. [Google Scholar] [CrossRef] [PubMed]
- Alder, J.K.; Barkauskas, C.E.; Limjunyawong, N.; Stanley, S.E.; Kembou, F.; Tuder, R.M.; Hogan, B.L.M.; Mitzner, W.; Armanios, M. Telomere dysfunction causes alveolar stem cell failure. Proc. Natl. Acad. Sci. USA 2015, 112, 5099–5104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.T. Epigenetic regulation by long noncoding RNAs. Science 2012, 338, 1435–1439. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Zhang, J.; Wang, M.; Song, X.; Liu, W.; Mao, C.; Lv, C. Differential expression of long non-coding RNAs in bleomycin-induced lung fibrosis. Int. J. Mol. Med. 2013, 32, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhang, J.; Liu, Y.; Zhang, S.; Wang, Y.; Liu, B.; Liu, H.; Li, R.; Lv, C.; Song, X. Regulation of TERRA on telomeric and mitochondrial functions in IPF pathogenesis. BMC Pulm. Med. 2017, 17, 163. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Yu, H.; Ding, L.; Wu, Z.; Gonzalez De Los Santos, F.; Liu, J.; Ullenbruch, M.; Hu, B.; Martins, V.; Phan, S.H. Conditional Knockout of Telomerase Reverse Transcriptase in Mesenchymal Cells Impairs Mouse Pulmonary Fibrosis. PLoS ONE 2015, 10, e0142547. [Google Scholar] [CrossRef] [PubMed]
- Naikawadi, R.P.; Disayabutr, S.; Mallavia, B.; Donne, M.L.; Green, G.; La, J.L.; Rock, J.R.; Looney, M.R.; Wolters, P.J. Telomere dysfunction in alveolar epithelial cells causes lung remodeling and fibrosis. JCI Insight 2016, 1, e86704. [Google Scholar] [CrossRef] [PubMed]
- Povedano, J.M.; Martinez, P.; Flores, J.M.; Mulero, F.; Blasco, M.A. Mice with Pulmonary Fibrosis Driven by Telomere Dysfunction. Cell Rep. 2015, 12, 286–299. [Google Scholar] [CrossRef] [Green Version]
- Fridlender, Z.G.; Cohen, P.Y.; Golan, O.; Arish, N.; Wallach-Dayan, S.; Breuer, R. Telomerase activity in bleomycin-induced epithelial cell apoptosis and lung fibrosis. Eur. Respir. J. 2007, 30, 205–213. [Google Scholar] [CrossRef] [Green Version]
- Arish, N.; Cohen, P.Y.; Golan-Gerstl, R.; Fridlender, Z.; Dayan, M.R.; Zisman, P.; Breuer, R.; Wallach-Dayan, S.B. Overexpression of Telomerase Protects Human and Murine Lung Epithelial Cells from Fas- and Bleomycin-Induced Apoptosis via FLIP Upregulation. PLoS ONE 2015, 10, e0126730. [Google Scholar] [CrossRef]
- Le Saux, C.J.; Davy, P.; Brampton, C.; Ahuja, S.S.; Fauce, S.; Shivshankar, P.; Nguyen, H.; Ramaseshan, M.; Tressler, R.; Pirot, Z.; et al. A novel telomerase activator suppresses lung damage in a murine model of idiopathic pulmonary fibrosis. PLoS ONE 2013, 8, e58423. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.R.; Wood, E.; Collins, K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature 1999, 402, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Stuart, B.D.; Choi, J.; Zaidi, S.; Xing, C.; Holohan, B.; Chen, R.; Choi, M.; Dharwadkar, P.; Torres, F.; Girod, C.E.; et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat. Genet. 2015, 47, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, T.W.; van Moorsel, C.H.M.; Borie, R.; Crestani, B. Pulmonary phenotypes associated with genetic variation in telomere-related genes. Curr. Opin. Pulm. Med. 2018, 24, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Borie, R.; Bouvry, D.; Cottin, V.; Gauvain, C.; Cazes, A.; Debray, M.-P.; Cadranel, J.; Dieude, P.; Degot, T.; Dominique, S.; et al. Regulator of telomere length 1 (RTEL1) mutations are associated with heterogeneous pulmonary and extra-pulmonary phenotypes. Eur. Respir. J. 2019, 53, 1800508. [Google Scholar] [CrossRef] [PubMed]
- Borie, R.; Tabèze, L.; Thabut, G.; Nunes, H.; Cottin, V.; Marchand-Adam, S.; Prevot, G.; Tazi, A.; Cadranel, J.; Mal, H.; et al. Prevalence and characteristics of Tert and Terc mutations in suspected genetic pulmonary fibrosis. Eur. Respir. J. 2016, 48, 1721–1731. [Google Scholar] [CrossRef] [PubMed]
- Armanios, M. Telomeres and age-related disease: How telomere biology informs clinical paradigms. J. Clin. Investig. 2013, 123, 996–1002. [Google Scholar] [CrossRef] [PubMed]
- Armanios, M.Y.; Chen, J.J.-L.; Cogan, J.D.; Alder, J.K.; Ingersoll, R.G.; Markin, C.; Lawson, W.E.; Xie, M.; Vulto, I.; Phillips, J.A.; et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2007, 356, 1317–1326. [Google Scholar] [CrossRef]
- El-Chemaly, S.; Ziegler, S.G.; Calado, R.T.; Wilson, K.A.; Wu, H.P.; Haughey, M.; Peterson, N.R.; Young, N.S.; Gahl, W.A.; Moss, J.; et al. Natural history of pulmonary fibrosis in two subjects with the same telomerase mutation. Chest 2011, 139, 1203–1209. [Google Scholar] [CrossRef]
- Tsakiri, K.D.; Cronkhite, J.T.; Kuan, P.J.; Xing, C.; Raghu, G.; Weissler, J.C.; Rosenblatt, R.L.; Shay, J.W.; Garcia, C.K. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl. Acad. Sci. USA 2007, 104, 7552–7557. [Google Scholar] [CrossRef] [Green Version]
- Alder, J.K.; Chen, J.J.-L.; Lancaster, L.; Danoff, S.; Su, S.; Cogan, J.D.; Vulto, I.; Xie, M.; Qi, X.; Tuder, R.M.; et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13051–13056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cronkhite, J.T.; Xing, C.; Raghu, G.; Chin, K.M.; Torres, F.; Rosenblatt, R.L.; Garcia, C.K. Telomere shortening in familial and sporadic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Planas-Cerezales, L.; Arias-Salgado, E.G.; Buendia-Roldán, I.; Montes-Worboys, A.; López, C.E.; Vicens-Zygmunt, V.; Hernaiz, P.L.; Sanuy, R.L.; Leiro-Fernandez, V.; Vilarnau, E.B.; et al. Predictive factors and prognostic effect of telomere shortening in pulmonary fibrosis. Respirology 2019, 24, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Valdes, A.M.; Andrew, T.; Gardner, J.P.; Kimura, M.; Oelsner, E.; Cherkas, L.F.; Aviv, A.; Spector, T.D. Obesity, cigarette smoking, and telomere length in women. Lancet 2005, 366, 662–664. [Google Scholar] [CrossRef]
- Tsuji, T.; Aoshiba, K.; Nagai, A. Alveolar cell senescence in patients with pulmonary emphysema. Am. J. Respir. Crit. Care Med. 2006, 174, 886–893. [Google Scholar] [CrossRef]
- Stanley, S.E.; Chen, J.J.L.; Podlevsky, J.D.; Alder, J.K.; Hansel, N.N.; Mathias, R.A.; Qi, X.; Rafaels, N.M.; Wise, R.A.; Silverman, E.K.; et al. Telomerase mutations in smokers with severe emphysema. J. Clin. Investig. 2015, 125, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Stuart, B.D.; Lee, J.S.; Kozlitina, J.; Noth, I.; Devine, M.S.; Glazer, C.S.; Torres, F.; Kaza, V.; Girod, C.E.; Jones, K.D.; et al. Effect of telomere length on survival in patients with idiopathic pulmonary fibrosis: An observational cohort study with independent validation. Lancet Respir. Med. 2014, 2, 557–565. [Google Scholar] [CrossRef]
- Newton, C.A.; Batra, K.; Torrealba, J.; Kozlitina, J.; Glazer, C.S.; Aravena, C.; Meyer, K.; Raghu, G.; Collard, H.R.; Garcia, C.K. Telomere-related lung fibrosis is diagnostically heterogeneous but uniformly progressive. Eur. Respir. J. 2016, 48, 1710–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kropski, J.A.; Mitchell, D.B.; Markin, C.; Polosukhin, V.V.; Choi, L.; Johnson, J.E.; Lawson, W.E.; Phillips, J.A.; Cogan, J.D.; Blackwell, T.S.; et al. A novel dyskerin (DKC1) mutation is associated with familial interstitial pneumonia. Chest 2014, 146, e1–e7. [Google Scholar] [CrossRef]
- Alder, J.K.; Stanley, S.E.; Wagner, C.L.; Hamilton, M.; Hanumanthu, V.S.; Armanios, M. Exome sequencing identifies mutant TINF2 in a family with pulmonary fibrosis. Chest 2015, 147, 1361–1368. [Google Scholar] [CrossRef]
- Stanley, S.E.; Gable, D.L.; Wagner, C.L.; Carlile, T.M.; Hanumanthu, V.S.; Podlevsky, J.D.; Khalil, S.E.; DeZern, A.E.; Rojas-Duran, M.F.; Applegate, C.D.; et al. Loss-of-function mutations in the RNA biogenesis factor NAF1 predispose to pulmonary fibrosis-emphysema. Sci. Transl. Med. 2016, 8, 351ra107. [Google Scholar] [CrossRef] [PubMed]
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu, G.; Anstrom, K.J.; King, T.E.; Lasky, J.A.; Martinez, F.J. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N. Engl. J. Med. 2012, 366, 1968–1977. [Google Scholar] [PubMed]
- Newton, C.A.; Zhang, D.; Oldham, J.M.; Kozlitina, J.; Ma, S.-F.; Martinez, F.J.; Raghu, G.; Noth, I.; Garcia, C.K. Telomere Length and Use of Immunosuppressive Medications in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Justet, A.; Thabut, G.; Manali, E.; Molina Molina, M.; Kannengiesser, C.; Cadranel, J.; Cottin, V.; Gondouin, A.; Nunes, H.; Magois, E.; et al. Safety and efficacy of pirfenidone in patients carrying telomerase complex mutation. Eur. Respir. J. 2018, 51, 1701875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Townsley, D.M.; Dumitriu, B.; Young, N.S. Danazol Treatment for Telomere Diseases. N. Engl. J. Med. 2016, 375, 1095–1096. [Google Scholar] [CrossRef] [PubMed]
- Silhan, L.L.; Shah, P.D.; Chambers, D.C.; Snyder, L.D.; Riise, G.C.; Wagner, C.L.; Hellström-Lindberg, E.; Orens, J.B.; Mewton, J.F.; Danoff, S.K.; et al. Lung transplantation in telomerase mutation carriers with pulmonary fibrosis. Eur. Respir. J. 2014, 44, 178–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokman, S.; Singer, J.P.; Devine, M.S.; Westall, G.P.; Aubert, J.-D.; Tamm, M.; Snell, G.I.; Lee, J.S.; Goldberg, H.J.; Kukreja, J.; et al. Clinical outcomes of lung transplant recipients with telomerase mutations. J. Heart Lung Transplant. 2015, 34, 1318–1324. [Google Scholar] [CrossRef] [Green Version]
- Borie, R.; Kannengiesser, C.; Hirschi, S.; Le Pavec, J.; Mal, H.; Bergot, E.; Jouneau, S.; Naccache, J.-M.; Revy, P.; Boutboul, D.; et al. Severe hematologic complications after lung transplantation in patients with telomerase complex mutations. J. Heart Lung Transplant. 2015, 34, 538–546. [Google Scholar] [CrossRef]
- Swaminathan, A.C.; Neely, M.L.; Frankel, C.W.; Kelly, F.L.; Petrovski, S.; Durheim, M.T.; Bush, E.; Snyder, L.; Goldstein, D.B.; Todd, J.L.; et al. Lung Transplant Outcomes in Patients with Pulmonary Fibrosis with Telomere-Related Gene Variants. Chest 2019. [Google Scholar] [CrossRef]
- Popescu, I.; Mannem, H.; Winters, S.A.; Hoji, A.; Silveira, F.; McNally, E.; Pipeling, M.R.; Lendermon, E.A.; Morrell, M.R.; Pilewski, J.M.; et al. Impaired Cytomegalovirus Immunity in Idiopathic Pulmonary Fibrosis Lung Transplant Recipients with Short Telomeres. Am. J. Respir. Crit. Care Med. 2019, 199, 362–376. [Google Scholar] [CrossRef]
- Molina-Molina, M.; Borie, R. Clinical implications of telomere dysfunction in lung fibrosis. Curr. Opin. Pulm. Med. 2018, 24, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Courtwright, A.M.; El-Chemaly, S. Telomeres in Interstitial Lung Disease: The Short and the Long of It. Ann. Am. Thorac. Soc. 2019, 16, 175–181. [Google Scholar] [CrossRef]
- Newton, C.A.; Oldham, J.M.; Ley, B.; Anand, V.; Adegunsoye, A.; Liu, G.; Batra, K.; Torrealba, J.; Kozlitina, J.; Glazer, C.; et al. Telomere Length and Genetic Variant Associations with Interstitial Lung Disease Progression and Survival. Eur. Respir. J. 2019, 53, 1801641. [Google Scholar] [CrossRef] [PubMed]
- Armanios, M. Telomerase and idiopathic pulmonary fibrosis. Mutat. Res. 2012, 730, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Hockemeyer, D.; Collins, K. Control of telomerase action at human telomeres. Nat. Struct. Mol. Biol. 2015, 22, 848–852. [Google Scholar] [CrossRef] [Green Version]
- Frank, A.K.; Tran, D.C.; Qu, R.W.; Stohr, B.A.; Segal, D.J.; Xu, L. The Shelterin TIN2 Subunit Mediates Recruitment of Telomerase to Telomeres. PLoS Genet. 2015, 11, e1005410. [Google Scholar] [CrossRef] [PubMed]
- Fingerlin, T.E.; Murphy, E.; Zhang, W.; Peljto, A.L.; Brown, K.K.; Steele, M.P.; Loyd, J.E.; Cosgrove, G.P.; Lynch, D.; Groshong, S.; et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat. Genet. 2013, 45, 613–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, A.J.; Lev, A.; Zhang, Y.; Weiss, B.; Rylova, A.; Eyal, E.; Kol, N.; Barel, O.; Cesarkas, K.; Soudack, M.; et al. Mutations in STN1 cause Coats plus syndrome and are associated with genomic and telomere defects. J. Exp. Med. 2016, 213, 1429–1440. [Google Scholar] [CrossRef] [PubMed]
- Boccardi, V.; Razdan, N.; Kaplunov, J.; Mundra, J.J.; Kimura, M.; Aviv, A.; Herbig, U. Stn1 is critical for telomere maintenance and long-term viability of somatic human cells. Aging Cell 2015, 14, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.E. The Cajal body. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2008, 1783, 2108–2115. [Google Scholar] [CrossRef] [Green Version]
- Cristofari, G.; Adolf, E.; Reichenbach, P.; Sikora, K.; Terns, R.M.; Terns, M.P.; Lingner, J. Human telomerase RNA accumulation in Cajal bodies facilitates telomerase recruitment to telomeres and telomere elongation. Mol. Cell 2007, 27, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Hebert, M.D.; Poole, A.R. Towards an understanding of regulating Cajal body activity by protein modification. RNA Biol. 2017, 14, 761–778. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.B.; Graham, M.E.; Lovrecz, G.O.; Bache, N.; Robinson, P.J.; Reddel, R.R. Protein composition of catalytically active human telomerase from immortal cells. Science 2007, 315, 1850–1853. [Google Scholar] [CrossRef] [PubMed]
- Poole, A.R.; Hebert, M.D. SMN and coilin negatively regulate dyskerin association with telomerase RNA. Biol. Open 2016, 5, 726–735. [Google Scholar] [CrossRef] [Green Version]
- Pandita, T.K. ATM function and telomere stability. Oncogene 2002, 21, 611–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arias-Salgado, E.G.; Galvez, E.; Planas-Cerezales, L.; Pintado-Berninches, L.; Vallespin, E.; Martinez, P.; Carrillo, J.; Iarriccio, L.; Ruiz-Llobet, A.; Catalá, A.; et al. Genetic analyses of aplastic anemia and idiopathic pulmonary fibrosis patients with short telomeres, possible implication of DNA-repair genes. Orphanet J. Rare Dis. 2019, 14, 82. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.-Y.; Liu, H.-C.; Wu, L.-C.; Chen, C.-Y.; Chang, J.T.; Hsu, S.-L. Gallic acid induces apoptosis of lung fibroblasts via a reactive oxygen species-dependent ataxia telangiectasia mutated-p53 activation pathway. J. Agric. Food Chem. 2010, 58, 2943–2951. [Google Scholar] [CrossRef]
- Van Ly, D.; Low, R.R.J.; Frölich, S.; Bartolec, T.K.; Kafer, G.R.; Pickett, H.A.; Gaus, K.; Cesare, A.J. Telomere Loop Dynamics in Chromosome End Protection. Mol. Cell 2018, 71, 510–525. [Google Scholar] [CrossRef]
- Gobbini, E.; Trovesi, C.; Cassani, C.; Longhese, M.P. Telomere uncapping at the crossroad between cell cycle arrest and carcinogenesis. Mol. Cell. Oncol. 2014, 1, e29901. [Google Scholar] [CrossRef] [Green Version]
- Korthagen, N.M.; van Moorsel, C.H.M.; Barlo, N.P.; Kazemier, K.M.; Ruven, H.J.T.; Grutters, J.C. Association between variations in cell cycle genes and idiopathic pulmonary fibrosis. PLoS ONE 2012, 7, e30442. [Google Scholar] [CrossRef]
- Counter, C.M. The roles of telomeres and telomerase in cell life span. Mutat. Res. 1996, 366, 45–63. [Google Scholar] [CrossRef]
- Waters, D.W.; Blokland, K.E.C.; Pathinayake, P.S.; Burgess, J.K.; Mutsaers, S.E.; Prele, C.M.; Schuliga, M.; Grainge, C.L.; Knight, D.A. Fibroblast senescence in the pathology of idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 315, L162–L172. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Telomere-Related Mutation | Interstitial Lung Disease 1 | References |
|---|---|---|
| Tert | IPF; NSIP; AIP; COP; SR-ILD; HP; PPFE | [46,48,58,59] |
| Tert | IPF; NSIP; PPFE; HP | [46,48,59] |
| PARN | IPF; NSIP; HP | [43,59] |
| TINIF2 | IPF | [60] |
| NAF1 | IPF | [61] |
| DKC1 | IPF; NSIP | [59] |
| RTEL1 | IPF; NSIP; PPEF; HP | [43,48] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arish, N.; Petukhov, D.; Wallach-Dayan, S.B. The Role of Telomerase and Telomeres in Interstitial Lung Diseases: From Molecules to Clinical Implications. Int. J. Mol. Sci. 2019, 20, 2996. https://doi.org/10.3390/ijms20122996
Arish N, Petukhov D, Wallach-Dayan SB. The Role of Telomerase and Telomeres in Interstitial Lung Diseases: From Molecules to Clinical Implications. International Journal of Molecular Sciences. 2019; 20(12):2996. https://doi.org/10.3390/ijms20122996
Chicago/Turabian StyleArish, Nissim, Dmytro Petukhov, and Shulamit B. Wallach-Dayan. 2019. "The Role of Telomerase and Telomeres in Interstitial Lung Diseases: From Molecules to Clinical Implications" International Journal of Molecular Sciences 20, no. 12: 2996. https://doi.org/10.3390/ijms20122996