Role of the Ubiquitin Proteasome System (UPS) in the HIV-1 Life Cycle

Department of Microbiology, Immunology, and Genetics, University of North Texas, Health Science Center, Fort Worth, TX 76107, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(12), 2984; https://doi.org/10.3390/ijms20122984
Submission received: 21 May 2019
/
Revised: 10 June 2019
/
Accepted: 11 June 2019
/
Published: 19 June 2019
(This article belongs to the Special Issue Proteostasis and Proteasome Inhibitors)
{kind=link}
Abstract
:Given that the ubiquitin proteasome system (UPS) is the major protein degradation process in the regulation of a wide variety of cellular processes in eukaryotic cells, including alteration of cellular location, modulation of protein activity, and regulation of protein interaction, it is reasonable to suggest that the infecting HIV-1 and the invaded hosts exploit the UPS in a contest for survival and proliferation. However, to date, regulation of the HIV-1 life cycle has been mainly explained by the stage-specific expression of HIV-1 viral genes, not by elimination processes of the synthesized proteins after completion of their duties in the infected cells, which is also quintessential for understanding the molecular processes of the virus life cycle and thereby HIV-1 pathogenesis. In fact, several previous publications have indicated that the UPS plays a critical role in the regulation of the proteasomal degradation of viral and cellular counterparts at every step of the HIV-1 life cycle, from the virus entry to release of the assembled virus particles, which is integral for the regulation of survival and proliferation of the infecting HIV-1 and to replication restriction of the invading virus in the host. However, it is unknown whether and how these individual events taking place at different stages of the HIV-1 life cycle are orchestrated as an overall strategy to overcome the restrictions conferred by the host cells. Thus, in this review, we overview the interplay between HIV-1 viral and cellular proteins for restrictions/competitions for proliferation of the virus in the infected cell, which could open a new avenue for the development of therapeutics against HIV-1 via targeting a specific step of the proteasome degradation pathway during the HIV-1 life cycle.
1. Introduction
The ubiquitin proteasome system (UPS) is a major intracellular protein degradation system involved in various critical cellular processes in eukaryotes. The UPS consists of the concerted actions of three distinct enzymatic components: E1 ubiquitin-activating enzyme, E2 ubiquitin-conjugating enzyme, and E3 ubiquitin ligase [1,2]. Among these, E3 ubiquitin ligase is the most critical in protein degradation processes, since the enzyme recognizes a target protein to be degraded and transfers ubiquitin (Ub) to the target protein for degradation with specificity [2,3]. The polyubiquitinated proteins will then be recognized by the 19S regulatory particle in an ATP-dependent binding step [4], enter into the 26S proteasome complex, and degrade into amino acids or small peptides, unless the polyubiquitinated proteins are deubiquitinated by the deubiquitinating enzymes (DUBs), such as ubiquitin specific protease (USP) proteins [5]. That is, E3 ubiquitin ligase mediates the last step of the ubiquitination process by recognizing and selecting the target molecules with specificity, and thus most studies have focused on the interaction of the target protein with E3 ubiquitin ligases for understanding the molecular mechanisms of the protein degradation processes by the UPS.
The proteasomal degradation pathway plays a critical role for many cellular processes, including cell cycle [6,7,8], apoptosis [9,10,11], antigen-presentation [12,13,14], etc., and thus aberrations and dysregulations of the UPS can result in the pathogenesis of several important human diseases [15,16,17]. As such, given the general importance of the UPS in a wide variety of cellular processes by regulating protein fate and function, it is reasonable to suggest that HIV-1 and the invaded hosts exploit the UPS in a contest for their survival and proliferation. In fact, several previous reports have indicated that HIV-1 and HIV-1-infected cells exploit cellular machineries for viral and cellular protein decay at every step of the HIV-1 life cycle to restrict their proliferations reciprocally. For instance, Env/Vpu/Nef-mediated degradation of CD4 receptor molecules impedes virus entry and thereby superinfection [18,19,20,21,22,23,24], TRIM5α-triggered Gag is integral to species tropism [25,26,27,28,29,30,31], degradation of tetherin by Vpu is essential for the release of the assembled virus particles [32,33,34,35], and so on. In addition to these interactions between viral and cellular proteins, the interplay between HIV-1 viral proteins is also known to have an integral role in the regulation of the viral replication in the infected cells [36]. A recent publication even indicated that regulation of proteasomal degradation plays a key role in the establishment of viral latency [37]. Thus, we present a review on how the interplay between viral and viral/cellular proteins in their degradation impacts the HIV-1 life cycle, focusing on recent advances in this field of research.
2. Proteasomal Degradation in Regulation of the HIV-1 Life Cycle
2.1. HIV-1 Life Cycle
The HIV genome contains at least six regulatory genes, vif, vpr (vpx for HIV-2), vpu, tat, rev, and nef, in addition to three structural genes, gag, pol, and env, which present in all standard retroviruses. Molecular regulation for the stage-specific expression of the viral genes has been studied comprehensively; that is, regulatory genes to produce Tat, Rev, and Nef, are expressed at the early stage, while those for structural proteins, such as Gag, Pol, and Env, are expressed at the late stage of virus infection [38]. Specifically, the Tat expressed at the early stage of the virus infection accelerates the expression of viral genes, and the Rev selectively pumps out the structural genes containing Rev-responsive element (RRE) to the cytoplasm so that the structural proteins are encoded at the late stage of virus infection [38,39,40,41]. Hence, Rev protein plays a key role in the transition from the early to the late stage of the HIV-1 life cycle. Consequently, the HIV-1 life cycle would be dysregulated if the expressed Tat and Rev, including other viral proteins, were not degraded in a timely manner after they completed their duties—the transactivation of the viral gene expressions by Tat and transitioning from the early to the late stage by Rev; that is, elimination processes of these synthesized proteins are quintessential in understanding the regulation of the HIV-1 replication cycle and thereby HIV-1 pathogenesis. This review highlights the impacts of viral and viral/cellular protein degradation on the regulation of the major steps of the HIV-1 life cycle.
2.2. Impacts of the Interplay between Viral and Cellular Proteins on the Regulation of the Proteasomal Degradation for the HIV-1 Life Cycle
2.2.1. Virus Entry
According to recent research, the most notable cellular proteins involved in the regulation of HIV-1 entry via the proteasome degradation are SERINC (SERine INCorporator) and IFITM (Interferon-Induced TransMembrane). First, the SERINC family is composed of five members, from SERINC1 to SERINC5, and participate in the transport of serine amino acid through the lipid bilayer and in the biosynthesis of sphingolipids and phosphatidylserine by incorporating serine into membrane lipids [42]. Among the family, SERINC5 and, to a lesser extent, SERINC3 are involved in the impairment of viral infectivity by blocking viral entry [43,44,45], probably through restricting the expansion of the viral fusion pore and thus preventing the release of the viral core into the cytoplasm [43,45]. The SERINC-mediated antiviral activity is counteracted by Nef, wherein Nef induces downregulation of SERINC from the cell membrane by sequestering it in the endosomes for its subsequent degradation by the endo-lysosomal system [43,46,47]. However, the precise molecular mechanisms on how Nef, a virion protein within the virus particle [48,49,50,51], can enhance virus entry are unclear, albeit intracellular Nef from HIV-1-infected cells can ablate SERINC-mediated blockage of the virus entry [43,45,52,53]. With respect to HIV-1 replication, Nef is known to be dispensable or displays slightly, if at all, positive for an in vitro virus replication in the HIV-1 susceptible CD4+ cells [54,55,56], and Nef-mediated counteraction against SERINC is strain dependent (some strains of HIV-1 have Nef-dependent mechanisms to resist high levels of SERINCS, while other strains are independent) [52]. Further, a recent report addressed that the counteraction of SERINC5 by Nef did not increase (or only modestly increased) HIV-1 replication in CD4+ T cells and in ex vivo infected lymphoid tissue or the resting peripheral mononuclear cells (PBMC), respectively [57], casting implications of the significance of the interplay between Nef and SERINC in the enhancement of the virus entry and thus HIV-1 infectivity. In this context, a recent report indicated that HIV-1 envelope (Env), not Nef, is able to resist high levels of SERINC5 without excluding SERINC5 from incorporation into virions [52]. Since HIV-1 infection into its susceptible cells is initiated by the binding of Envelope (Env) protein to the receptor/co-receptor molecules [58,59,60,61], followed by fusion between viral and cytoplasmic membranes by gp41 of Env [62,63], SERINC-mediated inhibition of the correct fusion of the viral Env with the plasma membrane by promoting inactivation of Env [43,45,53], thereby preventing virus entry into host cells, is germane in the explanation of the role of SERINC in HIV-1 entry and thus replication.
IFITMs (IFITM1, 2, and 3) are one of the several interferon (IFN)-stimulated genes (ISGs) that restrict entry of a broad spectrum of enveloped viruses [64,65], including HIV-1 [66,67]. IFITMs-mediated restriction of the virus entry is known to be achieved by blocking the imprinting of the molecules into virions and the processing of Env. However, detailed mechanisms are unclear at this point. Recent reports indicated that IFITMs not only impede HIV-1 entry but also inhibit HIV-1 infectivity in viral producer cells [68,69,70] by negative imprinting of the incorporation of IFITMs into virions [68,69,71] and by impairment of the processing of HIV-1 Env [70]. Even if there were a consensus for the role of IFITMs in the inhibition of HIV-1 entry into the target cells, molecular details of IFITMs-mediated blockage of the virus entry mechanisms remain to be further elucidated. Specifically, it is controversial whether IFITMs distinguish co-receptors in the virus entry, if IFITM-mediated inhibition occurs in a co-receptor-dependent manner, and impair virus entry or viral infectivity of HIV-1 in producer cells [67]. It has been reported that IFITMs impeded HIV-1 differently with different tropism and that IFITMs-mediated inhibition of the virus entry is a co-receptor dependent process, wherein IFITM1 more potently restricts entry of CCR5 viruses, while IFITM2 and 3 are more effective in the blocking of CXCR4 entry [71,72], which is different from other reports [70,73].
Degradation of CD4 by Vpu/Env/Nef is also crucial for virus entry as well as superinfection, which has been well established in previous reports, even if the molecular mechanisms of these viral protein-mediated degradations of CD4 were disparate [18,19,20,21,22,23,24,74]. Since Vpu, Env, and Nef are expressed in a stage-specific manner of the HIV-1 life cycle—that is, Nef is expressed at early stages, while Env and Vpu are encoded from messages harboring Rev-responsive element (RRE) and thus at late stages of the HIV-1 life cycle—intracellular-expressed Nef at the early infection downregulate CD4 by enhancing its internalization and directing the receptor to lysosomes for degradation [20,75,76,77,78,79]. Nef is polyubiquitinated, and diubiquitination of the lysine 144 is required for Nef-mediated CD4 downregulation [80]. In contrast, Vpu and Env reduce the amount of CD4 on the infected cell surface by impairing transport of the newly synthesized CD4 to the cell surface at the late stage of the HIV-1 replication cycle [74,81,82]. Downregulation of the amount of CD4 on the surface of the infected cells by these intracellular viral proteins in HIV-1-infected cells impedes superinfection throughout the HIV-1 life cycle, which is reported to contribute to the prevention of premature cell death and thus to expand the period of effective virus production [21]. Taken together, all of these reports indicate that regulation of the amount of cellular proteins on the surface of the infected cells or of virions by the viral proteins via protein degradation processes plays an integral role in the regulation of virus entry, including superinfection, the initial step of the HIV-1 life cycle, into the host cells.
2.2.2. Intracellular Events
Uncoating: TRIM5α/Gag
It has been reported that HIV-1 does not replicate in nonhuman primates [83,84,85], and TRIM5α (TRIpartite Motif-containing protein 5α), a RING-type E3 ubiquitin ligase [86] interacting with the viral capsid, is responsible for the restriction of replication of the entered HIV-1 in a species-specific manner [25,87,88,89]. Specifically, TRIM5α of human or new world monkeys exerts little or no effect on HIV-1 replication, as opposed to that of rhesus (Macaca mulatta) or cynomolgus (Macaca fascicularis) monkeys [25,84,90,91], showing a key role of TRIM5α in the interspecies barrier for HIV-1 replication [26,27,28,29,30,31]. To elaborate the molecular process of TRIM5α-triggered inhibition of the virus replication, TRIM5α existing as a dimer in the target cell cytoplasm forms a complementary hexameric lattice upon interaction of the TRIM5α SPRY (SPIa and Ryanodine Receptors) domain with the capsid of a restriction-sensitive retrovirus, causing envelopment of the infectious core [92,93,94,95,96]. Then, TRIM5α increases intrinsic E3 ubiquitin ligase activity so that it is autocatalytic, covalently attaching ubiquitin to N-terminal of TRIM5α [95], and if avidity for the viral capsid protein were sufficient, the virion core uncoats prematurely and reverse transcription will be blocked [90,97,98], so that virus replication is ablated. These processes appear to be mediated by the UPS, since treatment with the proteasome inhibitors restores a normal decapsidation and reverse transcription, and TRIM5α and viral cores are co-localized with the proteasome [99,100,101].
Moreover, as noted above, interaction of TRIM5α with the viral capsid enhances its E3-Ub ligase activity and together with the E2 enzyme, UBC13/UVE1A (Ub-conjugating enzyme 13/Ub-conjugating enzyme variant 1A), stimulates TAK1 (Transforming growth factor β-Activated kinase 1), which in turn activates AP1 and NFκB signaling, which are critical for regulation of the expression of inflammatory chemokines and cytokines [94,102]. These reports indicate that in addition to direct antiviral activity, TRIM5α also plays an important role as an innate immune sensor for the retrovirus capsid lattice.
Reverse Transcription
(i) APOBEC/Vif
Apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like 3 (APOBEC3/A3), which induces the transition of cytosine to uracil on single-stranded DNA, is incorporated into budding HIV-1 virions and is thus carried over into the next infected cells [103]. During and after entry, viral genomic RNA is reverse transcribed to generate double-strand DNA, using viral reverse transcriptase, and in the process, APOBEC3G (A3G), prevents the production of functional viral protein by inducing hypermutations on single-stranded DNA, leading to C to U transitions [103,104,105,106,107,108]. These mutations can either be recognized by uracil DNA glycosylases, like the virion-associated UNG2 (Uracil N-Glycosylase 2), leading to the degradation of the provirus by abasic site endonucleases [109]. These mutations frequently produce mutated non-functional viral proteins and thereby impair HIV-1 replication.
HIV-1 Vif, a virion protein, essential for efficient virus replication by producing infectious progeny virus particles [110,111,112], is known to counteract cellular restriction factor APOBEC3G by precluding A3G incorporation into virions [105,113,114,115,116,117]. Specifically, binding of Vif to APOBEC3G induces the polyubiquitination of APOBEC3G followed by its proteasomal degradation, preventing its incorporation into HIV-1 virions [118,119]. In the process, Vif interacts with Cullin-RING Ub ligase (CRL) complex (Cullin5, Elongin-B and -C) to generate Vif-BC-Cul5 through a novel SOCS-box motif, and the Vif in the complex associates with APOBEC3G to induce the ubiquitination and degradation of APOBEC3G [115,120,121]. The Vif-BC-Cul5 complex is also known to trigger polyubiquitination followed by proteasomal degradation of another human cytidine deaminase APOBEC3F, another member of the APOBEC3 family, regulating HIV-1 replication [122].
Several previous reports have indicated that Vif-mediated degradation of the host restriction factor APOBEC3 for the inhibition of HIV-1 replication is also counter-regulated. Heat shock protein 70 (HSP70) disrupts interactions between Vif and APOBEC3G and thereby impedes ubiquitination followed by degradation of APOBEC3G [123], and HDAC6 enhances autophagic degradation of Vif to prevent Vif-triggered ubiquitination and degradation of APOBEC3G [124]. The stability of Vif itself is also regulated by various cellular proteins: Mdm2, an E3 Ub ligase, reduces the level of intracellular Vif by inducing ubiquitination and degradation [125,126], and core binding factor β (CBFβ), which is required for the formation of Vif-Cul5 E3 Ub ligase complex, reverses Mdm2-associated degradation of Vif by binding to Vif and thus blocking the interaction of Vif with Mdm2 [127,128,129] Further, proteasomal decay of Vif is also known to be regulated by cyclin F and two HECTs (Homologous to the E6-AP Carboxyl Terminuses)—AIP4 and NEDD [130].
(ii) SAMHD1/Vpr (Vpx)
Vpx is present only in HIV-2/SIVsmm/SIVmac, but not in HIV-1 and SIVcpz, while all primate immunodeficiency viruses harbor the vpr gene in their genomes [131,132,133]. Since it is believed that vpx has evolved from vpr, and these genes share many functional similarities, it is believed that lack of vpx in HIV-1 can be compensated by the presence of vpr [134], which is strongly supported by Gibbs et al., wherein deletion of either gene of SIVmac239 progresses with AIDS to a terminal stage, even if virus burden in the infected rhesus monkeys were lowered and declines in CD4+ lymphocytes were delayed, while deletion of both genes severely lowers virus burdens and hence shows no evidence of disease progressions [135]. One of the similarities is that both Vpx and Vpr play a critical role in the ubiquitination followed by degradation of SAMHD1 (Sterile Alpha Motif and Histidine Aspartate domain-containing protein 1), which inhibits reverse transcription by depleting the pool of cellular deoxy Nucleotide Triphosphates (dNTPs) [136,137,138]. For degradation of SAMHD1, Vpx or Vpr recruits the CUL4A-DDB1-DCAF1 (DDB1 and CUL4-associated factor 1) E3 ubiquitin ligase by a direct interaction with its substrate recognition protein, DCAF1 [139], which induces the ubiquitination followed by degradation of SAMHD loaded to the complex by interacting its C-terminal domain with Vpx and Vpr [139,140,141,142]; that is, Vpx and Vpr can enhance diverse primate immunodeficiency virus replication by inducing degradation of the host restriction factor, SAMHD1, and thereby increasing reverse transcription activity.
Recent report showed that Vpx and Vpr also associates with the human silencing hub (HUSH) complex [143,144], FAM208A (TASOR/RAP140), MPHOSPH8 (MPP8), PPHLN1 (PERIPHILIN), and MORC2 [145,146,147], which restricts the replication of primate immunodeficiency viruses [143,144]. It has been reported that Vpx and Vpr counteract the HUSH complex-mediated inhibition of the virus replication by reducing the steady-state level of these proteins in a DCAF1/CUL4A/B/proteasome-dependent manner [148,149], providing explanations of the impacts of Vpx and Vpr on the reporter gene expression, which is not illustrated by SAMHD1 degradation [150,151,152]. Taken together, these reports show that regulation of the stability of Vpx/Vpr and their corresponding cellular partners plays an integral role in the restriction of the infected primate immunodeficiency viruses and the infected cells in regard to their survival and proliferation.
Integration of Provirus into Host Chromosome
The reverse transcribed double-strand DNA is transported to the nucleus and incorporated into the host chromosome, wherein viral protein, integrase (IN), plays a main role. Since IN comprises the N-terminal phenylalanine, N-degron signals, the protein is susceptible to rapid degradation by 26S proteasome followed by the class of E3 Ub ligases [153,154]. Substitution of the N-terminal amino acid to methionine increases, but the protein is still short-lived [154,155,156,157,158], indicating that IN is degraded through the proteasomal pathway, independent of N-terminal recognition. In fact, Ali et al. reported that the E3 RING ligase, TRIM33, plays a major role in the determination of the stability of IN by binding to the carboxy-terminal domain of IN through its RING portion and determining its poly-ubiquitination, and lack of TRIM33 rescues the infectivity of HIV-1 [159]. These data indicate that HIV-1 infectivity is impeded by the TRIM33-mediated decrease of IN function by reducing IN stability.
HIV-1 Replication (Viral Gene Expression)
(i) Tat
After the establishment of HIV-1 infection by inserting the reverse-transcribed double-strand viral DNA into the host chromosome, HIV-1 Tat increases the steady-state levels of all the viral transcripts by interacting with the TAR (trans-activating responsive) element from the integrated proviral DNA and thereby HIV-1 replication [160,161,162,163,164,165]. Molecular mechanisms on how Tat augments viral gene expression have been thoroughly investigated, while elimination processes of the protein, after it completes its duty, have not been studied comprehensively; that is, regulation of the stability of HIV-1 Tat is key to understanding HIV-1 replication and thus HIV-1-associated pathogenicity. First, it has been reported that HIV-1 Rev causes specific degradation of cytoplasmic Tat and thus inhibits HIV-1 replication [166]. According to the report, Rev induces the degradation of Tat indirectly by down-modulating the expression of NQO1 [NAD(P)H:quinine oxidoreductase 1], which contributes to the stabilization of the Tat protein in a dose-dependent manner, and in the process, the nuclear export signal of Rev is critical [166]. Another report indicated that Nef suppresses the nuclear localization of Tat and thus triggers the degradation of Tat by the proteasome-dependent pathway in the cytoplasm through Hsp70 [36], which is also known to inhibit Vif-mediated ubiquitination and degradation of APOBEC3G [123]. In addition, nucleocapsid (NC) reduces the amount of the intracellular Tat, which is reversed by treatment of MG132 [167]. However, the level of ubiquitination of Tat by NC is unchanged, suggesting that NC-mediated degradation of Tat is ubiquitination-independent [167].
Besides viral proteins, the interplay between Tat and cellular elements involved in proteasomal degradation modulates the replicability of HIV-1. Tat stabilizes Mdm2, an E3 Ub ligase, by activating Akt, which enhances Tat-mediated viral replication [168]. PJA2, a RING finger E3 ligase, polyubiquitinates Tat in a non-degradative manner and regulates the elongation step of transcription of Tat in P-TEFb complex [169]. A recent report showed that a type of long noncoding RNA, called NRON, induces degradation of Tat by directly linking Tat to the Ub proteasome components, including CUL4B and PSMD11, and thus develops latent infection of HIV-1 [170].
(ii) Nef
Activation of the resting CD4+ T cells is critical for replication of the infected HIV-1 [171,172,173,174], and Nef is known to activate the cells by stimulating various signaling molecules [175,176,177,178]. That is, proteasomal degradation of key signaling proteins involved in the T cell activation process by Nef alters the replicability of the infected HIV-1 into its susceptible cells. Further, Simmons et al. showed that Nef, in conjunction with c-Cbl, excludes the E2 Ub-conjugating enzyme, UbcH7, from lipid rafts, and suppression of p85Cool-1/βPix in the ternary complex amongst Nef/c-Cbl/p85Coo1/βPix in the lipid rafts attenuates HIV-1 replication [179]. Our unpublished data indicate that UBE3A (Ub protein ligase E3A, also known as E6AP—an E6 associated protein) which causes E6-mediated p53 degradation in HPV (human papilloma virus) interacts with Nef and inhibits HIV-1 replication. Further, a previous report also showed that the addition of MG132 or lactacystin, each a specific inhibitor of cellular proteasome activity, preferentially increases cellular permissiveness of nef-deficient rather than wild type HIV-1 [180]. Taken together, all these reports denote the significance of the proteasomal degradation in the regulation of HIV-1 replication.
2.2.3. Release of Virus Particles—Budding:
(i) Tetherin/Vpu
At the last stage of the HIV-1 life cycle, the newly assembled virions remain tethered to the plasma membrane without being released, and are eventually endocytosed and degraded in the absence of Vpu [181,182], demonstrating that Vpu is required to release the assembled virus particles to the outside of the infected cells. Virion-tethering to the plasma membrane is mediated by two domains—a transmembrane domain proximal to the N-terminus and an extracellular C-terminal glycosyl-phosphatidylinositol anchor domain—of tetherin/BST-2 [182], and interaction of Vpu with tetherin is known to sequester BST-2 away from the virion budding sites in intracellular compartments, particularly the trans-Golgi-network, thereby allowing the release of the assembled virions [182,183,184,185,186]. In the process, Vpu recruits E3 ubiquitin ligase adaptor, β-TrCP, by the cytoplasmic DSGxxS motif [187], which may lead to ubiquitination of tetherin by the cellular E3 ubiquitin ligases, March8 and NEDD4 (Neural precursor cell Expressed Developmentally Down-regulated protein 4) [188] followed by degradation in the endo-lysosomal system [185,189].
Vpu-triggered downregulation of tetherin on the surface of the HIV-1-infected cells is also known to be achieved without involvement of β-TrCP [185,190,191]. Vpu binds and targets tetherin to ESCRT (Endosomal Sorting Complex Required for Transporter)-dependent endosomal degradation via interaction with clathrin adaptor AP-1 and -2, wherein phosphorylation of Vpu is required [192,193,194]. Reduction of the surface expression of tetherin by Vpu is also exploited by the autophagy pathway. Vpu induces the removal of tetherin from HIV-1 budding sites by interacting with ATG8 (Autophagy related protein 8), ortholog LC3, thereby leading to phagocytosis [195]. However, Vpu-associated degradation of tetherin might not be an absolute requirement for budding of the assembled virions, since Vpu is capable of intracellular sequestration of tetherin without degradation [34,186].
When Vpu is deficient, as is observed with SIVagm, SIVblu, and SIVmac, Nef takes over the role of tetherin counteraction [32,183,184], and in HIV-2, Env substitutes for Vpu function in releasing virions [196,197]. Thus, efficient virus budding in the vpu-deficient primate immunodeficiency viruses is implemented by other viral genes.
(ii) ESCRT/NEDD/Gag
Two late (L) domains, the PTAP and YPXnL, (where X refers to any amino acid and n = 1 to 3 residues) identified within the p6 region of the HIV-1 Gag play an essential role in the budding of the assembled virus, which is confirmed by the mutational analyses in the motifs [198,199]. The PTAP motif binds to the cellular protein Tsg101 [200,201,202], which functions in HIV-1 budding [200,203,204], while LYPXnL recruits Alix/AIP-1 [205,206]. Over-expression of a fragment containing Bro1 with the adjacent V domain of Alix inhibits release via both the PTAP/Tsg101 and the LYPXnL/Alix pathways in a nucleocapsid (NC)-dependent manner, and provision of a link to the ESCRT machinery through NEDD4-2s over-expression rescues NC mutant-associated release defects [207,208], indicating that the NC region of HIV-1 Gag cooperates with the two L domains to recruit the cellular machinery necessary for viral budding [209] and NEDD-like ligase is critical to function in virus release [207,208,209], illustrating the significance of the ESCRT/NEDD pathway in the release of the virus.
3. Concluding Remarks and Future Directions
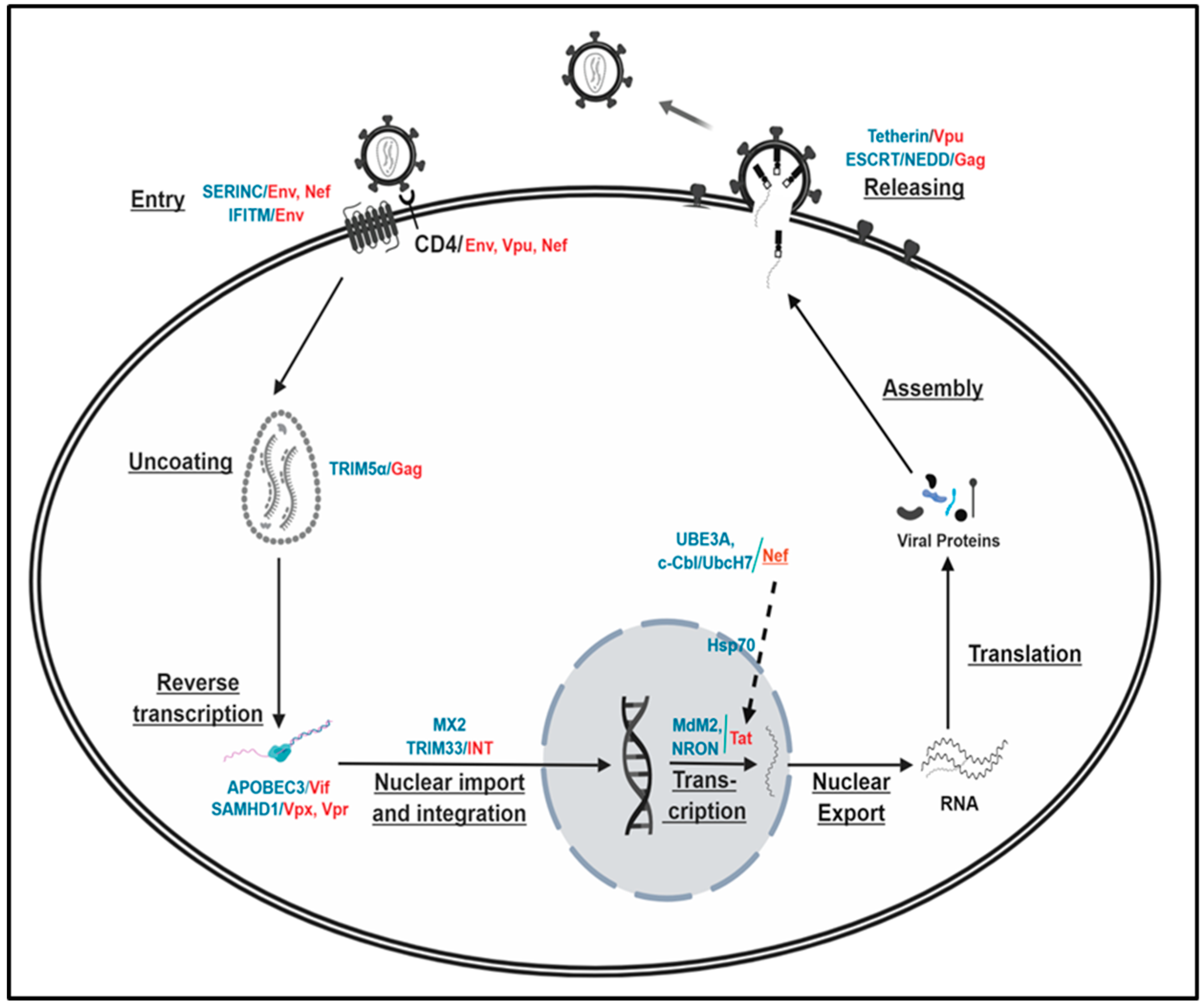
The above assessments reveal that the proteasomal degradation plays an integral role in regulating the degradation of viral and cellular proteins at every step of the HIV-1 life cycle, which is vital for the survival and proliferation of the virus and the HIV-1-infected host cells. However, even if the molecular processes of the individual degradation event in the HIV-1 life cycle were well elucidated, as described above and Figure 1, the significance of the degradation of certain viral and/or cellular proteins with respect to the virus replication is still elusive. One of the reasons is the dispensability of certain viral genes in the virus replication. For instance, vpr, vpx, or nef are basically dispensable for efficient virus replications in the T cell line [54,55,135,210,211], and thus the impact of the proteasomal decay of these proteins encoded from these genes on the replicability of the primate immunodeficiency viruses is obscure. Another important point is that multiple viral proteins are involved in the degradation of the identical target molecules, and thus the specific contribution of each viral protein to the virus replication by proteasomal degradation of the target protein could be murky. For instance, both Env and Nef affect SERINC-associated virus entry, Vpu/Env/Nef regulate the down-modulation of CD4 on the HIV-1-infected cell surface, and both Vpx and Vpr in HIV-2 and certain SIVs alter reverse transcription activity by interplaying with SAMDH, as described above. Finally, there is a need for the elucidation of the molecular mechanism(s) regarding how these viral proteins that localize different subcellular organelles in the infected cells and are expressed at different stages of HIV-1 replication cycle (early vs. late) coordinate or orchestrate protein degradation. It is also needed to answer questions about how the identical cellular partners regulate or are regulated by the same viral element. Elucidation of how these particular events are coordinated among different virus stages, and of which molecules play key roles in viral and cellular protein fate determination, will fortify our understanding of fine-scale life cycle stage transitions and proteasomal degradation-mediated HIV-1 pathogenicity, fostering antiviral therapeutics targeting a specific step in proteasome degradation processes.
Author Contributions
V.K.R. and I.-W.P. wrote and revised this manuscript for publication, and both authors read and approved the final version of the manuscript.
Funding
This work was supported by R01DK099055 (I.-W.P).
Conflicts of Interest
The authors have declared that no competing interests exist.
References
- Haas, A.L.; Warms, J.V.; Hershko, A.; Rose, I.A. Ubiquitin-activating enzyme. Mechanism and role in protein-ubiquitin conjugation. J. Biol. Chem. 1982, 257, 2543–2548. [Google Scholar] [PubMed]
- Thrower, J.S.; Hoffman, L.; Rechsteiner, M.; Pickart, C.M. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000, 19, 94–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsasser, S.; Finley, D. Delivery of ubiquitinated substrates to protein-unfolding machines. Nat. Cell Biol. 2005, 7, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.W.; Li, X.; Thompson, D.; Wooding, K.; Chang, T.L.; Tang, Z.; Yu, H.; Thomas, P.J.; DeMartino, G.N. Atp binding and atp hydrolysis play distinct roles in the function of 26s proteasome. Mol. Cell 2006, 24, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Nijman, S.M.; Luna-Vargas, M.P.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.; Sixma, T.K.; Bernards, R. A genomic and functional inventory of deubiquitinating enzymes. Cell 2005, 123, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Chesnel, F.; Bazile, F.; Pascal, A.; Kubiak, J.Z. Cyclin b dissociation from cdk1 precedes its degradation upon mpf inactivation in mitotic extracts of xenopus laevis embryos. Cell Cycle 2006, 5, 1687–1698. [Google Scholar] [CrossRef] [PubMed]
- Brito, D.A.; Rieder, C.L. Mitotic checkpoint slippage in humans occurs via cyclin b destruction in the presence of an active checkpoint. Curr. Biol. 2006, 16, 1194–1200. [Google Scholar] [CrossRef] [PubMed]
- Havens, C.G.; Ho, A.; Yoshioka, N.; Dowdy, S.F. Regulation of late g1/s phase transition and apc cdh1 by reactive oxygen species. Mol. Cell. Biol. 2006, 26, 4701–4711. [Google Scholar] [CrossRef]
- Gupta, I.; Singh, K.; Varshney, N.K.; Khan, S. Delineating crosstalk mechanisms of the ubiquitin proteasome system that regulate apoptosis. Front. Cell Dev. Biol. 2018, 6, 11. [Google Scholar] [CrossRef]
- VerPlank, J.J.S.; Goldberg, A.L. Regulating protein breakdown through proteasome phosphorylation. Biochem. J. 2017, 474, 3355–3371. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, Y.; Nakabayashi, O.; Nakano, H. Flip the switch: Regulation of apoptosis and necroptosis by cflip. Int. J. Mol. Sci. 2015, 16, 30321–30341. [Google Scholar] [CrossRef] [PubMed]
- Cascio, P.; Hilton, C.; Kisselev, A.F.; Rock, K.L.; Goldberg, A.L. 26s proteasomes and immunoproteasomes produce mainly n-Extended versions of an antigenic peptide. EMBO J. 2001, 20, 2357–2366. [Google Scholar] [CrossRef] [PubMed]
- Mallery, D.L.; McEwan, W.A.; Bidgood, S.R.; Towers, G.J.; Johnson, C.M.; James, L.C. Antibodies mediate intracellular immunity through tripartite motif-Containing 21 (trim21). Proc. Natl. Acad. Sci. USA 2010, 107, 19985–19990. [Google Scholar] [CrossRef] [PubMed]
- Murata, S.; Sasaki, K.; Kishimoto, T.; Niwa, S.; Hayashi, H.; Takahama, Y.; Tanaka, K. Regulation of CD8+ t cell development by thymus-specific proteasomes. Science 2007, 316, 1349–1353. [Google Scholar] [CrossRef]
- Kleiger, G.; Mayor, T. Perilous journey: A tour of the ubiquitin-proteasome system. Trends Cell Biol. 2014, 24, 352–359. [Google Scholar] [CrossRef]
- Ben-Neriah, Y. Regulatory functions of ubiquitination in the immune system. Nat. Immunol. 2002, 3, 20–26. [Google Scholar] [CrossRef]
- Predmore, J.M.; Wang, P.; Davis, F.; Bartolone, S.; Westfall, M.V.; Dyke, D.B.; Pagani, F.; Powell, S.R.; Day, S.M. Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies. Circulation 2010, 121, 997–1004. [Google Scholar] [CrossRef]
- Garcia, J.V.; Miller, A.D. Downregulation of cell surface CD4 by nef. Res. Virol. 1992, 143, 52–55. [Google Scholar] [CrossRef]
- Mariani, R.; Skowronski, J. Cd4 down-Regulation by nef alleles isolated from human immunodeficiency virus type 1-infected individuals. Proc. Natl. Acad. Sci. USA 1993, 90, 5549–5553. [Google Scholar] [CrossRef]
- Aiken, C.; Konner, J.; Landau, N.R.; Lenburg, M.E.; Trono, D. Nef induces CD4 endocytosis: Requirement for a critical dileucine motif in the membrane-Proximal CD4 cytoplasmic domain. Cell 1994, 76, 853–864. [Google Scholar] [CrossRef]
- Wildum, S.; Schindler, M.; Munch, J.; Kirchhoff, F. Contribution of vpu, env, and nef to CD4 down-modulation and resistance of human immunodeficiency virus type 1-Infected t cells to superinfection. J. Virol. 2006, 80, 8047–8059. [Google Scholar] [CrossRef] [PubMed]
- Lama, J.; Mangasarian, A.; Trono, D. Cell-Surface expression of CD4 reduces HIV-1 infectivity by blocking env incorporation in a nef-and vpu-Inhibitable manner. Curr. Biol. 1999, 9, 622–631. [Google Scholar] [CrossRef]
- Ruiz, A.; Guatelli, J.C.; Stephens, E.B. The vpu protein: New concepts in virus release and CD4 down-modulation. Curr. HIV Res. 2010, 8, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Levesque, K.; Finzi, A.; Binette, J.; Cohen, E.A. Role of CD4 receptor down-Regulation during HIV-1 infection. Curr. HIV Res. 2004, 2, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The cytoplasmic body component trim5alpha restricts HIV-1 infection in old world monkeys. Nature 2004, 427, 848–853. [Google Scholar] [CrossRef]
- Shi, J.; Aiken, C. Saturation of trim5 alpha-mediated restriction of HIV-1 infection depends on the stability of the incoming viral capsid. Virolgy 2006, 350, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, R.; Noser, J.A.; Ohmine, S.; Ikeda, Y. Rhesus monkey trim5alpha restricts HIV-1 production through rapid degradation of viral gag polyproteins. Nat. Med. 2007, 13, 631–635. [Google Scholar] [CrossRef]
- Chatterji, U.; Bobardt, M.D.; Gaskill, P.; Sheeter, D.; Fox, H.; Gallay, P.A. Trim5alpha accelerates degradation of cytosolic capsid associated with productive HIV-1 entry. J. Biol. Chem. 2006, 281, 37025–37033. [Google Scholar] [CrossRef]
- Zhang, F.; Perez-Caballero, D.; Hatziioannou, T.; Bieniasz, P.D. No effect of endogenous trim5alpha on HIV-1 production. Nat. Med. 2008, 14, 235–236. [Google Scholar] [CrossRef]
- Arriagada, G.; Muntean, L.N.; Goff, S.P. Sumo-interacting motifs of human trim5alpha are important for antiviral activity. PLoS Pathog. 2011, 7, e1002019. [Google Scholar] [CrossRef]
- Battivelli, E.; Lecossier, D.; Matsuoka, S.; Migraine, J.; Clavel, F.; Hance, A.J. Strain-specific differences in the impact of human trim5alpha, different trim5alpha alleles, and the inhibition of capsid-cyclophilin a interactions on the infectivity of HIV-1. J. Virol. 2010, 84, 11010–11019. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Miyagi, E.; Andrew, A.J.; Kao, S.; Strebel, K. Vpu enhances HIV-1 virus release in the absence of bst-2 cell surface down-Modulation and intracellular depletion. Proc. Natl. Acad. Sci. USA 2009, 106, 2868–2873. [Google Scholar] [CrossRef] [PubMed]
- McNatt, M.W.; Zang, T.; Hatziioannou, T.; Bartlett, M.; Fofana, I.B.; Johnson, W.E.; Neil, S.J.; Bieniasz, P.D. Species-specific activity of HIV-1 vpu and positive selection of tetherin transmembrane domain variants. PLoS Pathog. 2009, 5, e1000300. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Hultquist, J.F.; Evans, D.T. The restriction factors of human immunodeficiency virus. J. Biol. Chem. 2012, 287, 40875–40883. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, R.; Naganuma, H.; Nishitsuji, H.; Takaku, H. Human immunodeficiency virus-1 nef suppresses hsp70-Mediated tat activation. FEBS Lett. 2011, 585, 3367–3371. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wu, J.; Chavez, L.; Hoh, R.; Deeks, S.G.; Pillai, S.K.; Zhou, Q. Reiterative enrichment and authentication of crispri targets (react) identifies the proteasome as a key contributor to HIV-1 latency. PLoS Pathog. 2019, 15, e1007498. [Google Scholar] [CrossRef] [PubMed]
- Arens, M.; Joseph, T.; Nag, S.; Miller, J.P.; Powderly, W.G.; Ratner, L. Alterations in spliced and unspliced HIV-1-Specific rna detection in peripheral blood mononuclear cells of individuals with varying CD4-Positive lymphocyte counts. AIDS Res. Hum. Retrovir. 1993, 9, 1257–1263. [Google Scholar] [CrossRef]
- Pavlakis, G.N.; Felber, B.K. Regulation of expression of human immunodeficiency virus. New Biol. 1990, 2, 20–31. [Google Scholar]
- Malim, M.H.; Tiley, L.S.; McCarn, D.F.; Rusche, J.R.; Hauber, J.; Cullen, B.R. HIV-1 structural gene expression requires binding of the rev trans-activator to its rna target sequence. Cell 1990, 60, 675–683. [Google Scholar] [CrossRef]
- Vaishnav, Y.N.; Vaishnav, M.; Wong-Staal, F. Identification and characterization of a nuclear factor that specifically binds to the rev response element (rre) of human immunodeficiency virus type 1 (HIV-1). New Biol. 1991, 3, 142–150. [Google Scholar] [PubMed]
- Inuzuka, M.; Hayakawa, M.; Ingi, T. Serinc, an activity-regulated protein family, incorporates serine into membrane lipid synthesis. J. Biol. Chem. 2005, 280, 35776–35783. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.; Chande, A.; Ziglio, S.; De Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 nef promotes infection by excluding serinc5 from virion incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Chande, A.; Cuccurullo, E.C.; Rosa, A.; Ziglio, S.; Carpenter, S.; Pizzato, M. S2 from equine infectious anemia virus is an infectivity factor which counteracts the retroviral inhibitors serinc5 and serinc3. Proc. Natl. Acad. Sci. USA 2016, 113, 13197–13202. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Wu, Y.; Gottlinger, H.G. Serinc3 and serinc5 restrict HIV-1 infectivity and are counteracted by nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Pereira, E.A.; daSilva, L.L. HIV-1 nef: Taking control of protein trafficking. Traffic 2016, 17, 976–996. [Google Scholar] [CrossRef] [PubMed]
- Trautz, B.; Pierini, V.; Wombacher, R.; Stolp, B.; Chase, A.J.; Pizzato, M.; Fackler, O.T. The antagonism of HIV-1 nef to serinc5 particle infectivity restriction involves the counteraction of virion-associated pools of the restriction factor. J. Virol. 2016, 90, 10915–10927. [Google Scholar] [CrossRef] [PubMed]
- Welker, R.; Kottler, H.; Kalbitzer, H.R.; Krausslich, H.G. Human immunodeficiency virus type 1 nef protein is incorporated into virus particles and specifically cleaved by the viral proteinase. Virology 1996, 219, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Pandori, M.W.; Fitch, N.J.; Craig, H.M.; Richman, D.D.; Spina, C.A.; Guatelli, J.C. Producer-Cell modification of human immunodeficiency virus type 1: Nef is a virion protein. J. Virol. 1996, 70, 4283–4290. [Google Scholar]
- Bukovsky, A.A.; Dorfman, T.; Weimann, A.; Gottlinger, H.G. Nef association with human immunodeficiency virus type 1 virions and cleavage by the viral protease. J. Virol. 1997, 71, 1013–1018. [Google Scholar] [Green Version]
- Welker, R.; Harris, M.; Cardel, B.; Krausslich, H.G. Virion incorporation of human immunodeficiency virus type 1 nef is mediated by a bipartite membrane-Targeting signal: Analysis of its role in enhancement of viral infectivity. J. Virol. 1998, 72, 8833–8840. [Google Scholar] [PubMed]
- Beitari, S.; Ding, S.; Pan, Q.; Finzi, A.; Liang, C. Effect of HIV-1 env on serinc5 antagonism. J. Virol. 2017, 91, e02214-16. [Google Scholar] [CrossRef] [PubMed]
- Sood, C.; Marin, M.; Chande, A.; Pizzato, M.; Melikyan, G.B. Serinc5 protein inhibits HIV-1 fusion pore formation by promoting functional inactivation of envelope glycoproteins. J. Biol. Chem. 2017, 292, 6014–6026. [Google Scholar] [CrossRef] [PubMed]
- Basmaciogullari, S.; Pizzato, M. The activity of nef on HIV-1 infectivity. Front. Microbiol. 2014, 5, 232. [Google Scholar] [CrossRef] [PubMed]
- Hammes, S.R.; Dixon, E.P.; Malim, M.H.; Cullen, B.R.; Greene, W.C. Nef protein of human immunodeficiency virus type 1: Evidence against its role as a transcriptional inhibitor. Proc. Natl. Acad. Sci. USA 1989, 86, 9549–9553. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Ikeuchi, K.; Byrn, R.; Groopman, J.; Baltimore, D. Lack of a negative influence on viral growth by the nef gene of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA 1989, 86, 9544–9548. [Google Scholar] [CrossRef] [PubMed]
- Kmiec, D.; Akbil, B.; Ananth, S.; Hotter, D.; Sparrer, K.M.J.; Sturzel, C.M.; Trautz, B.; Ayouba, A.; Peeters, M.; Yao, Z.; et al. Sivcol nef counteracts serinc5 by promoting its proteasomal degradation but does not efficiently enhance HIV-1 replication in human CD4+ t cells and lymphoid tissue. PLoS Pathog. 2018, 14, e1007269. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Broder, C.C.; Kennedy, P.E.; Berger, E.A. HIV-1 entry cofactor: Functional cdna cloning of a seven-transmembrane, g protein-Coupled receptor. Science 1996, 272, 872–877. [Google Scholar] [CrossRef]
- Deng, H.; Liu, R.; Ellmeier, W.; Choe, S.; Unutmaz, D.; Burkhart, M.; Di Marzio, P.; Marmon, S.; Sutton, R.E.; Hill, C.M.; et al. Identification of a major co-Receptor for primary isolates of HIV-1. Nature 1996, 381, 661–666. [Google Scholar] [CrossRef]
- Simmons, G.; Wilkinson, D.; Reeves, J.D.; Dittmar, M.T.; Beddows, S.; Weber, J.; Carnegie, G.; Desselberger, U.; Gray, P.W.; Weiss, R.A.; et al. Primary, syncytium-Inducing human immunodeficiency virus type 1 isolates are dual-Tropic and most can use either lestr or ccr5 as coreceptors for virus entry. J. Virol. 1996, 70, 8355–8360. [Google Scholar]
- Moore, J.P.; Trkola, A.; Dragic, T. Co-Receptors for HIV-1 entry. Curr. Opin. Immunol. 1997, 9, 551–562. [Google Scholar] [CrossRef]
- Burny, A.; Bex, F.; Brasseur, R.; Khim, M.C.; Delchambre, M.; Horth, M.; Verdin, E. Human immunodeficiency virus cell entry: New insights into the fusion mechanism. J. Acquir. Immune Defic. Syndr. 1988, 1, 579–582. [Google Scholar] [PubMed]
- Bergeron, L.; Sullivan, N.; Sodroski, J. Target cell-Specific determinants of membrane fusion within the human immunodeficiency virus type 1 gp120 third variable region and gp41 amino terminus. J. Virol. 1992, 66, 2389–2397. [Google Scholar] [PubMed]
- Bailey, C.C.; Zhong, G.; Huang, I.C.; Farzan, M. Ifitm-Family proteins: The cell’s first line of antiviral defense. Annu. Rev. Virol. 2014, 1, 261–283. [Google Scholar] [CrossRef] [PubMed]
- Perreira, J.M.; Chin, C.R.; Feeley, E.M.; Brass, A.L. Ifitms restrict the replication of multiple pathogenic viruses. J. Mol. Biol. 2013, 425, 4937–4955. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Pan, Q.; Rong, L.; He, W.; Liu, S.L.; Liang, C. The ifitm proteins inhibit HIV-1 infection. J. Virol. 2011, 85, 2126–2137. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Liu, S.L. The inhibition of HIV-1 entry imposed by interferon inducible transmembrane proteins is independent of co-Receptor usage. Viruses 2018, 10, 413. [Google Scholar] [CrossRef] [PubMed]
- Tartour, K.; Appourchaux, R.; Gaillard, J.; Nguyen, X.N.; Durand, S.; Turpin, J.; Beaumont, E.; Roch, E.; Berger, G.; Mahieux, R.; et al. Ifitm proteins are incorporated onto HIV-1 virion particles and negatively imprint their infectivity. RetroVirology 2014, 11, 103. [Google Scholar] [CrossRef]
- Compton, A.A.; Bruel, T.; Porrot, F.; Mallet, A.; Sachse, M.; Euvrard, M.; Liang, C.; Casartelli, N.; Schwartz, O. Ifitm proteins incorporated into HIV-1 virions impair viral fusion and spread. Cell Host Microbe 2014, 16, 736–747. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, M.; Wilkins, J.; Ding, S.; Swartz, T.H.; Esposito, A.M.; Zheng, Y.M.; Freed, E.O.; Liang, C.; Chen, B.K.; et al. Ifitm proteins restrict HIV-1 infection by antagonizing the envelope glycoprotein. Cell Rep. 2015, 13, 145–156. [Google Scholar] [CrossRef]
- Wang, Y.; Pan, Q.; Ding, S.; Wang, Z.; Yu, J.; Finzi, A.; Liu, S.L.; Liang, C. The v3 loop of HIV-1 env determines viral susceptibility to ifitm3 impairment of viral infectivity. J. Virol. 2017, 91, e02441-16. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.L.; Wilson, H.; Iyer, S.S.; Coss, K.; Doores, K.; Smith, S.; Kellam, P.; Finzi, A.; Borrow, P.; Hahn, B.H.; et al. Resistance of transmitted founder HIV-1 to ifitm-Mediated restriction. Cell Host Microbe 2016, 20, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Le Duff, Y.; Wang, Y.; Pan, Q.; Ding, S.; Zheng, Y.M.; Liu, S.L.; Liang, C. Primate lentiviruses are differentially inhibited by interferon-induced transmembrane proteins. Virology 2015, 474, 10–18. [Google Scholar] [CrossRef]
- Willey, R.L.; Maldarelli, F.; Martin, M.A.; Strebel, K. Human immunodeficiency virus type 1 vpu protein induces rapid degradation of CD4. J. Virol. 1992, 66, 7193–7200. [Google Scholar] [PubMed]
- Bresnahan, P.A.; Yonemoto, W.; Ferrell, S.; Williams-Herman, D.; Geleziunas, R.; Greene, W.C. A dileucine motif in HIV-1 nef acts as an internalization signal for CD4 downregulation and binds the ap-1 clathrin adaptor. Curr. Biol. 1998, 8, 1235–1238. [Google Scholar] [CrossRef]
- Craig, H.M.; Pandori, M.W.; Guatelli, J.C. Interaction of HIV-1 nef with the cellular dileucine-based sorting pathway is required for CD4 down-regulation and optimal viral infectivity. Proc. Natl. Acad. Sci. USA 1998, 95, 11229–11234. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.; DeTulleo, L.; Rapoport, I.; Skowronski, J.; Kirchhausen, T. A dileucine motif in HIV-1 nef is essential for sorting into clathrin-coated pits and for downregulation of CD4. Curr. Biol. 1998, 8, 1239–1242. [Google Scholar] [CrossRef]
- Mangasarian, A.; Foti, M.; Aiken, C.; Chin, D.; Carpentier, J.L.; Trono, D. The HIV-1 nef protein acts as a connector with sorting pathways in the golgi and at the plasma membrane. Immunity 1997, 6, 67–77. [Google Scholar] [CrossRef]
- Piguet, V.; Chen, Y.L.; Mangasarian, A.; Foti, M.; Carpentier, J.L.; Trono, D. Mechanism of nef-induced CD4 endocytosis: Nef connects CD4 with the mu chain of adaptor complexes. EMBO J. 1998, 17, 2472–2481. [Google Scholar] [CrossRef]
- Jin, Y.J.; Cai, C.Y.; Zhang, X.; Burakoff, S.J. Lysine 144, a ubiquitin attachment site in HIV-1 nef, is required for nef-Mediated CD4 down-Regulation. J. Immunol. 2008, 180, 7878–7886. [Google Scholar] [CrossRef]
- Geleziunas, R.; Bour, S.; Wainberg, M.A. Cell surface down-Modulation of CD4 after infection by HIV-1. FASEB J. 1994, 8, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.H.; Plemenitas, A.; Fielding, C.J.; Peterlin, B.M. Nef increases the synthesis of and transports cholesterol to lipid rafts and HIV-1 progeny virions. Proc. Natl. Acad. Sci. USA 2003, 100, 8460–8465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balzarini, J.; De Clercq, E.; Uberla, K. Siv/HIV-1 hybrid virus expressing the reverse transcriptase gene of HIV-1 remains sensitive to HIV-1-Specific reverse transcriptase inhibitors after passage in rhesus macaques. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1997, 15, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Himathongkham, S.; Luciw, P.A. Restriction of HIV-1 (subtype b) replication at the entry step in rhesus macaque cells. Virology 1996, 219, 485–488. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, W.; Schubert, D.; LaBonte, J.; Munson, L.; Gibson, S.; Scammell, J.; Ferrigno, P.; Sodroski, J. Species-Specific, postentry barriers to primate immunodeficiency virus infection. J. Virol. 1999, 73, 10020–10028. [Google Scholar]
- Han, K.; Lou, D.I.; Sawyer, S.L. Identification of a genomic reservoir for new trim genes in primate genomes. PLoS Genet. 2011, 7, e1002388. [Google Scholar] [CrossRef] [PubMed]
- Besnier, C.; Takeuchi, Y.; Towers, G. Restriction of lentivirus in monkeys. Proc. Natl. Acad. Sci. USA 2002, 99, 11920–11925. [Google Scholar] [CrossRef] [Green Version]
- Cowan, S.; Hatziioannou, T.; Cunningham, T.; Muesing, M.A.; Gottlinger, H.G.; Bieniasz, P.D. Cellular inhibitors with fv1-like activity restrict human and simian immunodeficiency virus tropism. Proc. Natl. Acad. Sci. USA 2002, 99, 11914–11919. [Google Scholar] [CrossRef]
- Munk, C.; Brandt, S.M.; Lucero, G.; Landau, N.R. A dominant block to HIV-1 replication at reverse transcription in simian cells. Proc. Natl. Acad. Sci. USA 2002, 99, 13843–13848. [Google Scholar] [CrossRef] [Green Version]
- Stremlau, M.; Perron, M.; Lee, M.; Li, Y.; Song, B.; Javanbakht, H.; Diaz-Griffero, F.; Anderson, D.J.; Sundquist, W.I.; Sodroski, J. Specific recognition and accelerated uncoating of retroviral capsids by the trim5alpha restriction factor. Proc. Natl. Acad. Sci. USA 2006, 103, 5514–5519. [Google Scholar] [CrossRef]
- Shibata, R.; Sakai, H.; Kawamura, M.; Tokunaga, K.; Adachi, A. Early replication block of human immunodeficiency virus type 1 in monkey cells. J. Gen. Virol. 1995, 76, 2723–2730. [Google Scholar] [CrossRef] [PubMed]
- Ganser-Pornillos, B.K.; Chandrasekaran, V.; Pornillos, O.; Sodroski, J.G.; Sundquist, W.I.; Yeager, M. Hexagonal assembly of a restricting trim5alpha protein. Proc. Natl. Acad. Sci. USA 2011, 108, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Pornillos, O.; Ganser-Pornillos, B.K.; Kelly, B.N.; Hua, Y.; Whitby, F.G.; Stout, C.D.; Sundquist, W.I.; Hill, C.P.; Yeager, M. X-Ray structures of the hexameric building block of the HIV capsid. Cell 2009, 137, 1282–1292. [Google Scholar] [CrossRef] [PubMed]
- Pertel, T.; Hausmann, S.; Morger, D.; Zuger, S.; Guerra, J.; Lascano, J.; Reinhard, C.; Santoni, F.A.; Uchil, P.D.; Chatel, L.; et al. Trim5 is an innate immune sensor for the retrovirus capsid lattice. Nature 2011, 472, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, A.J.; Vaysburd, M.; Maslen, S.; Zeng, J.; Skehel, J.M.; Towers, G.J.; James, L.C. Trivalent ring assembly on retroviral capsids activates trim5 ubiquitination and innate immune signaling. Cell Host Microbe 2018, 24, 761–775. [Google Scholar] [CrossRef] [PubMed]
- Morger, D.; Zosel, F.; Buhlmann, M.; Zuger, S.; Mittelviefhaus, M.; Schuler, B.; Luban, J.; Grutter, M.G. The Three-fold axis of the HIV-1 capsid lattice is the species-specific binding interface for trim5alpha. J. Virol. 2018, 92. [Google Scholar]
- Byeon, I.J.; Meng, X.; Jung, J.; Zhao, G.; Yang, R.; Ahn, J.; Shi, J.; Concel, J.; Aiken, C.; Zhang, P.; et al. Structural convergence between Cryo-EM and NMR reveals intersubunit interactions critical for HIV-1 capsid function. Cell 2009, 139, 780–790. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Ke, D.; Vu, T.; Ahn, J.; Shah, V.B.; Yang, R.; Aiken, C.; Charlton, L.M.; Gronenborn, A.M.; Zhang, P. Rhesus trim5alpha disrupts the HIV-1 capsid at the inter-Hexamer interfaces. PLoS Pathog. 2011, 7, e1002009. [Google Scholar] [CrossRef] [PubMed]
- Danielson, C.M.; Cianci, G.C.; Hope, T.J. Recruitment and dynamics of proteasome association with rhtrim5alpha cytoplasmic complexes during HIV-1 infection. Traffic 2012, 13, 1206–1217. [Google Scholar] [CrossRef] [PubMed]
- Lukic, Z.; Hausmann, S.; Sebastian, S.; Rucci, J.; Sastri, J.; Robia, S.L.; Luban, J.; Campbell, E.M. Trim5alpha associates with proteasomal subunits in cells while in complex with HIV-1 virions. RetroVirology 2011, 8, 93. [Google Scholar] [CrossRef] [PubMed]
- Rold, C.J.; Aiken, C. Proteasomal degradation of trim5alpha during retrovirus restriction. PLoS Pathog. 2008, 4, e1000074. [Google Scholar] [CrossRef] [PubMed]
- Yudina, Z.; Roa, A.; Johnson, R.; Biris, N.; de Souza Aranha Vieira, D.A.; Tsiperson, V.; Reszka, N.; Taylor, A.B.; Hart, P.J.; Demeler, B.; et al. Ring dimerization links higher-Order assembly of trim5alpha to synthesis of k63-Linked polyubiquitin. Cell Rep. 2015, 12, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, B.; Pomerantz, R.J.; Zhang, C.; Arunachalam, S.C.; Gao, L. The cytidine deaminase cem15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 2003, 424, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Mangeat, B.; Turelli, P.; Caron, G.; Friedli, M.; Perrin, L.; Trono, D. Broad antiretroviral defence by human apobec3g through lethal editing of nascent reverse transcripts. Nature 2003, 424, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Ito, F.; Fu, Y.; Kao, S.A.; Yang, H.; Chen, X.S. Family-Wide comparative analysis of cytidine and methylcytidine deamination by eleven human apobec proteins. J. Mol. Biol. 2017, 429, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Liddament, M.T.; Brown, W.L.; Schumacher, A.J.; Harris, R.S. Apobec3f properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr. Biol. 2004, 14, 1385–1391. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Bishop, K.N.; Sheehy, A.M.; Craig, H.M.; Petersen-Mahrt, S.K.; Watt, I.N.; Neuberger, M.S.; Malim, M.H. DNA deamination mediates innate immunity to retroviral infection. Cell 2003, 113, 803–809. [Google Scholar] [CrossRef]
- Yang, B.; Chen, K.; Zhang, C.; Huang, S.; Zhang, H. Virion-associated uracil DNA glycosylase-2 and apurinic/apyrimidinic endonuclease are involved in the degradation of apobec3g-edited nascent HIV-1 DNA. J. Biol. Chem. 2007, 282, 11667–11675. [Google Scholar] [CrossRef] [PubMed]
- Gabuzda, D.H.; Lawrence, K.; Langhoff, E.; Terwilliger, E.; Dorfman, T.; Haseltine, W.A.; Sodroski, J. Role of vif in replication of human immunodeficiency virus type 1 in CD4+ t lymphocytes. J. Virol. 1992, 66, 6489–6495. [Google Scholar] [PubMed]
- Kao, S.; Akari, H.; Khan, M.A.; Dettenhofer, M.; Yu, X.F.; Strebel, K. Human immunodeficiency virus type 1 vif is efficiently packaged into virions during productive but not chronic infection. J. Virol. 2003, 77, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- von Schwedler, U.; Song, J.; Aiken, C.; Trono, D. Vif is crucial for human immunodeficiency virus type 1 proviral DNA synthesis in infected cells. J. Virol. 1993, 67, 4945–4955. [Google Scholar] [PubMed]
- Mariani, R.; Chen, D.; Schrofelbauer, B.; Navarro, F.; Konig, R.; Bollman, B.; Munk, C.; Nymark-McMahon, H.; Landau, N.R. Species-specific exclusion of apobec3g from HIV-1 virions by vif. Cell 2003, 114, 21–31. [Google Scholar] [CrossRef]
- Stopak, K.; de Noronha, C.; Yonemoto, W.; Greene, W.C. HIV-1 vif blocks the antiviral activity of apobec3g by impairing both its translation and intracellular stability. Mol. Cell 2003, 12, 591–601. [Google Scholar] [CrossRef]
- Yu, X.; Yu, Y.; Liu, B.; Luo, K.; Kong, W.; Mao, P.; Yu, X.F. Induction of apobec3g ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 2003, 302, 1056–1060. [Google Scholar] [CrossRef]
- Desimmie, B.A.; Delviks-Frankenberrry, K.A.; Burdick, R.C.; Qi, D.; Izumi, T.; Pathak, V.K. Multiple apobec3 restriction factors for HIV-1 and one vif to rule them all. J. Mol. Biol. 2014, 426, 1220–1245. [Google Scholar] [CrossRef]
- Ehrlich, E.S.; Yu, X.F. Lentiviral vif: Viral hijacker of the ubiquitin-Proteasome system. Int. J. Hematol. 2006, 83, 208–212. [Google Scholar] [CrossRef]
- Conticello, S.G.; Harris, R.S.; Neuberger, M.S. The vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase apobec3g. Curr. Biol. 2003, 13, 2009–2013. [Google Scholar] [CrossRef]
- Shao, Q.; Wang, Y.; Hildreth, J.E.; Liu, B. Polyubiquitination of apobec3g is essential for its degradation by HIV-1 vif. J. Virol. 2010, 84, 4840–4844. [Google Scholar] [CrossRef]
- Kobayashi, M.; Takaori-Kondo, A.; Miyauchi, Y.; Iwai, K.; Uchiyama, T. Ubiquitination of apobec3g by an HIV-1 vif-Cullin5-Elongin b-Elongin c complex is essential for vif function. J. Biol. Chem. 2005, 280, 18573–18578. [Google Scholar] [CrossRef]
- Mehle, A.; Goncalves, J.; Santa-Marta, M.; McPike, M.; Gabuzda, D. Phosphorylation of a novel socs-Box regulates assembly of the HIV-1 vif-Cul5 complex that promotes apobec3g degradation. Genes Dev. 2004, 18, 2861–2866. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Sarkis, P.T.; Luo, K.; Yu, Y.; Yu, X.F. Regulation of apobec3f and human immunodeficiency virus type 1 vif by vif-Cul5-Elonb/c e3 ubiquitin ligase. J. Virol. 2005, 79, 9579–9587. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, R.; Nishitsuji, H.; Furukawa, A.; Katahira, M.; Habu, Y.; Takeuchi, H.; Ryo, A.; Takaku, H. Heat shock protein 70 inhibits HIV-1 vif-Mediated ubiquitination and degradation of apobec3g. J. Biol. Chem. 2011, 286, 10051–10057. [Google Scholar] [CrossRef] [PubMed]
- Valera, M.S.; de Armas-Rillo, L.; Barroso-Gonzalez, J.; Ziglio, S.; Batisse, J.; Dubois, N.; Marrero-Hernandez, S.; Borel, S.; Garcia-Exposito, L.; Biard-Piechaczyk, M.; et al. The hdac6/apobec3g complex regulates HIV-1 infectiveness by inducing vif autophagic degradation. RetroVirology 2015, 12, 53. [Google Scholar] [CrossRef] [PubMed]
- Izumi, T.; Takaori-Kondo, A.; Shirakawa, K.; Higashitsuji, H.; Itoh, K.; Io, K.; Matsui, M.; Iwai, K.; Kondoh, H.; Sato, T.; et al. Mdm2 is a novel e3 ligase for HIV-1 vif. RetroVirology 2009, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Akari, H.; Sakurai, A.; Yoshida, A.; Chiba, T.; Tanaka, K.; Strebel, K.; Adachi, A. Expression of HIV-1 accessory protein vif is controlled uniquely to be low and optimal by proteasome degradation. Microbes Infect. 2004, 6, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Du, J.; Evans, S.L.; Yu, Y.; Yu, X.F. T-Cell differentiation factor cbf-Beta regulates HIV-1 vif-Mediated evasion of host restriction. Nature 2011, 481, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Jager, S.; Kim, D.Y.; Hultquist, J.F.; Shindo, K.; LaRue, R.S.; Kwon, E.; Li, M.; Anderson, B.D.; Yen, L.; Stanley, D.; et al. Vif hijacks cbf-Beta to degrade apobec3g and promote HIV-1 infection. Nature 2011, 481, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Matsui, Y.; Shindo, K.; Nagata, K.; Yoshinaga, N.; Shirakawa, K.; Kobayashi, M.; Takaori-Kondo, A. Core binding factor beta protects HIV, type 1 accessory protein viral infectivity factor from mdm2-Mediated degradation. J. Biol. Chem. 2016, 291, 24892–24899. [Google Scholar] [CrossRef] [PubMed]
- Dussart, S.; Courcoul, M.; Bessou, G.; Douaisi, M.; Duverger, Y.; Vigne, R.; Decroly, E. The vif protein of human immunodeficiency virus type 1 is posttranslationally modified by ubiquitin. Biochem. Biophys. Res. Earch Commun. 2004, 315, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Nomaguchi, M.; Doi, N.; Matsumoto, Y.; Sakai, Y.; Fujiwara, S.; Adachi, A. Species tropism of HIV-1 modulated by viral accessory proteins. Front. Microbiol. 2012, 3, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, P.M.; Bailes, E.; Stevenson, M.; Emerman, M.; Hahn, B.H. Gene acquisition in HIV and siv. Nature 1996, 383, 586–587. [Google Scholar] [CrossRef] [PubMed]
- Tristem, M.; Purvis, A.; Quicke, D.L. Complex evolutionary history of primate lentiviral vpr genes. Virology 1998, 240, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Seissler, T.; Marquet, R.; Paillart, J.C. Hijacking of the ubiquitin/proteasome pathway by the HIV auxiliary proteins. Viruses 2017, 9, 322. [Google Scholar] [CrossRef]
- Gibbs, J.S.; Lackner, A.A.; Lang, S.M.; Simon, M.A.; Sehgal, P.K.; Daniel, M.D.; Desrosiers, R.C. Progression to aids in the absence of a gene for vpr or vpx. J. Virol. 1995, 69, 2378–2383. [Google Scholar] [PubMed]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Segeral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. Samhd1 is the dendritic-and myeloid-Cell-Specific HIV-1 restriction factor counteracted by vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Lahouassa, H.; Daddacha, W.; Hofmann, H.; Ayinde, D.; Logue, E.C.; Dragin, L.; Bloch, N.; Maudet, C.; Bertrand, M.; Gramberg, T.; et al. Samhd1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 2012, 13, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Nguyen, L.A.; Daddacha, W.; Hollenbaugh, J.A. Tight interplay among samhd1 protein level, cellular dntp levels, and HIV-1 proviral DNA synthesis kinetics in human primary monocyte-derived macrophages. J. Biol. Chem. 2012, 287, 21570–21574. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, A.; Ayinde, D.; David, A.; Le Rouzic, E.; Morel, M.; Collin, G.; Descamps, D.; Damond, F.; Brun-Vezinet, F.; Nisole, S.; et al. The human immunodeficiency virus type 2 vpx protein usurps the cul4a-ddb1 dcaf1 ubiquitin ligase to overcome a postentry block in macrophage infection. J. Virol. 2009, 83, 4854–4860. [Google Scholar] [CrossRef] [PubMed]
- Brandariz-Nunez, A.; Valle-Casuso, J.C.; White, T.E.; Laguette, N.; Benkirane, M.; Brojatsch, J.; Diaz-Griffero, F. Role of samhd1 nuclear localization in restriction of HIV-1 and sivmac. RetroVirology 2012, 9, 49. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, H.; Logue, E.C.; Bloch, N.; Daddacha, W.; Polsky, S.B.; Schultz, M.L.; Kim, B.; Landau, N.R. The vpx lentiviral accessory protein targets samhd1 for degradation in the nucleus. J. Virol. 2012, 86, 12552–12560. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Ehrlich, E.; Yu, X.F. Ddb1 and cul4a are required for human immunodeficiency virus type 1 vpr-induced g2 arrest. J. Virol. 2007, 81, 10822–10830. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Oliveira, N.M.; Cheney, K.M.; Pade, C.; Dreja, H.; Bergin, A.M.; Borgdorff, V.; Beach, D.H.; Bishop, C.L.; Dittmar, M.T.; et al. A whole genome screen for HIV restriction factors. RetroVirology 2011, 8, 94. [Google Scholar] [CrossRef]
- Yurkovetskiy, L.; Guney, M.H.; Kim, K.; Goh, S.L.; McCauley, S.; Dauphin, A.; Diehl, W.E.; Luban, J. Primate immunodeficiency virus proteins vpx and vpr counteract transcriptional repression of proviruses by the hush complex. Nat. Microbiol. 2018, 3, 1354–1361. [Google Scholar] [CrossRef]
- Tchasovnikarova, I.A.; Timms, R.T.; Matheson, N.J.; Wals, K.; Antrobus, R.; Gottgens, B.; Dougan, G.; Dawson, M.A.; Lehner, P.J. Gene silencing. Epigenetic silencing by the hush complex mediates position-Effect variegation in human cells. Science 2015, 348, 1481–1485. [Google Scholar] [CrossRef]
- Tchasovnikarova, I.A.; Timms, R.T.; Douse, C.H.; Roberts, R.C.; Dougan, G.; Kingston, R.E.; Modis, Y.; Lehner, P.J. Hyperactivation of hush complex function by charcot-Marie-Tooth disease mutation in morc2. Nat. Genet. 2017, 49, 1035–1044. [Google Scholar] [CrossRef]
- Liu, N.; Lee, C.H.; Swigut, T.; Grow, E.; Gu, B.; Bassik, M.C.; Wysocka, J. Selective silencing of euchromatic l1s revealed by genome-wide screens for l1 regulators. Nature 2018, 553, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Le Rouzic, E.; Belaidouni, N.; Estrabaud, E.; Morel, M.; Rain, J.C.; Transy, C.; Margottin-Goguet, F. HIV1 vpr arrests the cell cycle by recruiting dcaf1/vprbp, a receptor of the cul4-ddb1 ubiquitin ligase. Cell Cycle 2007, 6, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Swanson, S.K.; Manel, N.; Florens, L.; Washburn, M.P.; Skowronski, J. Lentiviral vpx accessory factor targets vprbp/dcaf1 substrate adaptor for cullin 4 e3 ubiquitin ligase to enable macrophage infection. PLoS Pathog. 2008, 4, e1000059. [Google Scholar] [CrossRef]
- Pertel, T.; Reinhard, C.; Luban, J. Vpx rescues HIV-1 transduction of dendritic cells from the antiviral state established by type 1 interferon. RetroVirology 2011, 8, 49. [Google Scholar] [CrossRef]
- Reinhard, C.; Bottinelli, D.; Kim, B.; Luban, J. Vpx rescue of HIV-1 from the antiviral state in mature dendritic cells is independent of the intracellular deoxynucleotide concentration. RetroVirology 2014, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.M.; Akiyama, H.; Agosto, L.M.; Emery, A.; Ettinger, C.R.; Swanstrom, R.I.; Henderson, A.J.; Gummuluru, S. Virion-associated vpr alleviates a postintegration block to HIV-1 infection of dendritic cells. J. Virol. 2017, 91, e00051-17. [Google Scholar] [CrossRef] [PubMed]
- Mulder, L.C.; Muesing, M.A. Degradation of HIV-1 integrase by the n-end rule pathway. J. Biol. Chem. 2000, 275, 29749–29753. [Google Scholar] [CrossRef] [PubMed]
- Tasaki, T.; Mulder, L.C.; Iwamatsu, A.; Lee, M.J.; Davydov, I.V.; Varshavsky, A.; Muesing, M.; Kwon, Y.T. A family of mammalian e3 ubiquitin ligases that contain the ubr box motif and recognize n-Degrons. Mol. Cell. Biol. 2005, 25, 7120–7136. [Google Scholar] [CrossRef] [PubMed]
- Emiliani, S.; Mousnier, A.; Busschots, K.; Maroun, M.; Van Maele, B.; Tempe, D.; Vandekerckhove, L.; Moisant, F.; Ben-Slama, L.; Witvrouw, M.; et al. Integrase mutants defective for interaction with ledgf/p75 are impaired in chromosome tethering and HIV-1 replication. J. Biol. Chem. 2005, 280, 25517–25523. [Google Scholar] [CrossRef] [PubMed]
- Devroe, E.; Engelman, A.; Silver, P.A. Intracellular transport of human immunodeficiency virus type 1 integrase. J. Cell Sci. 2003, 116, 4401–4408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llano, M.; Delgado, S.; Vanegas, M.; Poeschla, E.M. Lens epithelium-derived growth factor/p75 prevents proteasomal degradation of HIV-1 integrase. J. Biol. Chem. 2004, 279, 55570–55577. [Google Scholar] [CrossRef]
- Gerard, A.; Soler, N.; Segeral, E.; Belshan, M.; Emiliani, S. Identification of low molecular weight nuclear complexes containing integrase during the early stages of HIV-1 infection. RetroVirology 2013, 10, 13. [Google Scholar] [CrossRef]
- Ali, H.; Mano, M.; Braga, L.; Naseem, A.; Marini, B.; Vu, D.M.; Collesi, C.; Meroni, G.; Lusic, M.; Giacca, M. Cellular trim33 restrains HIV-1 infection by targeting viral integrase for proteasomal degradation. Nat. Commun. 2019, 10, 926. [Google Scholar] [CrossRef]
- Sodroski, J.; Rosen, C.; Wong-Staal, F.; Salahuddin, S.Z.; Popovic, M.; Arya, S.; Gallo, R.C.; Haseltine, W.A. Trans-acting transcriptional regulation of human t-cell leukemia virus type iii long terminal repeat. Science 1985, 227, 171–173. [Google Scholar] [CrossRef]
- Sodroski, J.; Patarca, R.; Rosen, C.; Wong-Staal, F.; Haseltine, W. Location of the trans-activating region on the genome of human t-cell lymphotropic virus type iii. Science 1985, 229, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Kao, S.Y.; Calman, A.F.; Luciw, P.A.; Peterlin, B.M. Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature 1987, 330, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Holland, E.C. HIV-1 tat trans-activation requires the loop sequence within tar. Nature 1988, 334, 165–167. [Google Scholar] [CrossRef] [PubMed]
- Dingwall, C.; Ernberg, I.; Gait, M.J.; Green, S.M.; Heaphy, S.; Karn, J.; Lowe, A.D.; Singh, M.; Skinner, M.A.; Valerio, R. Human immunodeficiency virus 1 tat protein binds trans-activation-responsive region (tar) rna in vitro. Proc. Natl. Acad. Sci. USA 1989, 86, 6925–6929. [Google Scholar] [CrossRef] [PubMed]
- Madore, S.J.; Cullen, B.R. Genetic analysis of the cofactor requirement for human immunodeficiency virus type 1 tat function. J. Virol. 1993, 67, 3703–3711. [Google Scholar]
- Lata, S.; Ali, A.; Sood, V.; Raja, R.; Banerjea, A.C. HIV-1 rev downregulates tat expression and viral replication via modulation of nad(p)h:Quinine oxidoreductase 1 (nqo1). Nat. Commun. 2015, 6, 7244. [Google Scholar] [CrossRef]
- Hong, H.W.; Lee, S.W.; Myung, H. Induced degradation of tat by nucleocapsid (nc) via the proteasome pathway and its effect on HIV transcription. Viruses 2013, 5, 1143–1152. [Google Scholar] [CrossRef]
- Raja, R.; Ronsard, L.; Lata, S.; Trivedi, S.; Banerjea, A.C. HIV-1 tat potently stabilises mdm2 and enhances viral replication. Biochem. J. 2017, 474, 2449–2464. [Google Scholar] [CrossRef]
- Faust, T.B.; Li, Y.; Jang, G.M.; Johnson, J.R.; Yang, S.; Weiss, A.; Krogan, N.J.; Frankel, A.D. Pja2 ubiquitinates the HIV-1 tat protein with atypical chain linkages to activate viral transcription. Sci. Rep. 2017, 7, 45394. [Google Scholar] [CrossRef]
- Li, J.; Chen, C.; Ma, X.; Geng, G.; Liu, B.; Zhang, Y.; Zhang, S.; Zhong, F.; Liu, C.; Yin, Y.; et al. Long noncoding rna nron contributes to HIV-1 latency by specifically inducing tat protein degradation. Nat. Commun. 2016, 7, 11730. [Google Scholar] [CrossRef]
- Spina, C.A.; Guatelli, J.C.; Richman, D.D. Establishment of a stable, inducible form of human immunodeficiency virus type 1 DNA in quiescent CD4 lymphocytes in vitro. J. Virol. 1995, 69, 2977–2988. [Google Scholar] [PubMed]
- Zack, J.A.; Haislip, A.M.; Krogstad, P.; Chen, I.S. Incompletely reverse-Transcribed human immunodeficiency virus type 1 genomes in quiescent cells can function as intermediates in the retroviral life cycle. J. Virol. 1992, 66, 1717–1725. [Google Scholar] [PubMed]
- Stevenson, M.; Stanwick, T.L.; Dempsey, M.P.; Lamonica, C.A. HIV-1 replication is controlled at the level of t cell activation and proviral integration. EMBO J. 1990, 9, 1551–1560. [Google Scholar] [CrossRef] [PubMed]
- Zack, J.A.; Arrigo, S.J.; Weitsman, S.R.; Go, A.S.; Haislip, A.; Chen, I.S. HIV-1 entry into quiescent primary lymphocytes: Molecular analysis reveals a labile, latent viral structure. Cell 1990, 61, 213–222. [Google Scholar] [CrossRef]
- Wang, J.K.; Kiyokawa, E.; Verdin, E.; Trono, D. The nef protein of HIV-1 associates with rafts and primes t cells for activation. Proc. Natl. Acad. Sci. USA 2000, 97, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Schrager, J.A.; Marsh, J.W. HIV-1 nef increases t cell activation in a stimulus-Dependent manner. Proc. Natl. Acad. Sci. USA 1999, 96, 8167–8172. [Google Scholar] [CrossRef]
- Fackler, O.T.; Wolf, D.; Weber, H.O.; Laffert, B.; D’Aloja, P.; Schuler-Thurner, B.; Geffin, R.; Saksela, K.; Geyer, M.; Peterlin, B.M.; et al. A natural variability in the proline-rich motif of nef modulates HIV-1 replication in primary t cells. Curr. Biol. 2001, 11, 1294–1299. [Google Scholar] [CrossRef]
- Renkema, G.H.; Saksela, K. Interactions of HIV-1 nef with cellular signal transducing proteins. Front. Biosci. 2000, 5, D268–D283. [Google Scholar] [CrossRef]
- Simmons, A.; Gangadharan, B.; Hodges, A.; Sharrocks, K.; Prabhakar, S.; Garcia, A.; Dwek, R.; Zitzmann, N.; McMichael, A. Nef-mediated lipid raft exclusion of ubch7 inhibits cbl activity in t cells to positively regulate signaling. Immunity 2005, 23, 621–634. [Google Scholar] [CrossRef]
- Qi, M.; Aiken, C. Selective restriction of nef-Defective human immunodeficiency virus type 1 by a proteasome-Dependent mechanism. J. Virol. 2007, 81, 1534–1536. [Google Scholar] [CrossRef]
- Neil, S.J.; Eastman, S.W.; Jouvenet, N.; Bieniasz, P.D. HIV-1 vpu promotes release and prevents endocytosis of nascent retrovirus particles from the plasma membrane. PLoS Pathog. 2006, 2, e39. [Google Scholar] [CrossRef] [PubMed]
- Hinz, A.; Miguet, N.; Natrajan, G.; Usami, Y.; Yamanaka, H.; Renesto, P.; Hartlieb, B.; McCarthy, A.A.; Simorre, J.P.; Gottlinger, H.; et al. Structural basis of HIV-1 tethering to membranes by the bst-2/tetherin ectodomain. Cell Host Microbe 2010, 7, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein bst-2 restricts HIV-1 release and is downregulated from the cell surface by the viral vpu protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef] [PubMed]
- McNatt, M.W.; Zang, T.; Bieniasz, P.D. Vpu binds directly to tetherin and displaces it from nascent virions. PLoS Pathog. 2013, 9, e1003299. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.S.; Katsura, C.; Skasko, M.A.; Fitzpatrick, K.; Lau, D.; Ruiz, A.; Stephens, E.B.; Margottin-Goguet, F.; Benarous, R.; Guatelli, J.C. Vpu antagonizes bst-2-mediated restriction of HIV-1 release via beta-trcp and endo-lysosomal trafficking. PLoS Pathog. 2009, 5, e1000450. [Google Scholar] [CrossRef] [PubMed]
- Dube, M.; Roy, B.B.; Guiot-Guillain, P.; Binette, J.; Mercier, J.; Chiasson, A.; Cohen, E.A. Antagonism of tetherin restriction of HIV-1 release by vpu involves binding and sequestration of the restriction factor in a perinuclear compartment. PLoS Pathog. 2010, 6, e1000856. [Google Scholar] [CrossRef]
- Margottin, F.; Bour, S.P.; Durand, H.; Selig, L.; Benichou, S.; Richard, V.; Thomas, D.; Strebel, K.; Benarous, R. A novel human wd protein, h-Beta Trcp, that interacts with HIV-1 vpu connects CD4 to the er degradation pathway through an f-Box motif. Mol. Cell 1998, 1, 565–574. [Google Scholar] [CrossRef]
- Roy, N.; Pacini, G.; Berlioz-Torrent, C.; Janvier, K. Characterization of e3 ligases involved in lysosomal sorting of the HIV-1 restriction factor bst2. J. Cell Sci. 2017, 130, 1596–1611. [Google Scholar] [CrossRef]
- Iwabu, Y.; Fujita, H.; Kinomoto, M.; Kaneko, K.; Ishizaka, Y.; Tanaka, Y.; Sata, T.; Tokunaga, K. HIV-1 accessory protein vpu internalizes cell-surface bst-2/tetherin through transmembrane interactions leading to lysosomes. J. Biol. Chem. 2009, 284, 35060–35072. [Google Scholar] [CrossRef]
- Ramirez, P.W.; DePaula-Silva, A.B.; Szaniawski, M.; Barker, E.; Bosque, A.; Planelles, V. HIV-1 vpu utilizes both cullin-Ring ligase (crl) dependent and independent mechanisms to downmodulate host proteins. RetroVirology 2015, 12, 65. [Google Scholar] [CrossRef]
- Tervo, H.M.; Homann, S.; Ambiel, I.; Fritz, J.V.; Fackler, O.T.; Keppler, O.T. Beta-trcp is dispensable for vpu’s ability to overcome the CD317/tetherin-Imposed restriction to HIV-1 release. RetroVirology 2011, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Janvier, K.; Pelchen-Matthews, A.; Renaud, J.B.; Caillet, M.; Marsh, M.; Berlioz-Torrent, C. The escrt-0 component hrs is required for HIV-1 vpu-mediated bst-2/tetherin down-Regulation. PLoS Pathog. 2011, 7, e1001265. [Google Scholar] [CrossRef] [PubMed]
- Kueck, T.; Foster, T.L.; Weinelt, J.; Sumner, J.C.; Pickering, S.; Neil, S.J. Serine phosphorylation of HIV-1 vpu and its binding to tetherin regulates interaction with clathrin adaptors. PLoS Pathog. 2015, 11, e1005141. [Google Scholar] [CrossRef] [PubMed]
- Pujol, F.M.; Laketa, V.; Schmidt, F.; Mukenhirn, M.; Muller, B.; Boulant, S.; Grimm, D.; Keppler, O.T.; Fackler, O.T. HIV-1 vpu antagonizes CD317/tetherin by adaptor protein-1-Mediated exclusion from virus assembly sites. J. Virol. 2016, 90, 6709–6723. [Google Scholar] [CrossRef] [PubMed]
- Madjo, U.; Leymarie, O.; Fremont, S.; Kuster, A.; Nehlich, M.; Gallois-Montbrun, S.; Janvier, K.; Berlioz-Torrent, C. Lc3c contributes to vpu-mediated antagonism of bst2/tetherin restriction on HIV-1 release through a non-Canonical autophagy pathway. Cell Rep. 2016, 17, 2221–2233. [Google Scholar] [CrossRef] [PubMed]
- Bour, S.; Schubert, U.; Peden, K.; Strebel, K. The envelope glycoprotein of human immunodeficiency virus type 2 enhances viral particle release: A vpu-like factor? J. Virol. 1996, 70, 820–829. [Google Scholar] [PubMed]
- Bour, S.; Strebel, K. The human immunodeficiency virus (HIV) type 2 envelope protein is a functional complement to HIV type 1 vpu that enhances particle release of heterologous retroviruses. J. Virol. 1996, 70, 8285–8300. [Google Scholar]
- Gottlinger, H.G.; Dorfman, T.; Sodroski, J.G.; Haseltine, W.A. Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc. Natl. Acad. Sci. USA 1991, 88, 3195–3199. [Google Scholar] [CrossRef]
- Huang, M.; Orenstein, J.M.; Martin, M.A.; Freed, E.O. P6gag is required for particle production from full-Length human immunodeficiency virus type 1 molecular clones expressing protease. J. Virol. 1995, 69, 6810–6818. [Google Scholar]
- Garrus, J.E.; von Schwedler, U.K.; Pornillos, O.W.; Morham, S.G.; Zavitz, K.H.; Wang, H.E.; Wettstein, D.A.; Stray, K.M.; Cote, M.; Rich, R.L.; et al. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 2001, 107, 55–65. [Google Scholar] [CrossRef]
- Martin-Serrano, J.; Zang, T.; Bieniasz, P.D. HIV-1 and ebola virus encode small peptide motifs that recruit tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 2001, 7, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- VerPlank, L.; Bouamr, F.; LaGrassa, T.J.; Agresta, B.; Kikonyogo, A.; Leis, J.; Carter, C.A. Tsg101, a homologue of ubiquitin-Conjugating (e2) enzymes, binds the l domain in HIV type 1 pr55(gag). Proc. Natl. Acad. Sci. USA 2001, 98, 7724–7729. [Google Scholar] [CrossRef] [PubMed]
- Bouamr, F.; Houck-Loomis, B.R.; De Los Santos, M.; Casaday, R.J.; Johnson, M.C.; Goff, S.P. The c-Terminal portion of the hrs protein interacts with tsg101 and interferes with human immunodeficiency virus type 1 gag particle production. J. Virol. 2007, 81, 2909–2922. [Google Scholar] [CrossRef] [PubMed]
- Demirov, D.G.; Ono, A.; Orenstein, J.M.; Freed, E.O. Overexpression of the n-terminal domain of tsg101 inhibits HIV-1 budding by blocking late domain function. Proc. Natl. Acad. Sci. USA 2002, 99, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Martin-Serrano, J.; Yarovoy, A.; Perez-Caballero, D.; Bieniasz, P.D. Divergent retroviral late-budding domains recruit vacuolar protein sorting factors by using alternative adaptor proteins. Proc. Natl. Acad. Sci. USA 2003, 100, 12414–12419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strack, B.; Calistri, A.; Craig, S.; Popova, E.; Gottlinger, H.G. Aip1/alix is a binding partner for HIV-1 p6 and eiav p9 functioning in virus budding. Cell 2003, 114, 689–699. [Google Scholar] [CrossRef]
- Chung, H.Y.; Morita, E.; von Schwedler, U.; Muller, B.; Krausslich, H.G.; Sundquist, W.I. Nedd4l overexpression rescues the release and infectivity of human immunodeficiency virus type 1 constructs lacking ptap and ypxl late domains. J. Virol. 2008, 82, 4884–4897. [Google Scholar] [CrossRef] [PubMed]
- Sette, P.; Jadwin, J.A.; Dussupt, V.; Bello, N.F.; Bouamr, F. The escrt-associated protein alix recruits the ubiquitin ligase nedd4-1 to facilitate HIV-1 release through the lypxnl l domain motif. J. Virol. 2010, 84, 8181–8192. [Google Scholar] [CrossRef]
- Dussupt, V.; Javid, M.P.; Abou-Jaoude, G.; Jadwin, J.A.; de La Cruz, J.; Nagashima, K.; Bouamr, F. The nucleocapsid region of HIV-1 gag cooperates with the ptap and lypxnl late domains to recruit the cellular machinery necessary for viral budding. PLoS Pathog. 2009, 5, e1000339. [Google Scholar] [CrossRef]
- Park, I.W.; Sodroski, J. Functional analysis of the vpx, vpr, and nef genes of simian immunodeficiency virus. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1995, 8, 335–344. [Google Scholar] [CrossRef]
- Trono, D. HIV accessory proteins: Leading roles for the supporting cast. Cell 1995, 82, 189–192. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Schematic representation of the HIV-1 life cycle. Each step of the HIV-1 life cycle is underlined, and the viral and cellular proteins involved in each step of the proteasomal regulation are shown as red and teal, respectively. Proteasomal regulation between viral proteins is depicted as a dotted line. See the text for the detailed description of the role of the indicated viral and cellular proteins for the proteasomal degradation processes.
Figure 1.
Schematic representation of the HIV-1 life cycle. Each step of the HIV-1 life cycle is underlined, and the viral and cellular proteins involved in each step of the proteasomal regulation are shown as red and teal, respectively. Proteasomal regulation between viral proteins is depicted as a dotted line. See the text for the detailed description of the role of the indicated viral and cellular proteins for the proteasomal degradation processes.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Rojas, V.K.; Park, I.-W. Role of the Ubiquitin Proteasome System (UPS) in the HIV-1 Life Cycle. Int. J. Mol. Sci. 2019, 20, 2984. https://doi.org/10.3390/ijms20122984
AMA Style
Rojas VK, Park I-W. Role of the Ubiquitin Proteasome System (UPS) in the HIV-1 Life Cycle. International Journal of Molecular Sciences. 2019; 20(12):2984. https://doi.org/10.3390/ijms20122984
Chicago/Turabian StyleRojas, Vivian K., and In-Woo Park. 2019. "Role of the Ubiquitin Proteasome System (UPS) in the HIV-1 Life Cycle" International Journal of Molecular Sciences 20, no. 12: 2984. https://doi.org/10.3390/ijms20122984
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.