Comparative Transcriptomic Analysis of Biological Process and Key Pathway in Three Cotton (Gossypium spp.) Species Under Drought Stress

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Transcriptome Sequencing, Data Statistics and Evaluation, and Reads Mapping
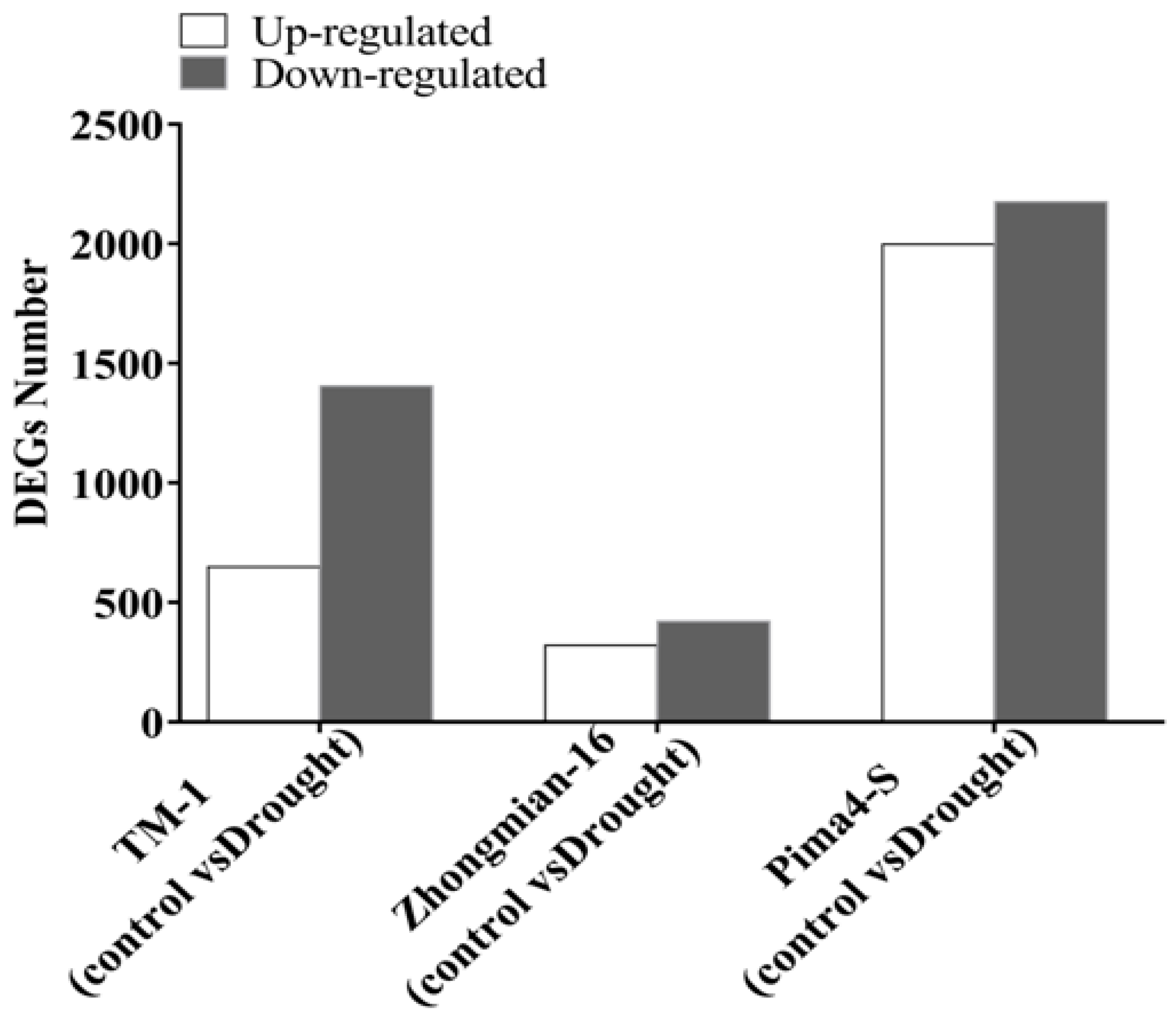
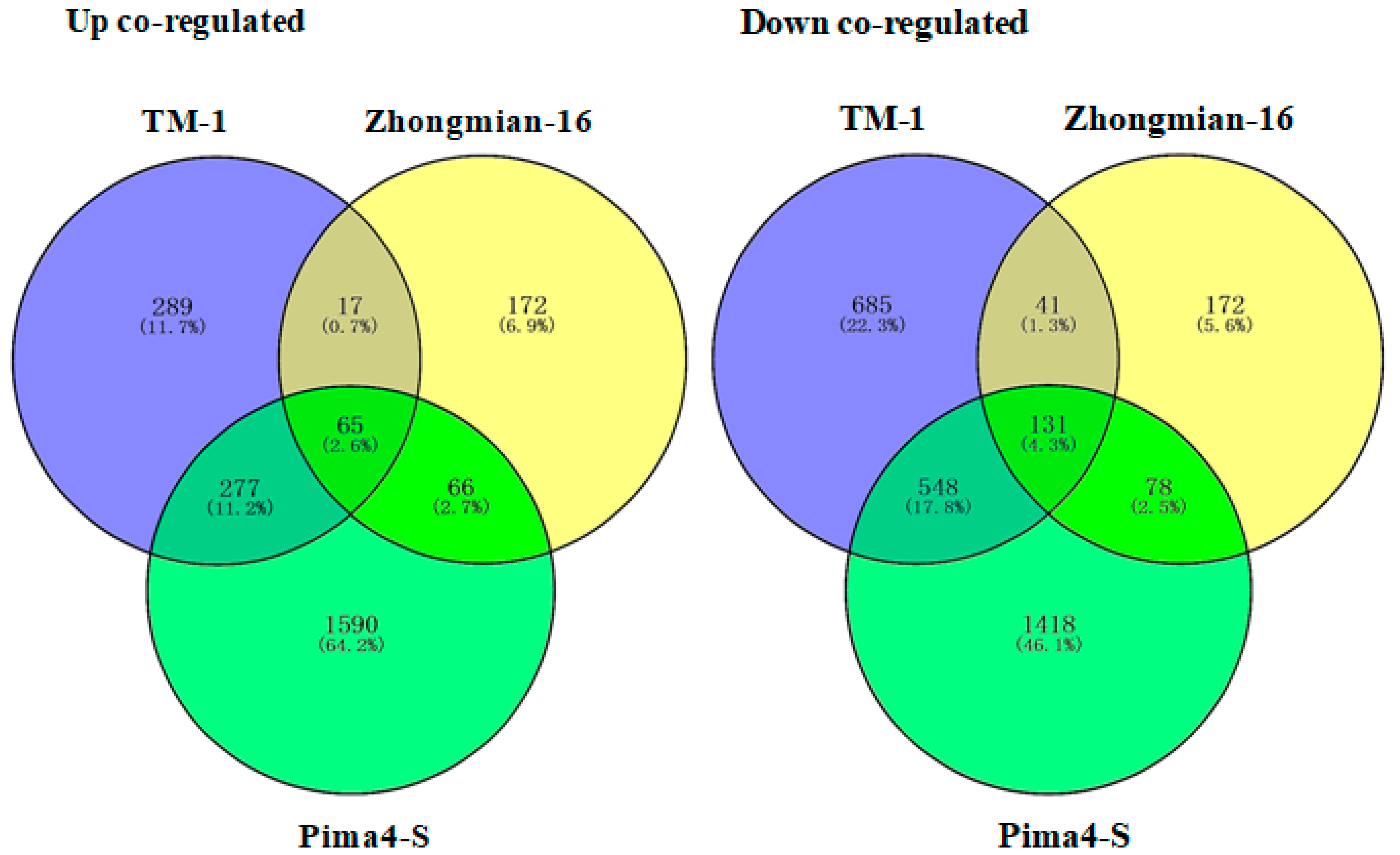
2.2. Study of Gene Expression in Three Cotton Species Under Drought Stress
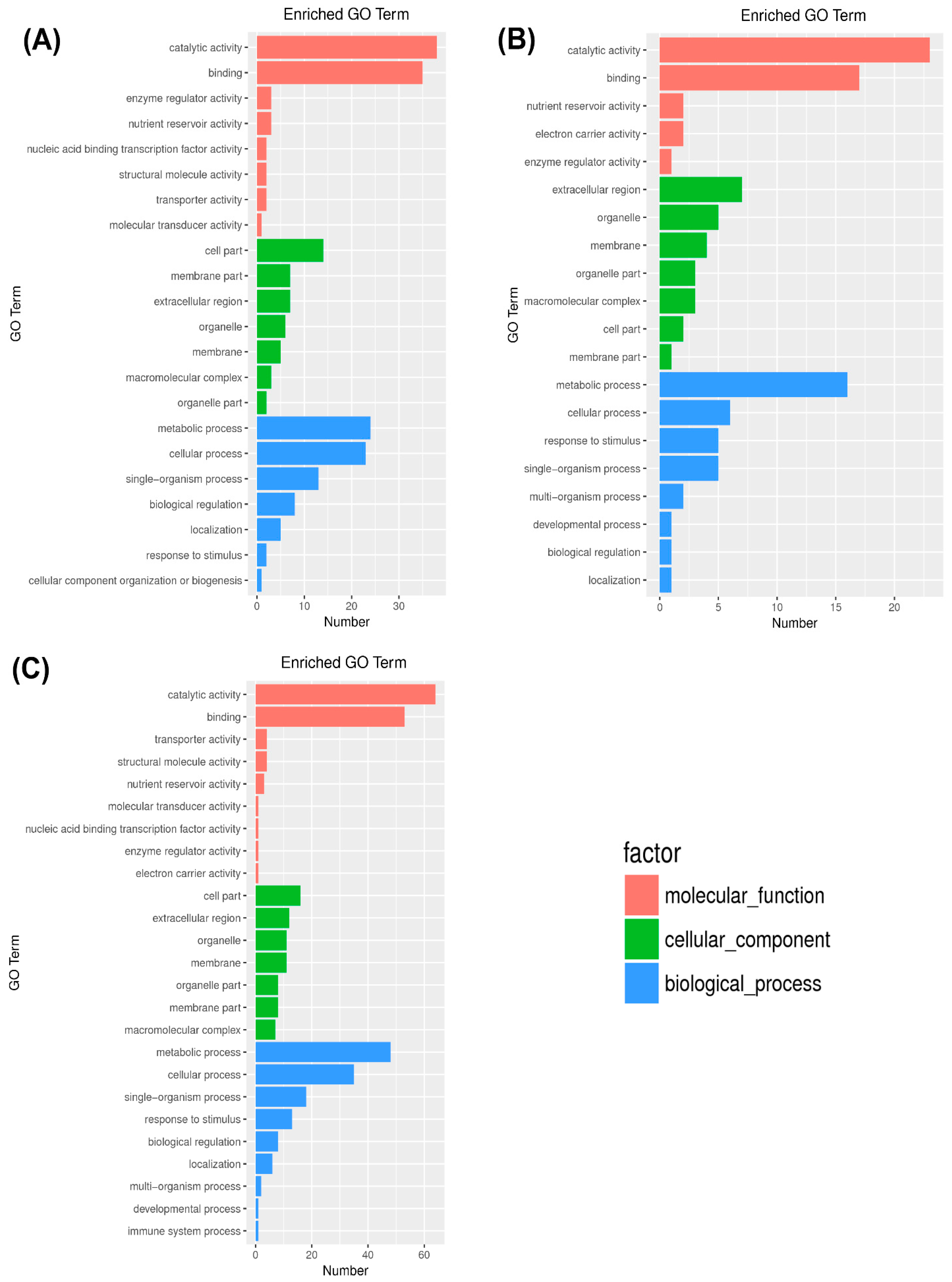
2.3. Gene Ontology (GO) Enrichment Analysis
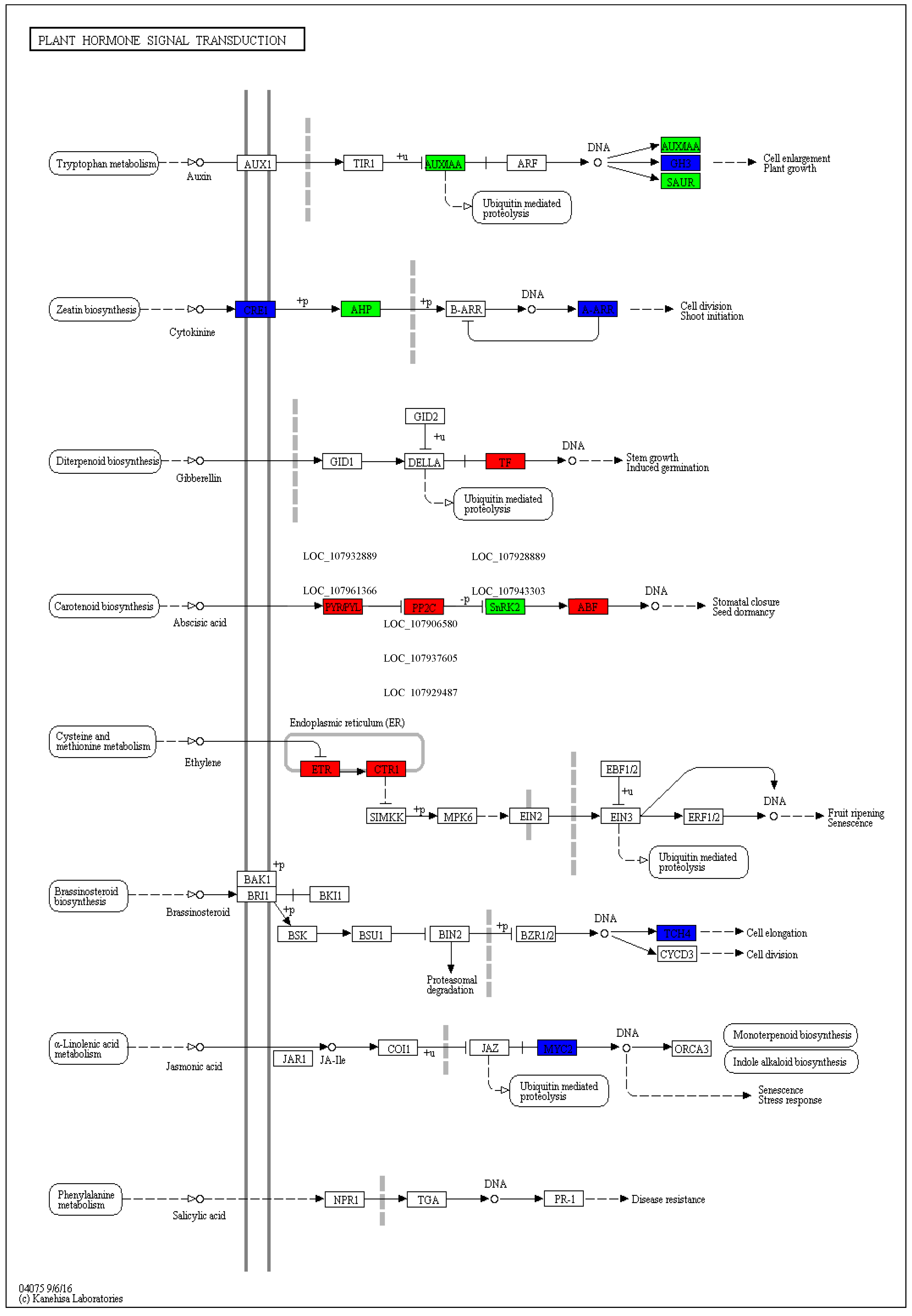
2.4. Kyoto Encyclopedia of Genes and Genomes and DEGs
2.5. Identification of Drought-Responsive Genes and Transcriptional Factors (TFs)
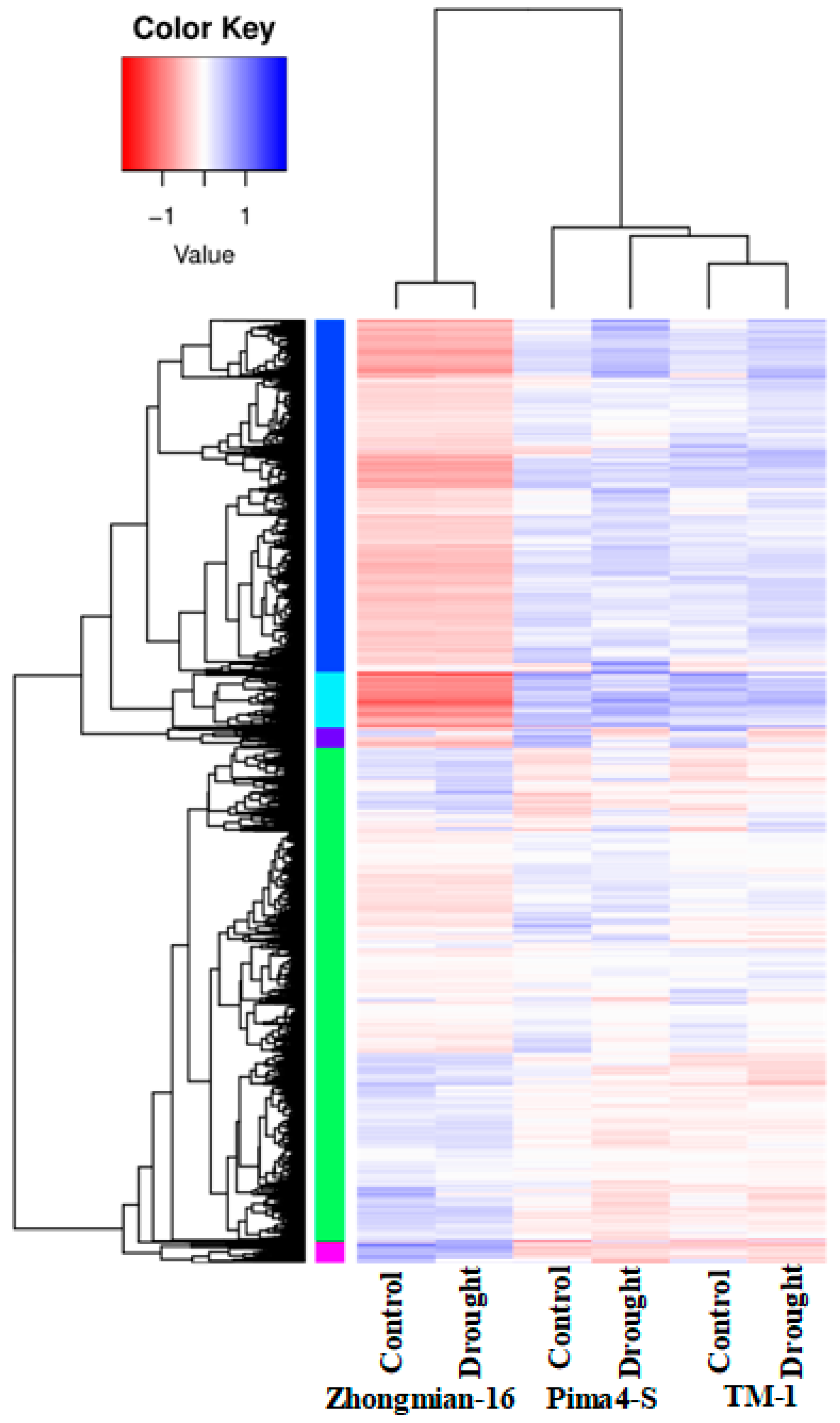
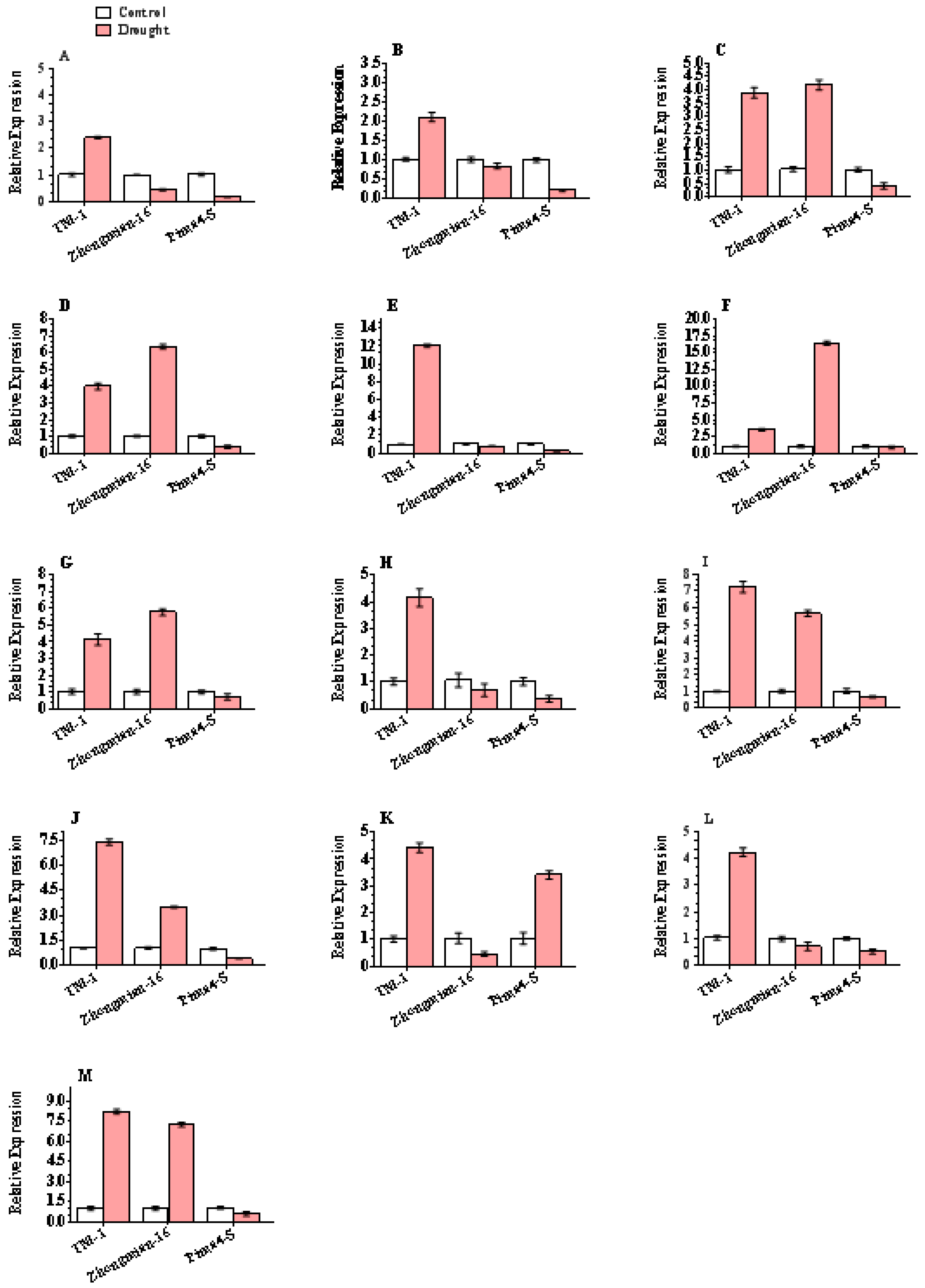
2.6. Justification of DEG-Based Gene Expression
3. Discussion
3.1. Drought-Related Genes in DEGs
3.2. Gene Ontology
3.3. KEGG Pathway Involved in Drought Stress
3.4. Drought-Responsive Genes and TFs Present in TM-1 Species
4. Materials and Methods
4.1. Plant Material and Growth Conditions
4.2. RNA Extraction and Illumina Sequencing
4.3. Differential Gene Expression (DGE) Analysis
4.4. GO and KEGG Pathway Analysis
4.5. Validation of DEGs by RT-qPCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Reynolds, M.; Tuberosa, R. Translational research impacting on crop productivity in drought-prone environments. Curr. Opin. Plant Biol. 2008, 11, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Babu, R.C.; Blum, A. Breeding for drought resistance in rice: Physiology and molecular genetics considerations. Crop Sci. 1997, 37, 1426–1434. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, X.; Jiao, Y.; Qin, Y.; Liu, X.; He, K.; Chen, C.; Ma, L.; Wang, J.; Xiong, L.; et al. Global genome expression analysis of rice in response to drought and high-salinity stresses in shoot, flag leaf, and panicle. Plant Mol. Biol. 2007, 63, 591–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, M.J. Role of Osmolytes and Antioxidant Enzymes for Drought Tolerance in Wheat. In Global Wheat Production; Fahad, S., Ed.; IntechOpen Limited: London, UK, 2018; pp. 172–174. [Google Scholar]
- Molina, C.; Rotter, B.; Horres, R.; Udupa, S.M.; Besser, B.; Bellarmino, L.; Baum, M.; Matsumura, H.; Terauchi, R.; Kahl, G.; et al. SuperSAGE: The drought stress-responsive transcriptome of chickpea roots. BMC Genom. 2008, 9, 553. [Google Scholar] [CrossRef] [PubMed]
- Stiller, W.N.; Read, J.J.; Constable, G.A.; Reid, P.E. Selection for water use efficiency traits in a cotton breeding program: Cultivar differences. Crop Sci. 2005, 45, 1107–1113. [Google Scholar] [CrossRef]
- Abid, M.; Ali, S.; Qi, L.K.; Zahoor, R.; Tian, Z.; Jiang, D.; Snider, J.L.; Dai, T. Physiological and biochemical changes during drought and recovery periods at tillering and jointing stages in wheat (Triticum aestivum L.). Sci. Rep. 2018, 8, 1–15. [Google Scholar]
- Park, W.; Scheffler, B.E.; Bauer, P.J.; Campbell, B.T. Genome-wide identification of differentially expressed genes under water deficit stress in upland cotton (Gossypium hirsutum L.). BMC Plant Biol. 2012, 12, 90. [Google Scholar] [CrossRef] [PubMed]
- Bowman, M.J.; Park, W.; Bauer, P.J.; Udall, J.A.; Page, J.T.; Raney, J.; Scheffler, B.E.; Jones, D.C.; Campbell, B.T. RNA-Seq transcriptome profiling of upland cotton (Gossypium hirsutum L.) root tissue under water-deficit stress. PLoS ONE 2013, 8, 19–21. [Google Scholar] [CrossRef]
- Ranjan, A.; Pathre, U.V.; Ranjan, S.; Mantri, S.; Trivedi, I.; Sawant, S.V.; Tuli, R.; Jena, S.N.; Asif, M.H.; Koul, B.; et al. Genome wide expression profiling of two accession of G. herbaceum L. in response to drought. BMC Genom. 2012, 13, 94. [Google Scholar] [CrossRef]
- Zhang, X.; Yao, D.; Wang, Q.; Xu, W.; Wei, Q.; Wang, C.; Liu, C.; Zhang, C.; Yan, H.; Ling, Y.; et al. mRNA-seq Analysis of the Gossypium arboreum transcriptome Reveals Tissue Selective Signaling in Response to Water Stress during Seedling Stage. PLoS ONE 2013, 8, e54762. [Google Scholar]
- Zhu, J.K. Abiotic Stress Signaling and Responses in Plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef]
- Moumeni, A.; Satoh, K.; Kondoh, H.; Asano, T.; Hosaka, A.; Venuprasad, R.; Serraj, R.; Kumar, A.; Leung, H.; Kikuchi, S. Comparative analysis of root transcriptome profiles of two pairs of drought-tolerant and susceptible rice near-isogenic lines under different drought stress. BMC Plant Biol. 2011, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Dubos, C.; Plomion, C. Identi cation of water-de cit responsive genes in maritime pine (Pinus pinaster Ait.) roots. Plant Mol. Biol. 2003, 51, 249–262. [Google Scholar] [CrossRef]
- Bruce, W.B.; Edmeades, G.O.; Barker, T.C. Molecular and physiological approaches to maize improvement for drought tolerance Wesley. J. Exp. Bot. 2002, 53, 13–25. [Google Scholar] [CrossRef]
- Rabbani, M.A.; Maruyama, K.; Abe, H.; Khan, M.A.; Katsura, K. Monitoring expression profiles of rice genes under cold, drought, and high-salinity stresses and and abscisic acid application using cDNA microarray and RNA gel-blot analyses. Plant Physiol. 2003, 133, 1755–1767. [Google Scholar] [CrossRef]
- Umezawa, T.; Fujita, M.; Fujita, Y.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Engineering drought tolerance in plants: Discovering and tailoring genes to unlock the future. Curr. Opin. Biotechnol. 2006, 17, 113–122. [Google Scholar] [CrossRef]
- Cushman, J.C.; Bohnert, H.J. Genomic approaches to plant stress tolerance. Curr. Opin. Plant Biol. 2000, 3, 117–124. [Google Scholar] [CrossRef]
- Ahmad, R.T.; Malik, T.A.; Khan, I.A.; Jaskani, M.J. Genetic analysis of some morpho-physiological traits related to drought stress in cotton (Gossypium hirsutum). Int. J. Agric. Biol. 2009, 11, 235–240. [Google Scholar]
- Basu, P.S.; Ali, M.; Chaturvedi, S.K. Osmotic adjustment increases water uptake, remobilization of assimilates and maintains photosynthesis in chickpea under drought. Indian J. Exp. Biol. 2007, 45, 261–267. [Google Scholar] [PubMed]
- Hasan, M.M.-U.; Ma, F.; Prodhan, Z.H.; Li, F.; Shen, H.; Chen, Y.; Wang, X. Molecular and physio-biochemical characterization of cotton species for assessing drought stress tolerance. Int. J. Mol. Sci. 2018, 19, 2636. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, M.A.; Iqbal, M.; Rasheed, R.; Hussain, I.; Perveen, S.; Mahmood, S. Dynamic Proline Metabolism: Importance and Regulation in Water-Limited Environments. In Plant Metabolites and Regulation under Environmental Stress; 2018; pp. 323–336. ISBN 9780128126905. [Google Scholar]
- Guo, Y.H.; Yu, Y.P.; Wang, D.; Wu, C.A.; Yang, G.D.; Huang, J.G.; Zheng, C.C. GhZFP1, a novel CCCH-type zinc finger protein from cotton, enhances salt stress tolerance and fungal disease resistance in transgenic tobacco by interacting with GZIRD21A and GZIPR5. New Phytol. 2009, 183, 62–75. [Google Scholar] [CrossRef]
- He, L.; Yang, X.; Wang, L.; Zhu, L.; Zhou, T.; Deng, J.; Zhang, X. Molecular cloning and functional characterization of a novel cotton CBL-interacting protein kinase gene (GhCIPK6) reveals its involvement in multiple abiotic stress tolerance in transgenic plants. Biochem. Biophys. Res. Commun. 2013, 435, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Gu, Z.; Xin, D.; Hao, L.; Liu, C.; Huang, J.; Ma, B.; Zhang, H. Identification and characterization of putative CIPK genes in maize. J. Genet. Genom. 2011, 2, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xi, D.; Li, S.; Gao, Z.; Zhao, S.; Shi, J.; Wu, C.; Guo, X. A cotton group C MAP kinase gene, GhMPK2, positively regulates salt and drought tolerance in tobacco. Plant Mol. Biol. 2011, 77, 17–31. [Google Scholar] [CrossRef]
- Taliercio, E.; Allen, R.D.; Essenberg, M.; Klueva, N.; Nguyen, H.; Patil, M.A.; Payton, P.; Millena, A.C.M.; Phillips, A.L.; Pierce, M.L.; et al. Analysis of ESTs from multiple Gossypium hirsutum tissues and identification of SSRs. Genome 2006, 49, 306–329. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Shinozaki, K.; Yamaguchi-Shinozaki, K. Gene networks involved in drought stress response and tolerance. J. Exp. Bot. 2007, 58, 221–227. [Google Scholar] [CrossRef]
- Gao, F.; Wang, J.; Wei, S.; Li, Z.; Wang, N.; Li, H.; Feng, J.; Li, H.; Zhou, Y.; Zhang, F. Transcriptomic analysis of drought stress responses in Ammopiptanthus mongolicus leaves using the RNA-seq technique. PLoS ONE 2015, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, M.; Canales, E.; Borrás-Hidalgo, O. Molecular aspects of abiotic stress in plants. Biotecnol. Apl. 2005, 22, 1–10. [Google Scholar]
- Manivannan, P.; Jaleel, C.A.; Sankar, B.; Kishorekumar, A.; Somasundaram, R.; Lakshmanan, G.M.A.; Panneerselvam, R. Growth, biochemical modifications and proline metabolism in Helianthus annuus L. as induced by drought stress. Colloids Surfaces B Biointerfaces 2007, 59, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Hasanuzzaman, M.; Nahar, K.; Gill, S.S.; Alharby, H.F.; Razafindrabe, B.H.N.; Fujita, M. Hydrogen peroxide pretreatment mitigates cadmium-induced oxidative stress in Brassica napus L.: An intrinsic study on antioxidant defense and glyoxalase systems. Front. Plant Sci. 2017, 8, 115. [Google Scholar] [CrossRef]
- Wang, R.; Chen, S.; Zhou, X.; Shen, X.; Deng, L.; Zhu, H.; Shao, J.; Shi, Y.; Dai, S.; Aloys, E.F.; et al. Ionic homeostasis and reactive oxygen species control in leaves and xylem sap of two poplars subjected to NaCl stress RUIGANG. Tree Physiol. 2008, 28, 947–957. [Google Scholar] [CrossRef]
- Caldana, C.; Degenkolbe, T.; Cuadros-Inostroza, A.; Klie, S.; Sulpice, R.; Leisse, A.; Steinhauser, D.; Fernie, A.R.; Willmitzer, L.; Hannah, M.A. High-density kinetic analysis of the metabolomic and transcriptomic response of Arabidopsis to eight environmental conditions. Plant J. 2011, 67, 869–884. [Google Scholar] [CrossRef]
- Vermeirssen, V.; De Clercq, I.; Van Parys, T.; Van Breusegem, F.; Van de Peer, Y. Arabidopsis\n Ensemble Reverse-Engineered Gene Regulatory Network Discloses Interconnected Transcription Factors in Oxidative Stress. Plant Cell 2014, 26, 4656–4679. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, C.; Li, L.; Meng, L.; Singh, J.; Jiang, N.; Deng, X.-W.; He, Z.-H.; Lemaux, P.G. Evolutionary expansion, gene structure, and expression of the rice wall-associated kinase gene family. Plant Physiol. 2005, 139, 1107–1124. [Google Scholar] [CrossRef]
- Lv, D.-W.; Subburaj, S.; Cao, M.; Yan, X.; Li, X.; Appels, R.; Sun, D.-F.; Ma, W.; Yan, Y.-M. Proteome and phosphoproteome characterization reveals new response and defense mechanisms of Brachypodium distachyon leaves under salt stress. Mol. Cell. Proteom. 2014, 13, 632–652. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Gan, S.-S. An abscisic acid-AtNAP transcription factor-SAG113 protein phosphatase 2C regulatory chain for controlling dehydration in senescing Arabidopsis leaves. Plant Physiol. 2011, 158, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Gaion, L.A.; Monteiro, C.C.; Cruz, F.J.R.; Rossatto, D.R.; López-Díaz, I.; Carrera, E.; Lima, J.E.; Peres, L.E.P.; Carvalho, R.F. Constitutive gibberellin response in grafted tomato modulates root-to-shoot signaling under drought stress. J. Plant Physiol. 2018, 221, 11–21. [Google Scholar] [CrossRef]
- Yu, C.; Zhan, Y.; Feng, X.; Huang, Z.A.; Sun, C. Identification and expression profiling of the auxin response factors in capsicum annuum L. Under abiotic stress and hormone treatments. Int. J. Mol. Sci. 2017, 18, 2719. [Google Scholar] [CrossRef]
- Guo, L.; Wang, Z.Y.; Lin, H.; Cui, W.E.; Chen, J.; Liu, M.; Chen, Z.L.; Qu, L.J.; Gu, H. Expression and functional analysis of the rice plasma-membrane intrinsic protein gene family. Cell Res. 2006, 16, 277–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, P.; Liu, P.; Yuan, G.; Jia, J.; Li, X.; Qi, D.; Ma, T.; Liu, P.; Yuan, G.; Ma, T.; et al. New Insights on Drought Stress Response by Global Investigation of Gene Expression Changes in Sheepgrass (Leymus chinensis). Front. Plant Sci. 2016, 7, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Xi, J.; Xu, P.; Xiang, C. Bin Loss of AtPDR11, a plasma membrane-localized ABC transporter, confers paraquat tolerance in Arabidopsis thaliana. Plant J. 2012, 69, 782–791. [Google Scholar] [CrossRef]
- Crouzet, J.; Roland, J.; Peeters, E.; Trombik, T.; Ducos, E.; Nader, J.; Boutry, M. NtPDR1, a plasma membrane ABC transporter from Nicotiana tabacum, is involved in diterpene transport. Plant Mol. Biol. 2013, 82, 181–192. [Google Scholar] [CrossRef]
- Bleecker, A.B.; Kende, H. Ethylene: A Gaseous Signal Molecule in Plants. Annu. Rev. Cell Dev. Biol. 2002, 16, 1–18. [Google Scholar] [CrossRef]
- Wu, Y. Modification of Expansin Transcript Levels in the Maize Primary Root at Low Water Potentials. Plant Physiol. 2002, 126, 1471–1479. [Google Scholar] [CrossRef]
- Liu, T.; Li, Z.; Hui, C.; Tang, M.; Zhang, H. Effect of Rhizophagus irregularis on osmotic adjustment, antioxidation and aquaporin PIP genes expression of Populus × canadensis ‘Neva’ under drought stress. Acta Physiol. Plant. 2016, 38, 191. [Google Scholar] [CrossRef]
- Sakuraba, Y.; Kim, Y.-S.; Han, S.-H.; Lee, B.-D.; Paek, N.-C. The Arabidopsis transcription factor NAC016 promotes drought stress responses by repressing AREB1 transcription through a trifurcate teed-forward regulatory loop involving NAP. Plant Cell 2015, 27, 1771–1787. [Google Scholar] [CrossRef]
- Singh, D.; Laxmi, A. Transcriptional regulation of drought response: A tortuous network of transcriptional factors. Front. Plant Sci. 2015, 6, 895. [Google Scholar] [CrossRef]
- Sofo, A.; Scopa, A.; Nuzzaci, M.; Vitti, A. Ascorbate peroxidase and catalase activities and their genetic regulation in plants subjected to drought and salinity stresses. Int. J. Mol. Sci. 2015, 16, 13561–13578. [Google Scholar] [CrossRef]
- Chen, L.; Ren, F.; Zhong, H.; Jiang, W.; Li, X. Identification and expression analysis of genes in response to high-salinity and drought stresses in Brassica napus. Acta Biochim. Biophys. Sin. (Shanghai) 2010, 42, 154–164. [Google Scholar] [CrossRef]
- Ramanjulu, S.; Bartels, D. Drought- and desiccation-induced modulation of gene expression in plants. Plant Cell Environ. 2002, 25, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Chinnusamy, V.; Schumaker, K.; Zhu, J.K. Molecular genetic perspectives on cross-talk and specificity in abiotic stress signalling in plants. J. Exp. Bot. 2004, 55, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Sunkar, R.; Bartels, D.; Kirch, H.H. Overexpression of a stress-inducible aldehyde dehydrogenase gene from Arabidopsis thaliana in transgenic plants improves stress tolerance. Plant J. 2003, 35, 452–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, H.; Chen, H.; Chen, C.; Shang, X.; Liu, X.; Xia, R.; Wei, S.; Sun, M.; Xie, Q.; Hao, D. Identification of Drought Tolerant Mechanisms in Maize Seedlings Based on Transcriptome Analysis of Recombination Inbred Lines. Front. Plant Sci. 2016, 7, 1080. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.K.; Jha, B. Transcription factors in plants and ABA dependent and independent abiotic stress signalling. Biol. Plant. 2010, 54, 201–212. [Google Scholar] [CrossRef]
- Wang, Z.-Y.; Xiong, L.; Li, W.; Zhu, J.-K.; Zhu, J. The Plant Cuticle Is Required for Osmotic Stress Regulation of Abscisic Acid Biosynthesis and Osmotic Stress Tolerance in Arabidopsis. Plant Cell 2011, 23, 1971–1984. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi-Shinozaki, K.; Shinozaki, K. Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Annu. Rev. Plant Biol. 2006, 57, 781–803. [Google Scholar] [CrossRef]
- Wenxuan, Q.; Guijie, D. Dynamic of Volatiles and Endogenous Hormones in Pinus massoniana Needles under Drought Stress. Sci. SILVAE Sin. 2017, 53, 49–55. [Google Scholar]
- Ma, Y.; Szostkiewicz, I.; Korte, A.; Moes, D.; Yang, Y.; Christmann, A.; Grill, E. Regulators of PP2C phosphatase activity function as abscisic acid sensors. Science 2009, 324, 1064–1068. [Google Scholar] [CrossRef]
- Liu, L.; Hu, X.; Song, J.; Zong, X.; Li, D.; Li, D. Over-expression of a Zea mays L. protein phosphatase 2C gene (ZmPP2C) in Arabidopsis thaliana decreases tolerance to salt and drought. J. Plant Physiol. 2009, 166, 531–542. [Google Scholar] [CrossRef]
- Zhang, H.; Mao, X.; Wang, C.; Jing, R. Overexpression of a common wheat gene Tasnrk2.8 enhances tolerance to drought, salt and low temperature in Arabidopsis. PLoS ONE 2010, 5, e16041. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.Q.; Chen, M.; Xu, Z.S.; Zhao, C.P.; Li, L.; Xu, H.J.; Tang, Y.M.; Zhao, X.; Ma, Y.Z. The soybean GmbZIP1 transcription factor enhances multiple abiotic stress tolerances in transgenic plants. Plant Mol. Biol. 2011, 75, 537–553. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Hu, W.; Liu, J.; Zhang, J.; Jia, C.; Miao, H.; Xu, B.; Jin, Z. A banana aquaporin gene, MaPIP1;1, is involved in tolerance to drought and salt stresses. BMC Plant Biol. 2014, 14, 59. [Google Scholar] [CrossRef]
- Mizoguchi, T.; Iriet, K.; Hirayama, T.; Hayashida, N.; Yamaguchi-Shinozaki, K.; Matsumotot, K.; Shinozaki, K.; Hahlbrock, K. A gene encoding a mitogen-activated protein kinase kinase kinase is induced simultaneously with genes for a mitogen-activated protein kinase and an S6 ribosomal protein kinase by touch, cold, and water stress in Arabidopsis thaliana. Plant Biol. 1996, 93, 765–769. [Google Scholar] [CrossRef]
- Yadav, K.; Jha, S.K.; Sharma, M.; Pandey, G.K. Mitogen activated protein kinase: A versatile signaling cascade in plants. J. Proteins Proteom. 2018, 9, 57–72. [Google Scholar]
- Yan, J.; Niu, F.; Liu, W.Z.; Zhang, H.; Wang, B.; Yang, B.; Jiang, Y.Q. Arabidopsis CIPK14 positively regulates glucose response. Biochem. Biophys. Res. Commun. 2014, 450, 1679–1683. [Google Scholar] [CrossRef]
- Ahmed, N.U.; Park, J.I.; Jung, H.J.; Kang, K.K.; Lim, Y.P.; Hur, Y.; Nou, I.S. Molecular characterization of thaumatin family genes related to stresses in Brassica rapa. Sci. Hortic. (Amsterdam) 2013, 152, 26–34. [Google Scholar] [CrossRef]
- Misra, R.C.; Sandeep; Kamthan, M.; Kumar, S.; Ghosh, S. A thaumatin-like protein of Ocimum basilicum confers tolerance to fungal pathogen and abiotic stress in transgenic Arabidopsis. Sci. Rep. 2016, 6, 25340. [Google Scholar] [CrossRef]
- Yoshida, S.; Ito, M.; Nishida, I.; Watanabe, A. Isolation and RNA gel blot analysis of genes that could serve as potential molecular markers for leaf senescence in Arabidopsis thaliana. Plant Cell Physiol. 2001, 42, 170–178. [Google Scholar] [CrossRef]
- Griffiths, C.A.; Gaff, D.F.; Neale, A.D. Drying without senescence in resurrection plants. Front. Plant Sci. 2014, 5, 36. [Google Scholar] [CrossRef]
- Rahman, M.A.; Ren, L.; Wu, W.; Yan, Y. Proteomic Analysis of PEG-Induced Drought Stress Responsive Protein in TERF1 Overexpressed Sugarcane (Saccharum officinarum) Leaves. Plant Mol. Biol. Report. 2015, 33, 716–730. [Google Scholar] [CrossRef]
- Bandehagh, A.; Uliaie, D.; Hosseini Salekdeh, G. Proteomic analysis of rapeseed (Brassica napus L.) seedling roots under salt stress. Sch. Res. Libr. Ann. Biol. Res. 2013, 212–221. [Google Scholar]
- Tran, L.-S.P. Isolation and Functional Analysis of Arabidopsis Stress-Inducible NAC Transcription Factors That Bind to a Drought-Responsive cis-Element in the early responsive to dehydration stress 1 Promoter. Plant Cell 2004, 16, 2481–2498. [Google Scholar] [CrossRef]
- Guo, Y.; Gan, S. AtNAP, a NAC family transcription factor, has an important role in leaf senescence. Plant J. 2006, 46, 601–612. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, K.; Tran, L.S.P.; Van Nguyen, D.; Fujita, M.; Maruyama, K.; Todaka, D.; Ito, Y.; Hayashi, N.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Functional analysis of a NAC-type transcription factor OsNAC6 involved in abiotic and biotic stress-responsive gene expression in rice. Plant J. 2007, 51, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Kiyosue, T.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Cloning of cDNAs for genes that are early-responsive to dehydration stress (ERDs) in Arabidopsis thaliana L.: Identification of three ERDs as HSP cognate genes. Plant Mol. Biol. 1994, 25, 791–798. [Google Scholar] [CrossRef]
- Xie, Y.; Lu, M.; Wang, G.; Peng, Y.; Cao, G.; Zhang, J.; Liu, X. Overexpression of a PLDα1 gene from Setaria italica enhances the sensitivity of Arabidopsis to abscisic acid and improves its drought tolerance. Plant Cell Rep. 2010, 29, 793–802. [Google Scholar]
- Harshavardhan, V.T.; Van Son, L.; Seiler, C.; Junker, A.; Weigelt-Fischer, K.; Klukas, C.; Altmann, T.; Sreenivasulu, N.; Bäumlein, H.; Kuhlmann, M. AtRD22 and AtUSPL1, members of the plant-specific BURP domain family involved in arabidopsis thaliana drought tolerance. PLoS ONE 2014, 9, e110065. [Google Scholar] [CrossRef]
- Hieno, A.; Naznin, H.A.; Hyakumachi, M.; Higuchi-Takeuchi, M.; Matsui, M.; Yamamoto, Y.Y. Possible involvement of MYB44 mediated stomatal regulation in systemic resistance induced by Penicillium simplicissimum GP17-2 in Arabidopsis. Microbes Environ. 2016, 31, 154–159. [Google Scholar] [CrossRef]
- Zhao, Y.; Xing, L.; Wang, X.; Hou, Y.-J.; Gao, J.; Wang, P.; Duan, C.-G.; Zhu, X.; Zhu, J.-K. The ABA receptor PYL8 promotes lateral root growth by enhancing MYB77-dependent transcription of Auxin- responsive genes. NIH Public Access 2015, 7, 1–25. [Google Scholar] [CrossRef]
- Zhu, J.-K.; Gao, J.; Zhao, Y.; Xing, L.; Xiang, C. The ABA receptor PYL9 together with PYL8 plays an important role in regulating lateral root growth. Sci. Rep. 2016, 6, 1–13. [Google Scholar]
- Dat, J.; Vandenabeele, S.; Vranová, E.; Van Montagu, M.; Inzé, D.; Van Breusegem, F. Dual action of the active oxygen species during plant stress responses. Cell. Mol. Life Sci. 2000, 57, 779–795. [Google Scholar] [CrossRef]
- Van Moerkercke, A.; Haring, M.A.; Schuurink, R.C. A model for combinatorial regulation of the petunia R2R3-MYB transcription factor ODORANT1. Plant Signal. Behav. 2012, 7, 518–520. [Google Scholar] [CrossRef]
- Shahnejat-Bushehri, S.; Tarkowska, D.; Sakuraba, Y.; Balazadeh, S. Arabidopsis NAC transcription factor JUB1 regulates GA/BR metabolism and signalling. Nat. Plants 2016, 2, 16013. [Google Scholar] [CrossRef]
- Chen, M.; Wang, Q.Y.; Cheng, X.G.; Xu, Z.S.; Li, L.C.; Ye, X.G.; Xia, L.Q.; Ma, Y.Z. GmDREB2, a soybean DRE-binding transcription factor, conferred drought and high-salt tolerance in transgenic plants. Biochem. Biophys. Res. Commun. 2007, 353, 299–305. [Google Scholar] [CrossRef]
- Zhang, G.; Chen, M.; Li, L.; Xu, Z.; Chen, X.; Guo, J.; Ma, Y. Overexpression of the soybean GmERF3 gene, an AP2/ERF type transcription factor for increased tolerances to salt, drought, and diseases in transgenic tobacco. J. Exp. Bot. 2009, 60, 3781–3796. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, F.; Li, D.; Zhang, H.; Huang, R. Expression of ethylene response factor JERF1 in rice improves tolerance to drought. Planta 2010, 232, 765–774. [Google Scholar] [CrossRef]
- Tak, H.; Negi, S.; Ganapathi, T.R. Banana NAC transcription factor MusaNAC042 is positively associated with drought and salinity tolerance. Protoplasma 2017, 254, 803–816. [Google Scholar] [CrossRef]
- Chaves, M.M.; Maroco, J.P.; Pereira, J.S. Understanding plant responses to drought—From genes to the whole plant. Funct. Plant Biol. 2003, 30, 239–264. [Google Scholar] [CrossRef]
- Li, H.; Wang, Y.; Wang, C.; Chen, C.; Li, C.; Song, W.; Li, L.; Yuan, J.; Wu, M.; Han, Z. Genome-Wide Identification of AP2/ERF Transcription Factors in Cauliflower and Expression Profiling of the ERF Family under Salt and Drought Stresses. Front. Plant Sci. 2017, 8, 946. [Google Scholar] [CrossRef]
- Dong, C.J.; Liu, J.Y. The Arabidopsis EAR-motif-containing protein RAP2.1 functions as an active transcriptional repressor to keep stress responses under tight control. BMC Plant Biol. 2010, 10, 47. [Google Scholar] [CrossRef]
- Chen, L.; Chen, Y.; Jiang, J.; Chen, S.; Chen, F.; Guan, Z.; Fang, W. The constitutive expression of Chrysanthemum dichrum ICE1 in Chrysanthemum grandiflorum improves the level of low temperature, salinity and drought tolerance. Plant Cell Rep. 2012, 31, 1747–1758. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-H.; Böhmer, M.; Hu, H.; Nishimura, N.; Schroeder, J.I. Guard cell signal transduction network: Advances in understanding abscisic acid, CO2, and Ca2+ signaling. Annu. Rev. Plant Biol. 2010, 61, 561–591. [Google Scholar] [CrossRef]
- MacAlister, C.A.; Ohashi-Ito, K.; Bergmann, D.C. Transcription factor control of asymmetric cell divisions that establish the stomatal lineage. Nature 2007, 445, 537–540. [Google Scholar] [CrossRef]
- Ohashi-Ito, K.; Bergmann, D.C. Arabidopsis FAMA Controls the final proliferation/differentiation switch during stomatal development. Plant Cell 2006, 18, 2493–2505. [Google Scholar] [CrossRef]
- Wu, F.; Wu, H.; Zhang, G.; Bachir, D.M.L. Differences in growth and yield in response to cadmium toxicity in cotton genotypes. J. Plant Nutr. Soil Sci. 2004, 167, 85–90. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.G.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Parallel mapping of transcriptomes to detect InDels, gene fusions, and more. 2012. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Summary | Min | Max | Total | Average |
|---|---|---|---|---|
| Total Raw reads | 49,311,644 | 64,712,390 | 326,938,772 | 54,489,795.3 |
| Total clean reads | 48,199,244 | 63,896,272 | 322,114,206 | 53,685,701 |
| Total clean reads/total Raw reads | 98% | 99% | 99% | 99% |
| Total clean bases | 7,111,623,297 | 9,450,136,133 | 47,633,258,301 | 7,938,876,384 |
| GC content | 41.88% | 43.60% | -- | 42.99% |
| Q20 | 96.26% | 96.76% | -- | 96.49% |
| Q30 | 91.18% | 92.17% | -- | 91.59% |
| Total mapped reads | 38,646,418 | 50,124,573 | 262,172,868 | 43,695,478 |
| Total mapped reads | 78.45% | 84.66% | 82% | |
| Multiple mapped | 4751,886 | 6,451,220 | 34,568,807 | 5,761,467.83 |
| Multiple mapped% | 9.67% | 12.10% | -- | 10.75% |
| Uniquely mapped | 33,372,748 | 43,788,024 | 227,604,061 | 37,934,010.2 |
| Uniquely mapped% | 68.49% | 74.98% | -- | 71% |
| Total mapped reads/Total clean reads | 80% | 78% | 81% | 81% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasan, M.M.-U.; Ma, F.; Islam, F.; Sajid, M.; Prodhan, Z.H.; Li, F.; Shen, H.; Chen, Y.; Wang, X. Comparative Transcriptomic Analysis of Biological Process and Key Pathway in Three Cotton (Gossypium spp.) Species Under Drought Stress. Int. J. Mol. Sci. 2019, 20, 2076. https://doi.org/10.3390/ijms20092076
Hasan MM-U, Ma F, Islam F, Sajid M, Prodhan ZH, Li F, Shen H, Chen Y, Wang X. Comparative Transcriptomic Analysis of Biological Process and Key Pathway in Three Cotton (Gossypium spp.) Species Under Drought Stress. International Journal of Molecular Sciences. 2019; 20(9):2076. https://doi.org/10.3390/ijms20092076
Chicago/Turabian StyleHasan, Md Mosfeq-Ul, Fanglu Ma, Faisal Islam, Muhammad Sajid, Zakaria H. Prodhan, Feng Li, Hao Shen, Yadong Chen, and Xuede Wang. 2019. "Comparative Transcriptomic Analysis of Biological Process and Key Pathway in Three Cotton (Gossypium spp.) Species Under Drought Stress" International Journal of Molecular Sciences 20, no. 9: 2076. https://doi.org/10.3390/ijms20092076