Comprehensive Analysis of lncRNAs and circRNAs Reveals the Metabolic Specialization in Oxidative and Glycolytic Skeletal Muscles

 ,
,
Abstract
:
1. Introduction
2. Results and Discussion
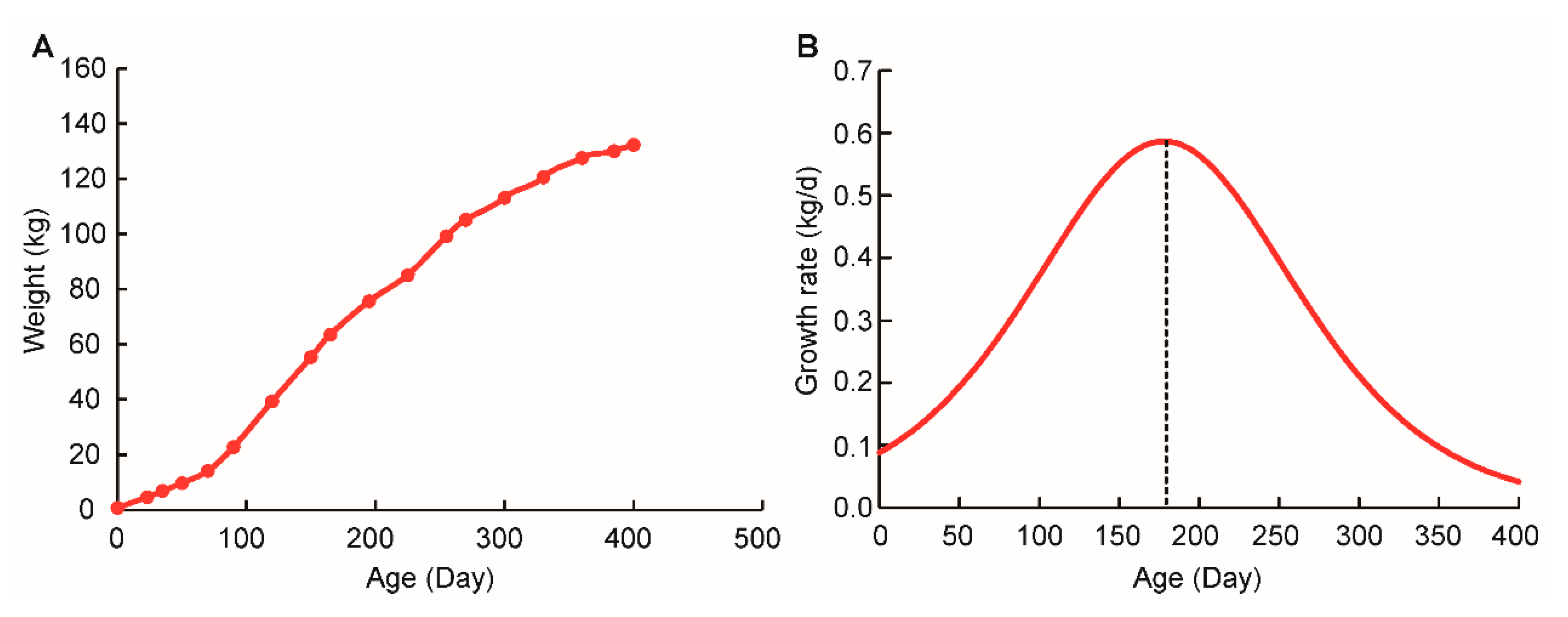
2.1. Sigmoidal Growth Curve of Qingyu Pigs
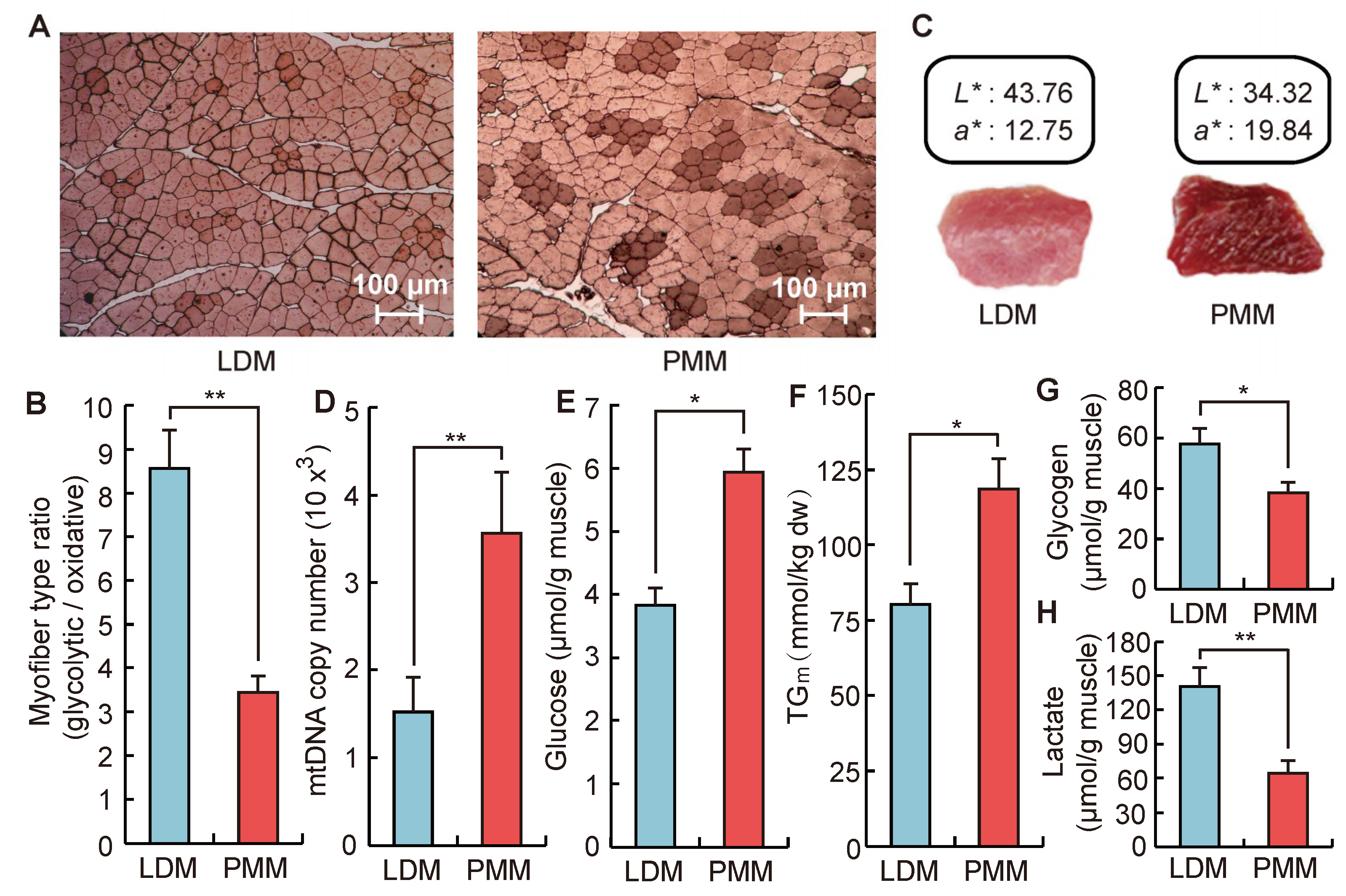
2.2. Differences in Phenotypic Traits between Oxidative and Glycolytic Skeletal Muscles
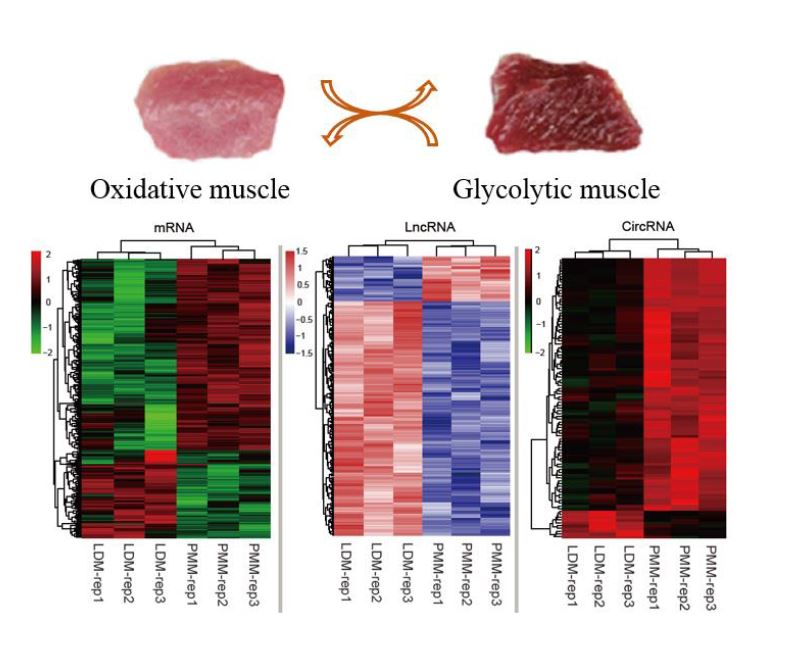
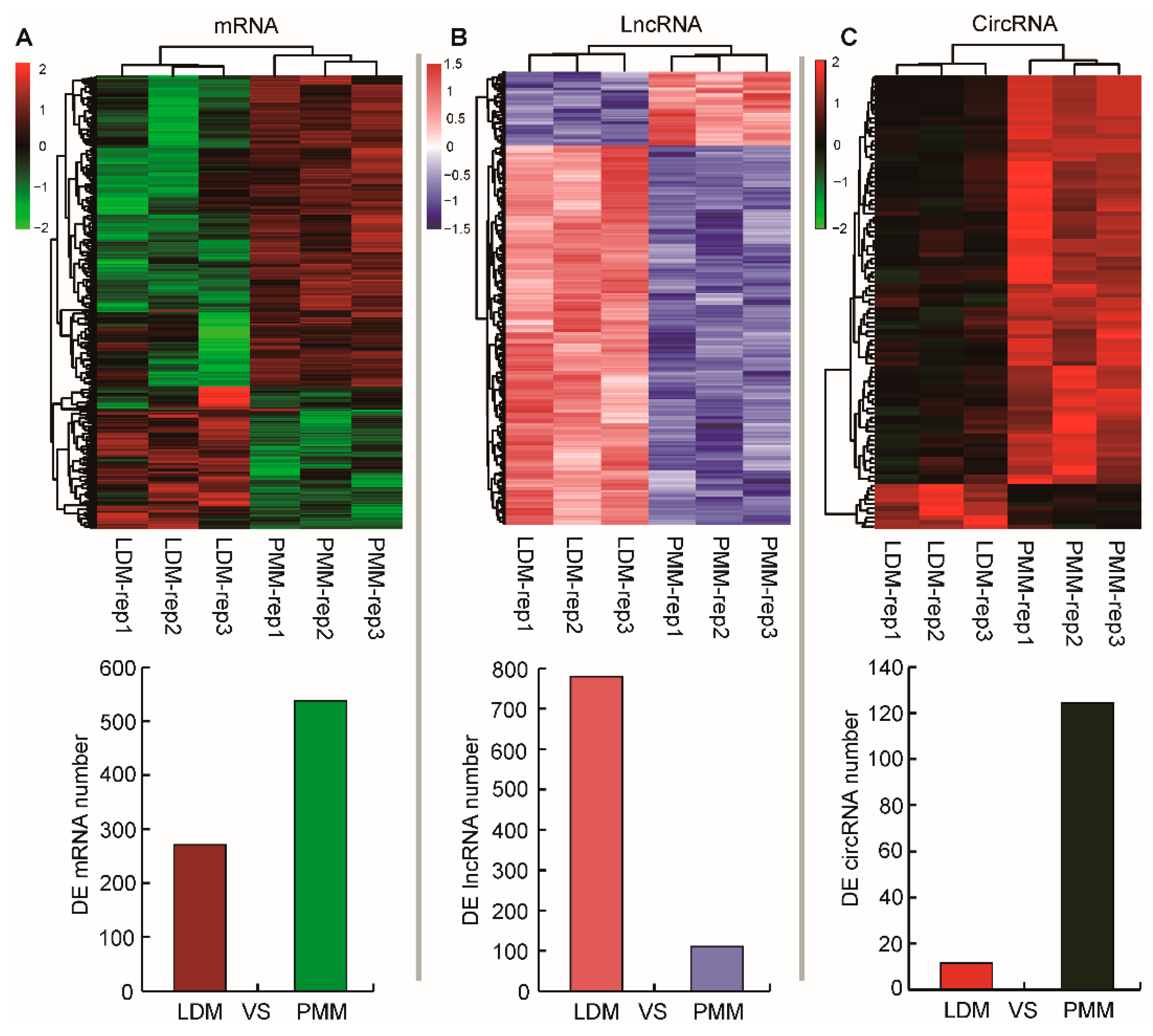
2.3. Identification of mRNAs, lncRNAs, and cricRNAs in Oxidative and Glycolytic Skeletal Muscles
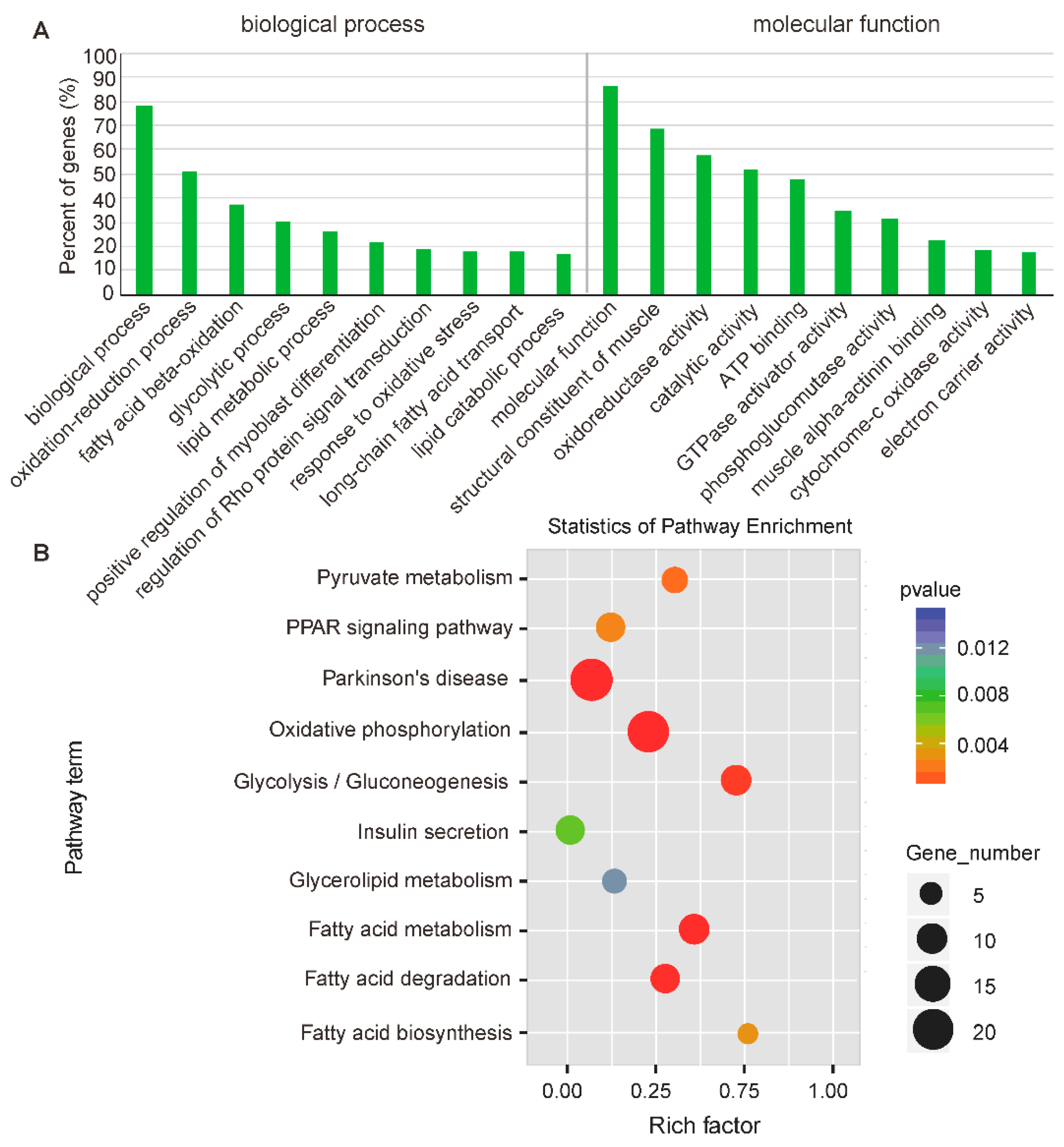
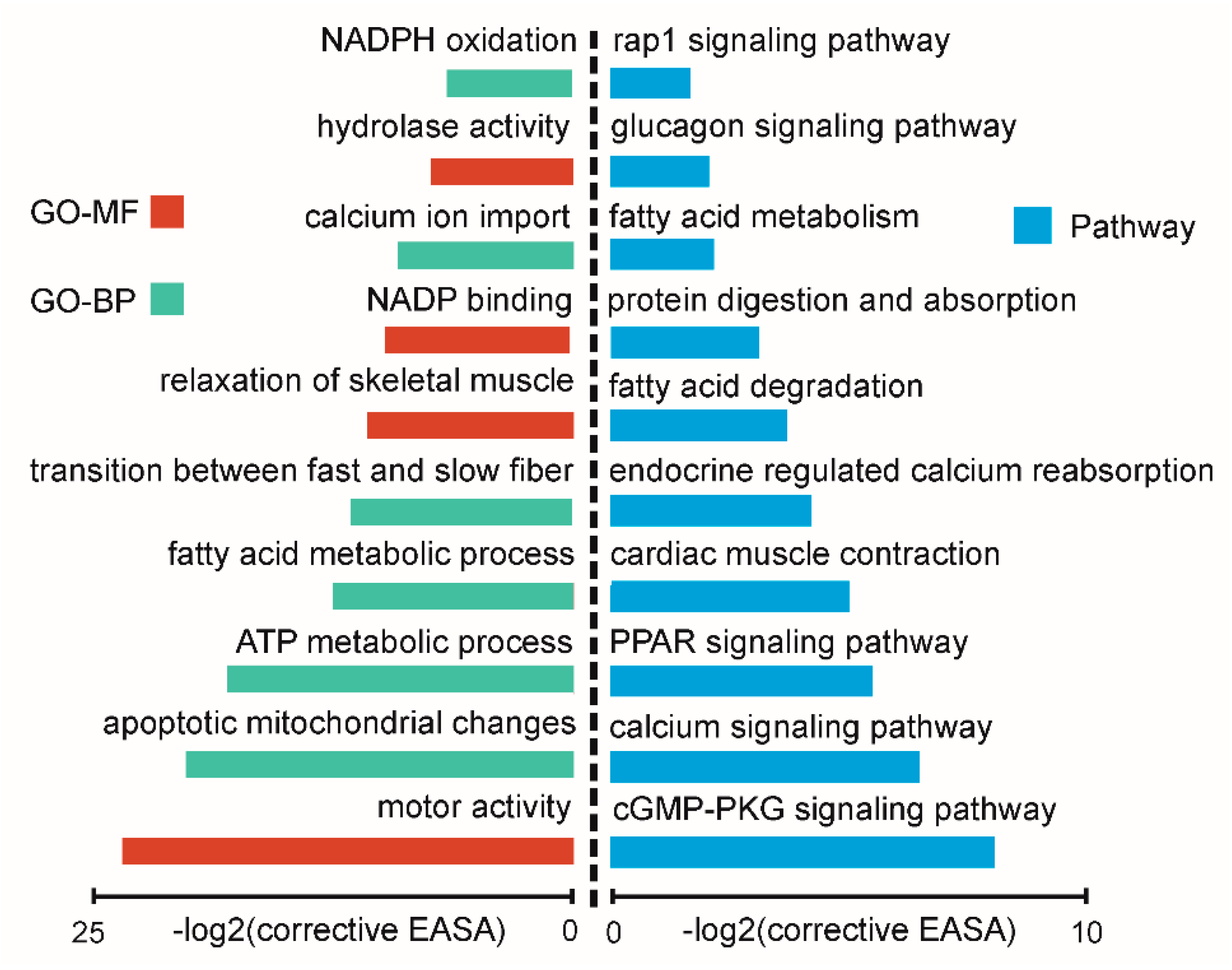
2.4. Function Enrichment Analysis of Nearest Neighbor Genes of Differentially Expressed lncRNAs
2.5. Function Analysis of Differentially Expressed circRNAs between Oxidative and Glycolytic Muscles
2.6. QTL Enrichment Analysis of Differentially Expressed Transcripts
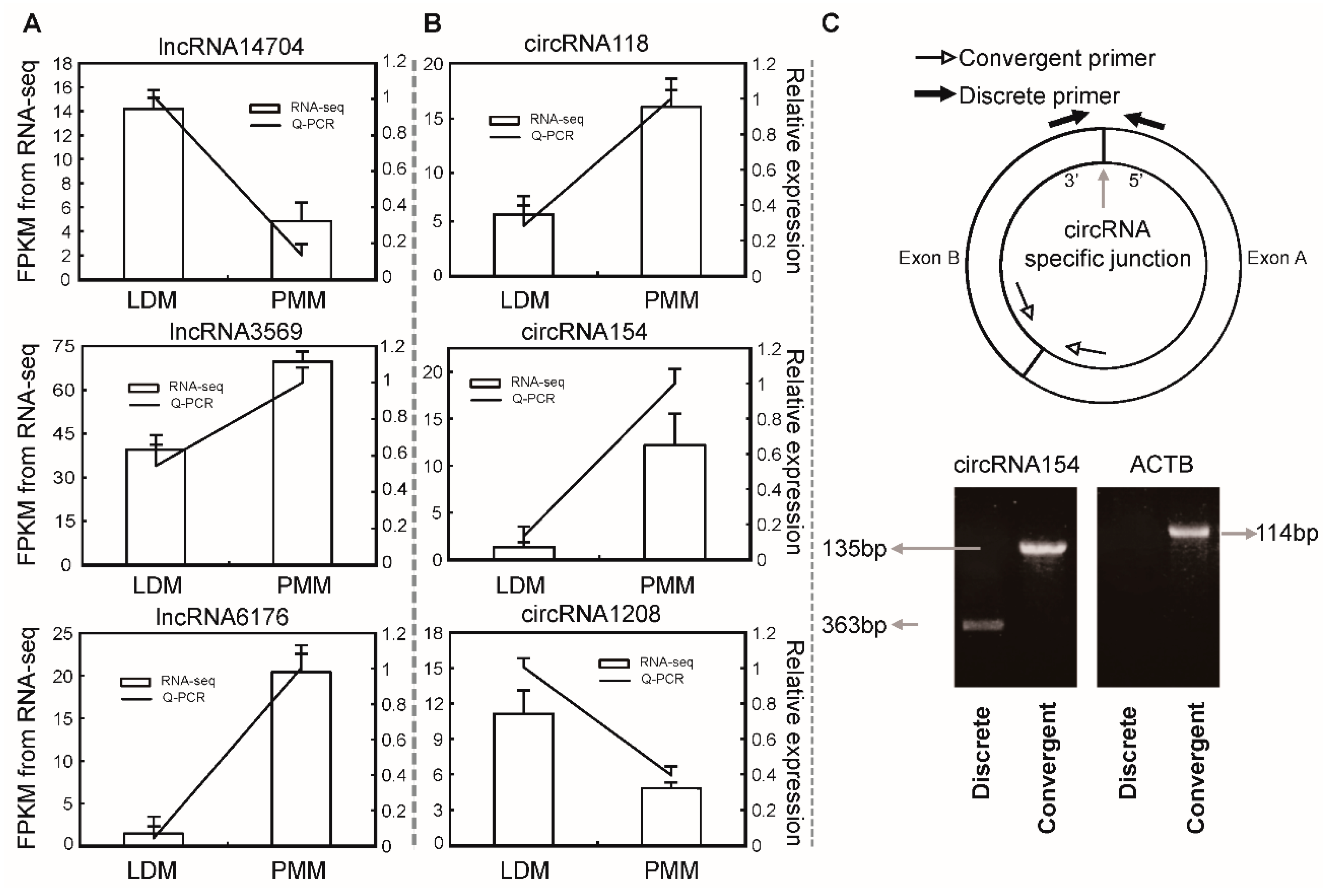
2.7. Validation of differentially expressed lncRNAs and circRNAs
3. Materials and Methods
3.1. Ethics Statement
3.2. Animal Materials and Tissue Collection
3.3. Growth Curve Models
3.4. Measurement of Phenotypic Traits
3.5. Transcriptome Library Construction and Sequencing
3.6. Identification of mRNAs, lncRNAs, and circRNAs
3.7. Functional Enrichment Analysis
3.8. Quantitative RT-PCR
3.9. Statistical Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Janssen, I.; Heymsfield, S.B.; Wang, Z.; Ross, R. Skeletal muscle mass and distribution in 468 men and women aged 18–88 yr. J. Appl. Physiol. 2000, 89, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [PubMed]
- Pandy, M.G.; Andriacchi, T.P. Muscle and joint function in human locomotion. Annu. Rev. Biomed. Eng. 2010, 12, 401–433. [Google Scholar] [CrossRef] [PubMed]
- Goodyear, L.J.; Hirshman, M.F.; Smith, R.J.; Horton, E.S. Glucose transporter number, activity, and isoform content in plasma membranes of red and white skeletal muscle. Am. J. Physiol. 1991, 261, E556–E561. [Google Scholar] [CrossRef] [PubMed]
- Albers, P.H.; Pedersen, A.J.; Birk, J.B.; Kristensen, D.E.; Vind, B.F.; Baba, O.; Nøhr, J.; Højlund, K.; Wojtaszewski, J.F. Human Muscle Fiber Type–Specific Insulin Signaling: Impact of Obesity and Type 2 Diabetes. Diabetes 2015, 64, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Lowell, B.B.; Shulman, G.I. Mitochondrial dysfunction and type 2 diabetes. Science 2005, 307, 384–387. [Google Scholar] [CrossRef]
- Westerblad, H.; Bruton, J.D.; Katz, A. Skeletal muscle: Energy metabolism, fiber types, fatigue and adaptability. Exp. Cell Res. 2010, 316, 3093–3099. [Google Scholar] [CrossRef]
- Sieck, G.C.; Prakash, Y.; Han, Y.-S.; Fang, Y.-H.; Geiger, P.C.; Zhan, W.-Z. Changes in actomyosin ATP consumption rate in rat diaphragm muscle fibers during postnatal development. J. Appl. Physiol. 2003, 94, 1896–1902. [Google Scholar] [CrossRef] [Green Version]
- Richter, E.A.; Hargreaves, M. Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol. Rev. 2013, 93, 993–1017. [Google Scholar] [CrossRef]
- Kamolrat, T.; Gray, S.R.; Thivierge, M.C. Fish oil positively regulates anabolic signalling alongside an increase in whole-body gluconeogenesis in ageing skeletal muscle. Eur. J. Nutr. 2013, 52, 647–657. [Google Scholar] [CrossRef]
- Juel, C.; Klarskov, C.; Nielsen, J.J.; Krustrup, P.; Mohr, M.; Bangsbo, J. Effect of high-intensity intermittent training on lactate and H+ release from human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E245–E251. [Google Scholar] [CrossRef] [PubMed]
- Chabowski, A.; Chatham, J.C.; Tandon, N.N.; Calles-Escandon, J.; Glatz, J.F.; Luiken, J.J.; Bonen, A. Fatty acid transport and FAT/CD36 are increased in red but not in white skeletal muscle of ZDF rats. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E675–E682. [Google Scholar] [CrossRef] [PubMed]
- Talbot, J.; Maves, L. Skeletal muscle fiber type: Using insights from muscle developmental biology to dissect targets for susceptibility and resistance to muscle disease. Rev. Dev. Biol. 2016, 5, 518–534. [Google Scholar] [CrossRef]
- Webster, C.; Silberstein, L.; Hays, A.P.; Blau, H.M. Fast muscle fibers are preferentially affected in Duchenne muscular dystrophy. Cell 1988, 52, 503–513. [Google Scholar] [CrossRef]
- Celegato, B.; Capitanio, D.; Pescatori, M.; Romualdi, C.; Pacchioni, B.; Cagnin, S.; Viganò, A.; Colantoni, L.; Begum, S.; Ricci, E. Parallel protein and transcript profiles of FSHD patient muscles correlate to the D4Z4 arrangement and reveal a common impairment of slow to fast fibre differentiation and a general deregulation of MyoD-dependent genes. Proteomics 2006, 6, 5303–5321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisani, V.; Panico, M.B.; Terracciano, C.; Bonifazi, E.; Meola, G.; Novelli, G.; Bernardi, G.; Angelini, C.; Massa, R. Preferential central nucleation of type 2 myofibers is an invariable feature of myotonic dystrophy type 2. Muscle Nerve 2008, 38, 1405–1411. [Google Scholar] [CrossRef] [PubMed]
- Clarke, N.F.; Waddell, L.B.; Cooper, S.T.; Perry, M.; Smith, R.L.; Kornberg, A.J. Recessive mutations in RYR1 are a common cause of congenital fiber type disproportion. Hum. Mutat. 2010, 31, E1544–E1550. [Google Scholar] [CrossRef] [PubMed]
- Nilwik, R.; Snijders, T.; Leenders, M.; Groen, B.B.; van Kranenburg, J.; Verdijk, L.B.; van Loon, L.J. The decline in skeletal muscle mass with aging is mainly attributed to a reduction in type II muscle fiber size. Exp. Gerontol. 2013, 48, 492–498. [Google Scholar] [CrossRef]
- Ryu, Y.; Kim, B. The relationship between muscle fiber characteristics, postmortem metabolic rate, and meat quality of pig longissimus dorsi muscle. Meat Sci. 2005, 71, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Du, J.; Xia, Y.; Tan, Z.; Fu, Y.; Yang, Q.; Li, X.; Tang, G.; Jiang, Y.; Wang, J. Genome-wide landscape of DNA methylomes and their relationship with mRNA and miRNA transcriptomes in oxidative and glycolytic skeletal muscles. Sci. Rep. 2016, 6, 32186. [Google Scholar] [CrossRef]
- Campbell, W.; Gordon, S.; Carlson, C.; Pattison, J.; Hamilton, M.; Booth, F. Differential global gene expression in red and white skeletal muscle. Am. J. Physiol.-Cell Physiol. 2001, 280, C763–C768. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, Z.; Li, H.; Xiong, Y.; Zuo, B. Differential transcriptional analysis between red and white skeletal muscle of Chinese Meishan pigs. Int. J. Biol. Sci. 2010, 6, 350. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Dong, M.; Fan, D.; Hou, P.; Li, H.; Liu, L.; Lin, C.; Liu, J.; Su, L.; Wu, L. LncRNA ANCR down-regulation promotes TGF-β-induced EMT and metastasis in breast cancer. Oncotarget 2017, 8, 67329. [Google Scholar] [CrossRef] [PubMed]
- Ouimet, M.; Drouin, S.; Lajoie, M.; Caron, M.; St-Onge, P.; Gioia, R.; Richer, C.; Sinnett, D. A childhood acute lymphoblastic leukemia-specific lncRNA implicated in prednisolone resistance, cell proliferation, and migration. Oncotarget 2017, 8, 7477. [Google Scholar] [CrossRef] [PubMed]
- Lasda, E.; Parker, R. Circular RNAs: Diversity of form and function. RNA 2014, 20, 1829–1842. [Google Scholar] [CrossRef] [PubMed]
- Salzman, J. Circular RNA expression: Its potential regulation and function. Trends Genet. 2016, 32, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Luo, J.; Du, J.; Liu, C.; Wu, X.; Pu, Q.; Fu, Y.; Tang, Q.; Liu, Y.; Li, Q. Transcriptome Analysis of Liangshan Pig Muscle Development at the Growth Curve Inflection Point and Asymptotic Stages Using Digital Gene Expression Profiling. PLoS ONE 2015, 10, e0135978. [Google Scholar] [CrossRef]
- Li, M.; Wu, H.; Luo, Z.; Xia, Y.; Guan, J.; Wang, T.; Gu, Y.; Chen, L.; Zhang, K.; Ma, J. An atlas of DNA methylomes in porcine adipose and muscle tissues. Nat. Commun. 2012, 3, 850. [Google Scholar] [CrossRef]
- Nešetřilová, H.; Šařecová, P.; Louda, F.; Nová, V. Multiphasic growth models for cattle. Czech J. Anim. Sci. 2005, 50, 347–354. [Google Scholar] [CrossRef]
- Tsoularis, A.; Wallace, J. Analysis of logistic growth models. Math. Biosci. 2002, 179, 21–55. [Google Scholar] [CrossRef] [Green Version]
- Strathe, A.B.; Danfær, A.; Sørensen, H.; Kebreab, E. A multilevel nonlinear mixed-effects approach to model growth in pigs. J. Anim. Sci. 2010, 88, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, M.; Ma, J.; Zhang, J.; Zhou, C.; Wang, T.; Gao, X.; Li, X. Identification of differences in microRNA transcriptomes between porcine oxidative and glycolytic skeletal muscles. BMC Mol. Biol. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, P.M.; Lowry, O.H. Myoglobin levels in individual human skeletal muscle fibers of different types. J. Histochem. Cytochem. 1984, 32, 1211–1216. [Google Scholar] [CrossRef] [PubMed]
- Schuler, M.; Ali, F.; Chambon, C.; Duteil, D.; Bornert, J.-M.; Tardivel, A.; Desvergne, B.; Wahli, W.; Chambon, P.; Metzger, D. PGC1α expression is controlled in skeletal muscles by PPARβ, whose ablation results in fiber-type switching, obesity, and type 2 diabetes. Cell Metab. 2006, 4, 407–414. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Watkins, S.; Kelley, D.E. Skeletal muscle lipid content and oxidative enzyme activity in relation to muscle fiber type in type 2 diabetes and obesity. Diabetes 2001, 50, 817–823. [Google Scholar] [CrossRef]
- Yu, L.; Tai, L.; Zhang, L.; Chu, Y.; Li, Y.; Zhou, L. Comparative analyses of long non-coding RNA in lean and obese pigs. Oncotarget 2017, 8, 41440. [Google Scholar] [CrossRef]
- Ren, H.; Wang, G.; Chen, L.; Jiang, J.; Liu, L.; Li, N.; Zhao, J.; Sun, X.; Zhou, P. Genome-wide analysis of long non-coding RNAs at early stage of skin pigmentation in goats (Capra hircus). BMC Genomics 2016, 17, 67. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, X.; Han, K.; Zhang, G.; Wang, J.; Xie, K.; Xue, Q. Genome-wide analysis of lncRNA and mRNA expression during differentiation of abdominal preadipocytes in the chicken. G3 (Bethesda) 2017, 7, 953–966. [Google Scholar] [CrossRef]
- Xia, J.; Xin, L.; Zhu, W.; Li, L.; Li, C.; Wang, Y.; Mu, Y.; Yang, S.; Li, K. Characterization of long non-coding RNA transcriptome in high-energy diet induced nonalcoholic steatohepatitis minipigs. Sci. Rep. 2016, 6, 30709. [Google Scholar] [CrossRef]
- Ørom, U.A.; Derrien, T.; Beringer, M.; Gumireddy, K.; Gardini, A.; Bussotti, G.; Lai, F.; Zytnicki, M.; Notredame, C.; Huang, Q. Long noncoding RNAs with enhancer-like function in human cells. Cell 2010, 143, 46–58. [Google Scholar] [CrossRef]
- Zhang, B.; Arun, G.; Mao, Y.S.; Lazar, Z.; Hung, G.; Bhattacharjee, G.; Xiao, X.; Booth, C.J.; Wu, J.; Zhang, C. The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep. 2012, 2, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Luo, J.; Lei, H.; Jiang, Y.; Bai, L.; Li, M.; Tang, G.; Li, X.; Zhang, S.; Zhu, L. Effects of muscle fiber type on glycolytic potential and meat quality traits in different Tibetan pig muscles and their association with glycolysis-related gene expression. Genet. Mol. Res. 2015, 14, 14366–14378. [Google Scholar] [CrossRef] [PubMed]
- Gollnick, P.; Pernow, B.; Essen, B.; Jansson, E.; Saltin, B. Availability of glycogen and plasma FFA for substrate utilization in leg muscle of man during exercise. Clin. Physiol. 1981, 1, 27–42. [Google Scholar] [CrossRef]
- Howald, H.; Hoppeler, H.; Claassen, H.; Mathieu, O.; Straub, R. Influences of endurance training on the ultrastructural composition of the different muscle fiber types in humans. Pflügers Arch. 1985, 403, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.S.; Chabowski, A.; Snook, L.A.; Schwenk, R.W.; Glatz, J.F.; Luiken, J.J.; Bonen, A. Additive effects of insulin and muscle contraction on fatty acid transport and fatty acid transporters, FAT/CD36, FABPpm, FATP1, 4 and 6. FEBS Lett. 2009, 583, 2294–2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Wang, J.; Zhang, C.; Lin, C.; Zhang, J.; Zhang, W.; Zhang, W.; Lu, Y.; Zheng, L.; Li, X. Circular RNA circITGA7 inhibits colorectal cancer growth and metastasis by modulating the Ras pathway and upregulating transcription of its host gene ITGA7. J. Pathol. 2018, 246, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Siede, D.; Rapti, K.; Gorska, A.; Katus, H.; Altmüller, J.; Boeckel, J.; Meder, B.; Maack, C.; Völkers, M.; Müller, O. Identification of circular RNAs with host gene-independent expression in human model systems for cardiac differentiation and disease. J. Mol. Cell. Cardiol. 2017, 109, 48–56. [Google Scholar] [CrossRef]
- Huang, M.; Zhong, Z.; Lv, M.; Shu, J.; Tian, Q.; Chen, J. Comprehensive analysis of differentially expressed profiles of lncRNAs and circRNAs with associated co-expression and ceRNA networks in bladder carcinoma. Oncotarget 2016, 7, 47186. [Google Scholar] [CrossRef]
- Rong, D.; Sun, H.; Li, Z.; Liu, S.; Dong, C.; Fu, K.; Tang, W.; Cao, H. An emerging function of circRNA-miRNAs-mRNA axis in human diseases. Oncotarget 2017, 8, 73271. [Google Scholar] [CrossRef]
- Shen, L.; Chen, L.; Zhang, S.; Zhang, Y.; Wang, J.; Zhu, L. MicroRNA-23a reduces slow myosin heavy chain isoforms composition through myocyte enhancer factor 2C (MEF2C) and potentially influences meat quality. Meat Sci. 2016, 116, 201–206. [Google Scholar] [CrossRef]
- Shen, L.; Chen, L.; Zhang, S.; Du, J.; Bai, L.; Zhang, Y.; Jiang, Y.; Li, X.; Wang, J.; Zhu, L. MicroRNA-27b regulates mitochondria biogenesis in myocytes. PLoS ONE 2016, 11, e0148532. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-E.; Kemper, J.K. Regulation of SIRT1 by microRNAs. Mol. Cells 2013, 36, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Gurd, B.J. Deacetylation of PGC-1α by SIRT1: Importance for skeletal muscle function and exercise-induced mitochondrial biogenesis. Appl. Physiol. Nutr. Metab. 2011, 36, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Joo, S.; Ryu, Y. Skeletal muscle fiber type and myofibrillar proteins in relation to meat quality. Meat Sci. 2010, 86, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Koohmaraie, M.; Shackelford, S.; Wheeler, T.; Lonergan, S.M.; Doumit, M. A muscle hypertrophy condition in lamb (callipyge): Characterization of effects on muscle growth and meat quality traits. J. Anim. Sci. 1995, 73, 3596–3607. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.; Huang, W.; Bhandari, S.; Basu, A. Goodness-of-fit testing in growth curve models: A general approach based on finite differences. Comput. Stat. Data Anal. 2011, 55, 1086–1098. [Google Scholar] [CrossRef]
- Gollnick, P.; Parsons, D.; Oakley, C. Differentiation of fiber types in skeletal muscle from the sequential inactivation of myofibrillar actomyosin ATPase during acid preincubation. Histochemistry 1983, 77, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Sloane-Stanley, G. A simple method for the isolation and purification of total lipids from animal tissues. J. Boil. Chem. 1957, 226, 497–509. [Google Scholar]
- Shen, L.; Zhang, S.; Li, Q.; Fu, Y.; Tang, G.; Jiang, Y.; Li, M.; Wang, J.; Li, X.; Che, L. The Landscape of Non-Coding RNA in an Adult Pig Model of Intrauterine Growth Restriction. Cell. Physiol. Biochem. 2018, 50, 1764–1778. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.-Q.; Liu, X.-Q.; Zhao, S.-Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Yue, H.; Zhang, M.; Luo, J.; Liu, L.; Wu, W.; Xiao, T.; Chen, X.; Chen, X.; Zhang, D. Transcriptional profiling analysis and functional prediction of long noncoding RNAs in cancer. Oncotarget 2016, 7, 8131. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ma, J.; Long, K.; Jin, L.; Liu, Y.; Zhou, C.; Tian, S.; Chen, L.; Luo, Z.; Tang, Q. Dynamic gene expression profiles during postnatal development of porcine subcutaneous adipose. PeerJ 2016, 4, e1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Data (Reads) | Clean Data (Reads) | Data Size (Gb) | Clean Data Q20 (%) | GC Content (%) | rRNA Reads (%) |
|---|---|---|---|---|---|---|
| LDM_1 | 90164014 | 89626552 | 13.52 | 96.75 | 52.52 | 0.56 |
| LDM_2 | 89601234 | 89357710 | 13.44 | 96.88 | 50.84 | 0.26 |
| LDM_3 | 89028092 | 88835098 | 13.35 | 96.75 | 53.75 | 0.21 |
| PMM_1 | 90586046 | 90269524 | 13.59 | 96.77 | 51.7 | 0.33 |
| PMM_2 | 90321580 | 89987360 | 13.55 | 96.44 | 51.41 | 0.35 |
| PMM_3 | 90497574 | 90141574 | 13.57 | 96.5 | 51.46 | 0.38 |
| Accession | Fold Change (LDM/PMM) | Host Gene | Sponge miRNAs |
|---|---|---|---|
| ciRNA290 | −16.97 | PAN2 | miR-133a, miR-150, miR-221, miR-23a, miR-23b, miR-27a, miR-27b, miR-30b |
| circRNA8413 | −7.08 | AQP4 | miR-9823 |
| circRNA17323 | −5.94 | C11orf70 | miR-9859 |
| circRNA16211 | −5.38 | PANK1 | miR-22, miR-339, miR-339, miR-676 |
| circRNA5396 | −5.28 | PANK1 | miR-22, miR-676 |
| circRNA10701 | 4.99 | SLC6A20 | miR-2320, miR-4335, miR-885 |
| ciRNA41 | −4.07 | MYH7 | miR-217 |
| ciRNA296 | −4.00 | CCDC88A | miR-421 |
| circRNA12969 | −3.82 | CLIP4 | miR-221, miR-23a, miR-23b, miR-27a, |
| circRNA9210 | −3.77 | CLIP4 | miR-133a, miR-145, miR-150, miR-23a, miR-23b, miR-27a, miR-27b |
| Chromosome | DE Transcript Number in QTL Region/QTL Region Length (Mb) | ||
|---|---|---|---|
| mRNA | LncRNA | CircRNA | |
| 1 | 25/15.39 | 31/19.56 | 1/0.31 |
| 2 | 32/15.45 | 32/13.69 | 4/2.82 |
| 3 | 21/10.07 | 17/9.72 | 13/1.70 |
| 4 | 31/11.78 | 27/13.26 | 4/2.09 |
| 5 | 18/5.87 | 6/4.53 | 17/1.43 |
| 6 | 35/18.78 | 18/9.88 | 2/1.73 |
| 7 | 41/26.39 | 33/22.27 | 1/0.92 |
| 8 | 14/12.24 | 29/13.27 | 0/0 |
| 9 | 19/13.90 | 17/13.27 | 2/2.13 |
| 10 | 16/10.93 | 24/8.18 | 5/2.04 |
| 11 | 4/10.93 | 1/1.73 | 1/1.00 |
| 12 | 21/13.89 | 19/12.07 | 0/0 |
| 13 | 25/14.34 | 32/13.19 | 4/0 |
| 14 | 19/10.95 | 21/11.51 | 3/1.22 |
| 15 | 17/6.68 | 21/8.56 | 8/2.13 |
| 16 | 8/3.95 | 16/7.80 | 5/2.04 |
| 17 | 6/1.30 | 4/2.13 | 4/1.81 |
| 18 | 6/2.52 | 8/3.56 | 0/0 |
| X | 4/3.07 | 1/0.72 | 0/0 |
| MT | 0/0 | 0/0 | 0/0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, L.; Gan, M.; Tang, Q.; Tang, G.; Jiang, Y.; Li, M.; Chen, L.; Bai, L.; Shuai, S.; Wang, J.; et al. Comprehensive Analysis of lncRNAs and circRNAs Reveals the Metabolic Specialization in Oxidative and Glycolytic Skeletal Muscles. Int. J. Mol. Sci. 2019, 20, 2855. https://doi.org/10.3390/ijms20122855
Shen L, Gan M, Tang Q, Tang G, Jiang Y, Li M, Chen L, Bai L, Shuai S, Wang J, et al. Comprehensive Analysis of lncRNAs and circRNAs Reveals the Metabolic Specialization in Oxidative and Glycolytic Skeletal Muscles. International Journal of Molecular Sciences. 2019; 20(12):2855. https://doi.org/10.3390/ijms20122855
Chicago/Turabian StyleShen, Linyuan, Mailin Gan, Qianzi Tang, Guoqing Tang, Yanzhi Jiang, Mingzhou Li, Lei Chen, Lin Bai, Surong Shuai, Jinyong Wang, and et al. 2019. "Comprehensive Analysis of lncRNAs and circRNAs Reveals the Metabolic Specialization in Oxidative and Glycolytic Skeletal Muscles" International Journal of Molecular Sciences 20, no. 12: 2855. https://doi.org/10.3390/ijms20122855