Dual-Specificity Phosphatase Regulation in Neurons and Glial Cells

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
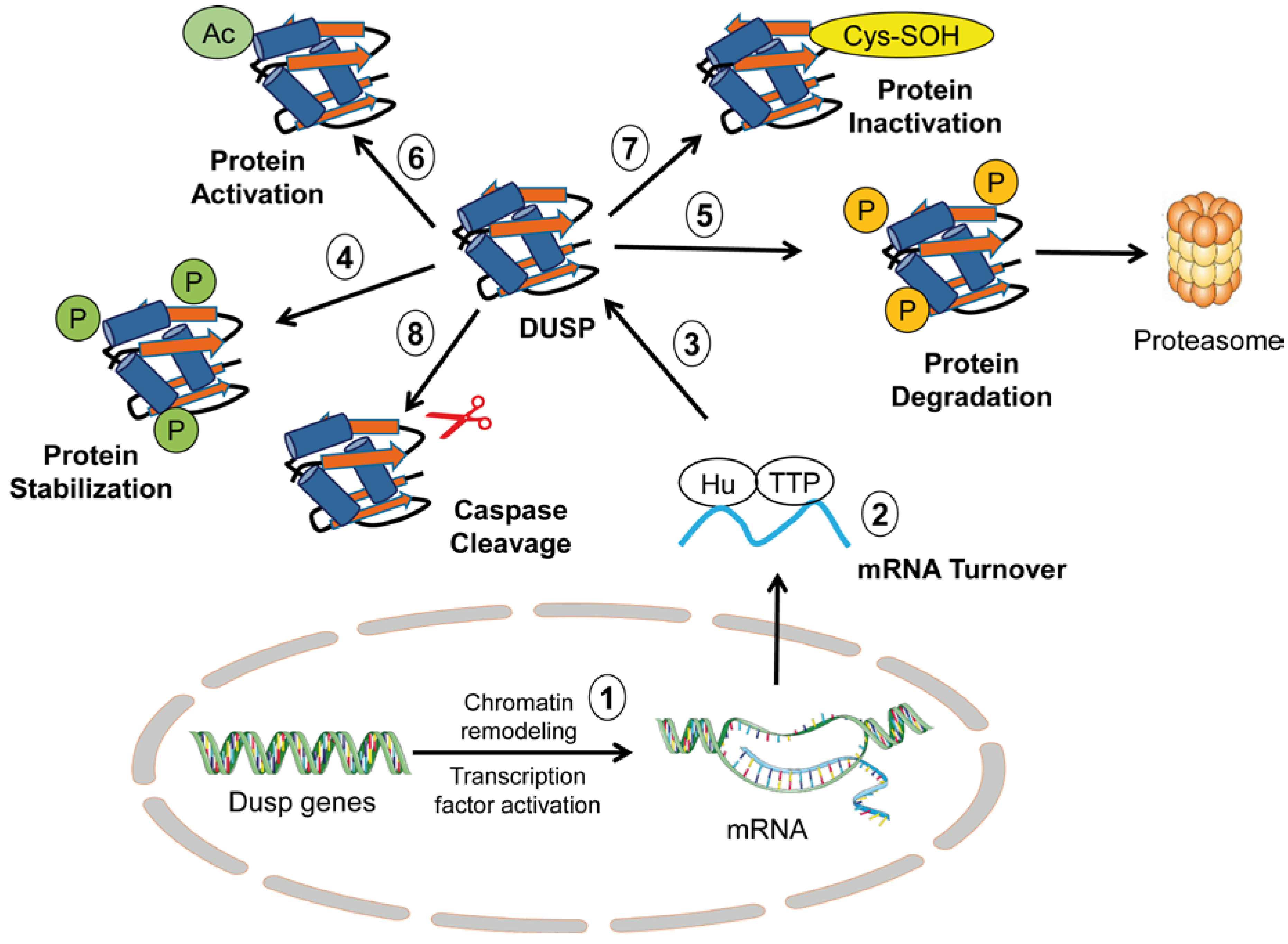
2. Overview of Regulatory Mechanisms of DUSP Activity
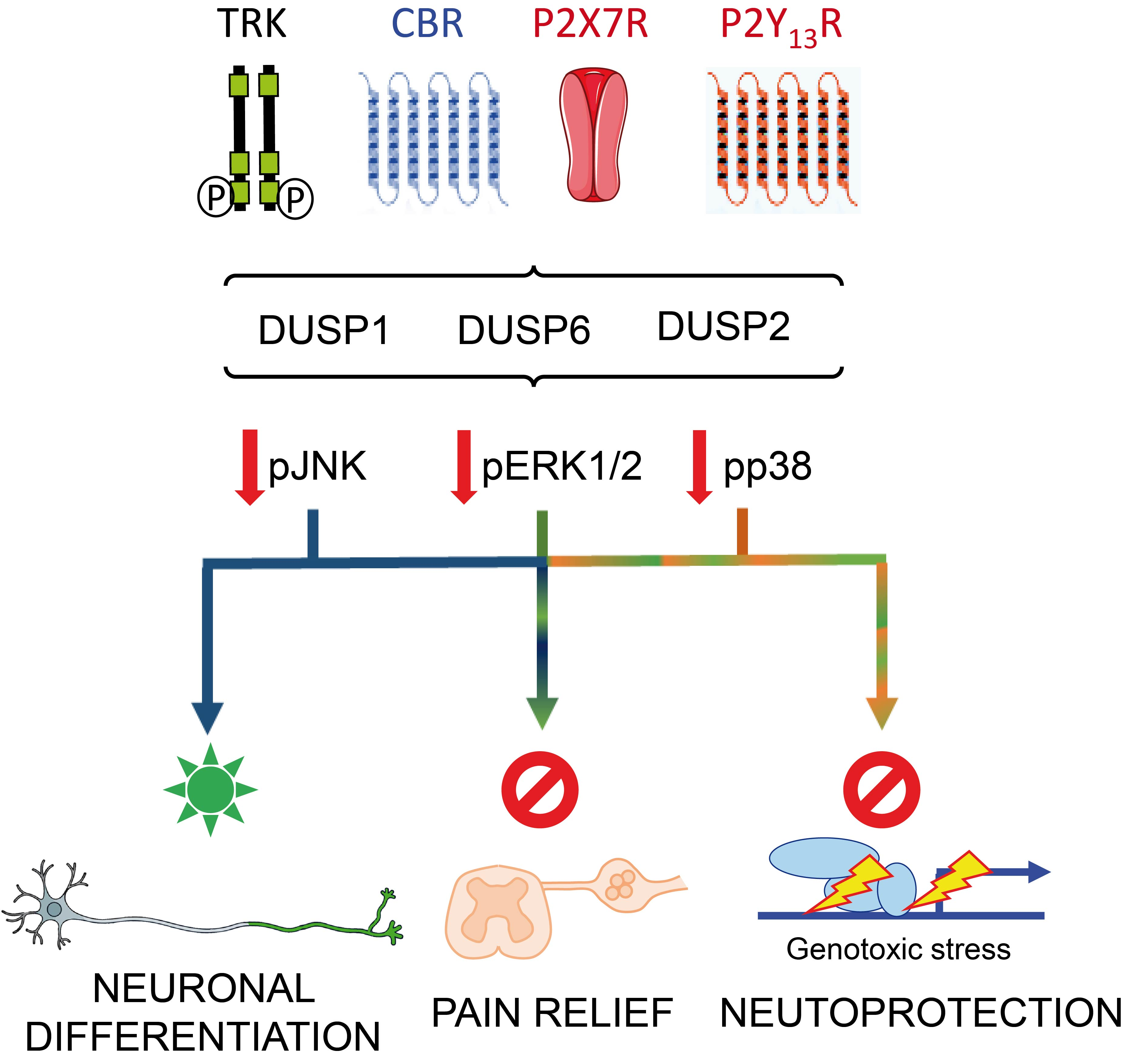
3. DUSP in Neuronal Differentiation and Nervous System Development
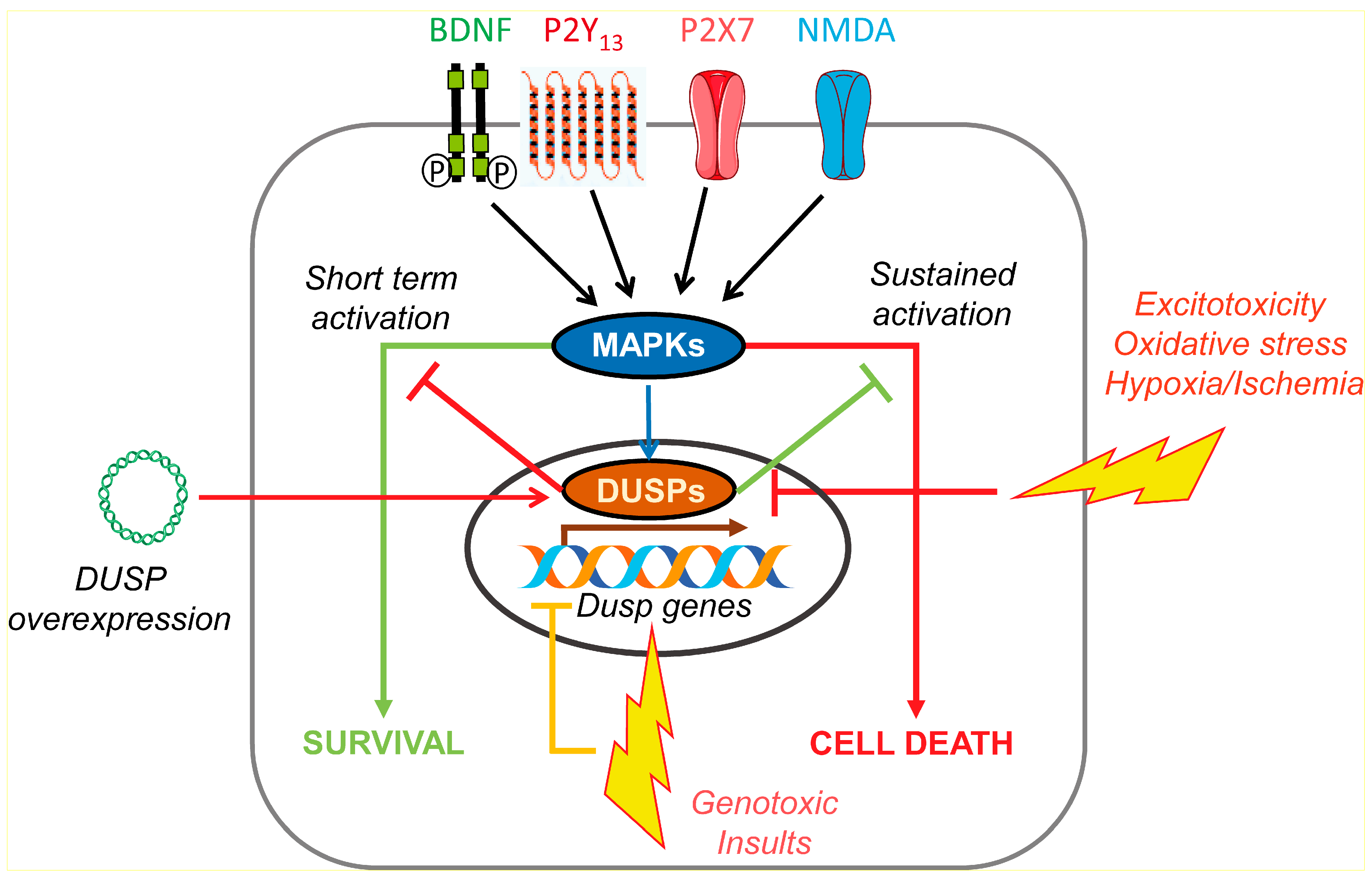
4. DUSP in Neuroprotection; Role of Neurotrophins and Nucleotides
4.1. DUSP Regulation in Excitotoxicity and Oxidative Stress
4.2. DUSP Regulation in Genotoxic Stress
4.3. DUSP in Hypoxia and Ischemia
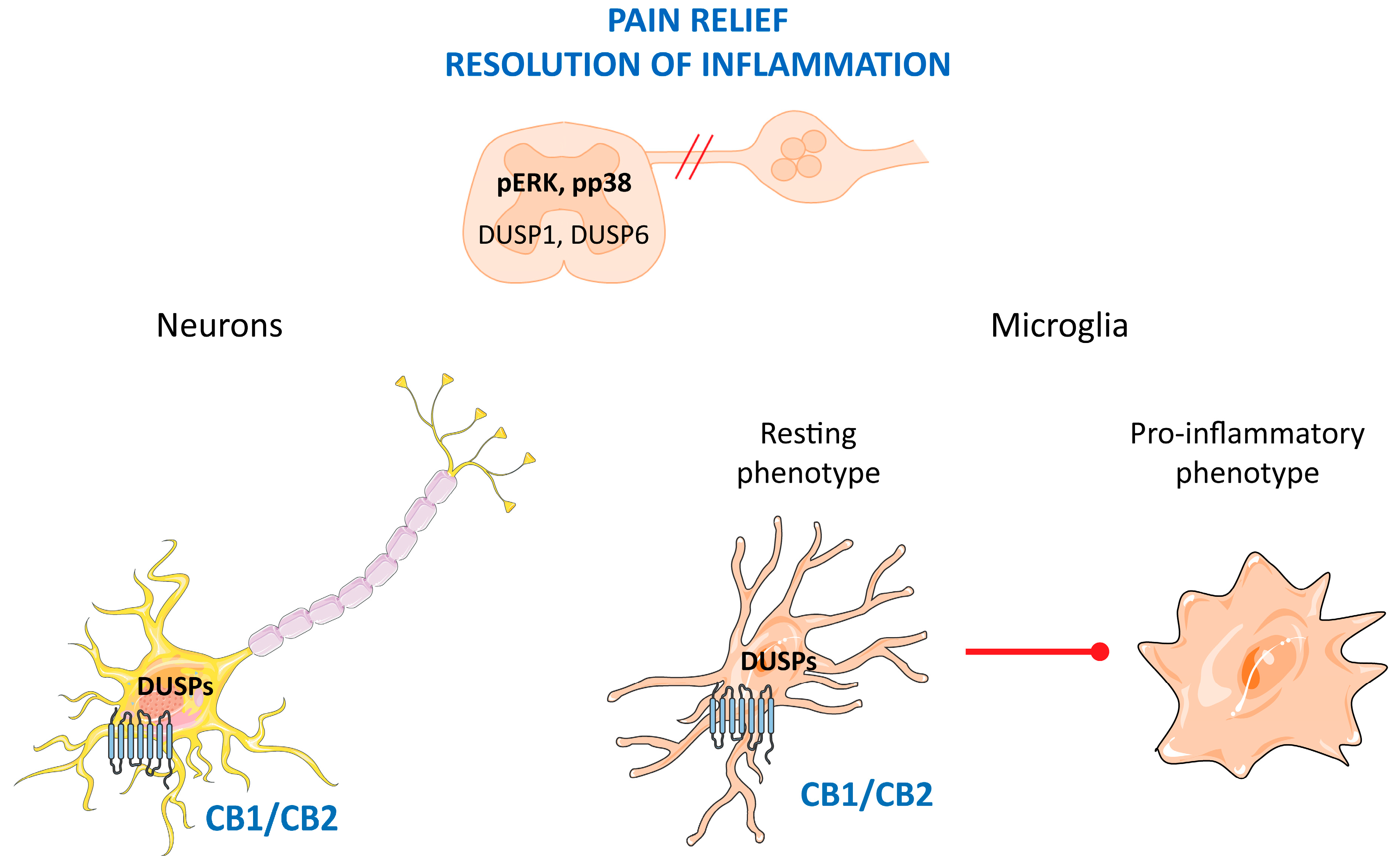
5. DUSP in Pain and Inflammation; Role of Cannabinoids
6. DUSP in Brain Diseases
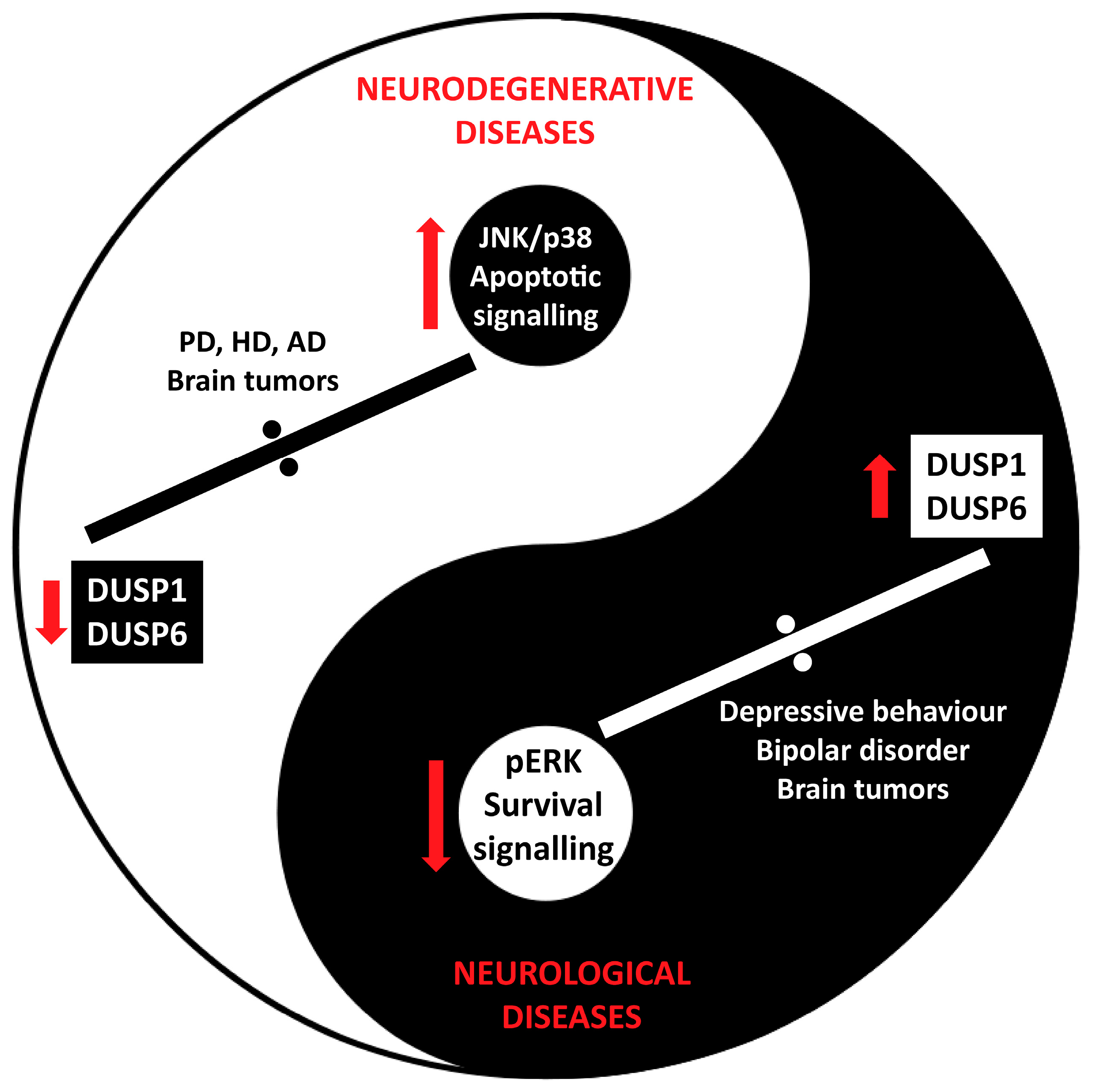
6.1. DUSP in Neurodegenerative Diseases
6.2. DUSP in Neurological Disorders
6.3. DUSPs in Brain Tumors
7. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer′s disease |
| BD | Bipolar disorder |
| BDNF | Brain-derived neurotrophic factor |
| BzATP | 2′(3′)-O-(4-Benzoylbenzoyl)adenosine 5′-triphosphate |
| DUSP | Dual-specificity protein phosphatase |
| EGF | Epidermal growth factor |
| ERK | Extracellular signal regulated protein kinase |
| ETS | E twenty-six |
| GSK3 | Glycogen synthase kinase 3 |
| H2O2 | Hydrogen peroxide |
| HD | Huntington′s disease |
| JNKs | c-Jun N-terminal kinase |
| MAPK | Mitogen-activated protein kinase |
| MEK | Mitogen extracellular activated kinase |
| NGF | Nerve growth factor |
| PD | Parkinson′s disease |
| PI3K | Phosphatidylinositol 3-kinase |
| PKA | Protein kinase A |
| PKC | Protein kinase C |
| PTP | Protein tyrosine phosphatase |
| TTP | Tristetraprolin |
References
- Dickinson, R.J.; Keyse, S.M. Diverse physiological functions for dual-specificity MAP kinase phosphatases. J. Cell Sci. 2006, 119, 4607–4615. [Google Scholar] [CrossRef]
- Junttila, M.R.; Li, S.P.; Westermarck, J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J. 2008, 22, 954–965. [Google Scholar] [CrossRef]
- Patterson, K.I.; Brummer, T.; O’Brien, P.M.; Daly, R.J. Dual-specificity phosphatases: Critical regulators with diverse cellular targets. Biochem. J. 2009, 418, 475–489. [Google Scholar] [CrossRef]
- Bermudez, O.; Pages, G.; Gimond, C. The dual-specificity MAP kinase phosphatases: Critical roles in development and cancer. Am. J. Physiol. Cell Physiol. 2010, 299, C189–C202. [Google Scholar] [CrossRef]
- Nunes-Xavier, C.; Roma-Mateo, C.; Rios, P.; Tarrega, C.; Cejudo-Marin, R.; Tabernero, L.; Pulido, R. Dual-specificity MAP kinase phosphatases as targets of cancer treatment. Anticancer Agents Med. Chem. 2011, 11, 109–132. [Google Scholar] [CrossRef]
- Owens, D.M.; Keyse, S.M. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene 2007, 26, 3203–3213. [Google Scholar] [CrossRef]
- Lang, R.; Hammer, M.; Mages, J. DUSP meet immunology: Dual specificity MAPK phosphatases in control of the inflammatory response. J. Immunol. 2006, 177, 7497–7504. [Google Scholar] [CrossRef]
- Jeffrey, K.L.; Camps, M.; Rommel, C.; Mackay, C.R. Targeting dual-specificity phosphatases: Manipulating MAP kinase signalling and immune responses. Nat. Rev. Drug Discov. 2007, 6, 391–403. [Google Scholar] [CrossRef]
- Wu, Z.; Jiao, P.; Huang, X.; Feng, B.; Feng, Y.; Yang, S.; Hwang, P.; Du, J.; Nie, Y.; Xiao, G.; et al. MAPK phosphatase-3 promotes hepatic gluconeogenesis through dephosphorylation of forkhead box O1 in mice. J. Clin. Investig. 2010, 120, 3901–3911. [Google Scholar] [CrossRef]
- Coddou, C.; Yan, Z.; Obsil, T.; Huidobro-Toro, J.P.; Stojilkovic, S.S. Activation and regulation of purinergic P2X receptor channels. Pharmacol. Rev. 2011, 63, 641–683. [Google Scholar] [CrossRef]
- von Kugelgen, I.; Hoffmann, K. Pharmacology and structure of P2Y receptors. Neuropharmacology 2016, 104, 50–61. [Google Scholar] [CrossRef]
- Perez-Sen, R.; Queipo, M.J.; Morente, V.; Ortega, F.; Delicado, E.G.; Miras-Portugal, M.T. Neuroprotection Mediated by P2Y13 Nucleotide Receptors in Neurons. Comput. Struct. Biotechnol. J. 2015, 13, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Perez-Sen, R.; Gomez-Villafuertes, R.; Ortega, F.; Gualix, J.; Delicado, E.G.; Miras-Portugal, M.T. An Update on P2Y13 Receptor Signalling and Function. Adv. Exp. Med. Biol. 2017, 1051, 139–168. [Google Scholar] [CrossRef]
- Miras-Portugal, M.T.; Gomez-Villafuertes, R.; Gualix, J.; Diaz-Hernandez, J.I.; Artalejo, A.R.; Ortega, F.; Delicado, E.G.; Perez-Sen, R. Nucleotides in neuroregeneration and neuroprotection. Neuropharmacology 2016, 104, 243–254. [Google Scholar] [CrossRef]
- Miras-Portugal, M.T.; Queipo, M.J.; Gil-Redondo, J.C.; Ortega, F.; Gomez-Villafuertes, R.; Gualix, J.; Delicado, E.G.; Perez-Sen, R. P2 receptor interaction and signalling cascades in neuroprotection. Brain Res. Bull. 2018. [Google Scholar] [CrossRef]
- Kondoh, K.; Nishida, E. Regulation of MAP kinases by MAP kinase phosphatases. Biochim. Biophys. Acta 2007, 1773, 1227–1237. [Google Scholar] [CrossRef]
- Caunt, C.J.; Keyse, S.M. Dual-specificity MAP kinase phosphatases (MKPs): Shaping the outcome of MAP kinase signalling. FEBS J. 2013, 280, 489–504. [Google Scholar] [CrossRef]
- Huang, C.Y.; Tan, T.H. DUSPs, to MAP kinases and beyond. Cell Biosci. 2012, 2, 24. [Google Scholar] [CrossRef]
- Ekerot, M.; Stavridis, M.P.; Delavaine, L.; Mitchell, M.P.; Staples, C.; Owens, D.M.; Keenan, I.D.; Dickinson, R.J.; Storey, K.G.; Keyse, S.M. Negative-feedback regulation of FGF signalling by DUSP6/MKP-3 is driven by ERK1/2 and mediated by Ets factor binding to a conserved site within the DUSP6/MKP-3 gene promoter. Biochem. J. 2008, 412, 287–298. [Google Scholar] [CrossRef]
- Furukawa, T.; Tanji, E.; Xu, S.; Horii, A. Feedback regulation of DUSP6 transcription responding to MAPK1 via ETS2 in human cells. Biochem. Biophys. Res. Commun. 2008, 377, 317–320. [Google Scholar] [CrossRef]
- Staples, C.J.; Owens, D.M.; Maier, J.V.; Cato, A.C.; Keyse, S.M. Cross-talk between the p38alpha and JNK MAPK pathways mediated by MAP kinase phosphatase-1 determines cellular sensitivity to UV radiation. J. Biol. Chem. 2010, 285, 25928–25940. [Google Scholar] [CrossRef]
- Franklin, C.C.; Srikanth, S.; Kraft, A.S. Conditional expression of mitogen-activated protein kinase phosphatase-1, MKP-1, is cytoprotective against UV-induced apoptosis. Proc. Natl. Acad. Sci. USA 1998, 95, 3014–3019. [Google Scholar] [CrossRef] [PubMed]
- Kassel, O.; Sancono, A.; Kratzschmar, J.; Kreft, B.; Stassen, M.; Cato, A.C. Glucocorticoids inhibit MAP kinase via increased expression and decreased degradation of MKP-1. EMBO J. 2001, 20, 7108–7116. [Google Scholar] [CrossRef] [PubMed]
- Shipp, L.E.; Lee, J.V.; Yu, C.Y.; Pufall, M.; Zhang, P.; Scott, D.K.; Wang, J.C. Transcriptional regulation of human dual specificity protein phosphatase 1 (DUSP1) gene by glucocorticoids. PLoS ONE 2010, 5, e13754. [Google Scholar] [CrossRef]
- Li, J.; Gorospe, M.; Hutter, D.; Barnes, J.; Keyse, S.M.; Liu, Y. Transcriptional induction of MKP-1 in response to stress is associated with histone H3 phosphorylation-acetylation. Mol. Cell. Biol. 2001, 21, 8213–8224. [Google Scholar] [CrossRef]
- Kuwano, Y.; Kim, H.H.; Abdelmohsen, K.; Pullmann, R., Jr.; Martindale, J.L.; Yang, X.; Gorospe, M. MKP-1 mRNA stabilization and translational control by RNA-binding proteins HuR and NF90. Mol. Cell. Biol. 2008, 28, 4562–4575. [Google Scholar] [CrossRef]
- Kuwano, Y.; Gorospe, M. Protecting the stress response, guarding the MKP-1 mRNA. Cell Cycle 2008, 7, 2640–2642. [Google Scholar] [CrossRef]
- Camps, M.; Nichols, A.; Gillieron, C.; Antonsson, B.; Muda, M.; Chabert, C.; Boschert, U.; Arkinstall, S. Catalytic activation of the phosphatase MKP-3 by ERK2 mitogen-activated protein kinase. Science 1998, 280, 1262–1265. [Google Scholar] [CrossRef]
- Chen, P.; Hutter, D.; Yang, X.; Gorospe, M.; Davis, R.J.; Liu, Y. Discordance between the binding affinity of mitogen-activated protein kinase subfamily members for MAP kinase phosphatase-2 and their ability to activate the phosphatase catalytically. J. Biol. Chem. 2001, 276, 29440–29449. [Google Scholar] [CrossRef]
- Zhang, Q.; Muller, M.; Chen, C.H.; Zeng, L.; Farooq, A.; Zhou, M.M. New insights into the catalytic activation of the MAPK phosphatase PAC-1 induced by its substrate MAPK ERK2 binding. J. Mol. Biol. 2005, 354, 777–788. [Google Scholar] [CrossRef]
- Liu, R.M.; Choi, J.; Wu, J.H.; Gaston Pravia, K.A.; Lewis, K.M.; Brand, J.D.; Mochel, N.S.; Krzywanski, D.M.; Lambeth, J.D.; Hagood, J.S.; et al. Oxidative modification of nuclear mitogen-activated protein kinase phosphatase 1 is involved in transforming growth factor beta1-induced expression of plasminogen activator inhibitor 1 in fibroblasts. J. Biol. Chem. 2010, 285, 16239–16247. [Google Scholar] [CrossRef]
- Tephly, L.A.; Carter, A.B. Differential expression and oxidation of MKP-1 modulates TNF-alpha gene expression. Am. J. Respir. Cell Mol. Biol. 2007, 37, 366–374. [Google Scholar] [CrossRef]
- Tonks, N.K. Protein tyrosine phosphatases: From genes, to function, to disease. Nat. Rev. Mol. Cell. Biol. 2006, 7, 833–846. [Google Scholar] [CrossRef]
- Seth, D.; Rudolph, J. Redox regulation of MAP kinase phosphatase 3. Biochemistry 2006, 45, 8476–8487. [Google Scholar] [CrossRef]
- Brondello, J.M.; Pouyssegur, J.; McKenzie, F.R. Reduced MAP kinase phosphatase-1 degradation after p42/p44MAPK-dependent phosphorylation. Science 1999, 286, 2514–2517. [Google Scholar] [CrossRef]
- Lin, Y.W.; Yang, J.L. Cooperation of ERK and SCFSkp2 for MKP-1 destruction provides a positive feedback regulation of proliferating signaling. J. Biol. Chem. 2006, 281, 915–926. [Google Scholar] [CrossRef]
- Cagnol, S.; Rivard, N. Oncogenic KRAS and BRAF activation of the MEK/ERK signaling pathway promotes expression of dual-specificity phosphatase 4 (DUSP4/MKP2) resulting in nuclear ERK1/2 inhibition. Oncogene 2013, 32, 564–576. [Google Scholar] [CrossRef]
- Marchetti, S.; Gimond, C.; Chambard, J.C.; Touboul, T.; Roux, D.; Pouyssegur, J.; Pages, G. Extracellular signal-regulated kinases phosphorylate mitogen-activated protein kinase phosphatase 3/DUSP6 at serines 159 and 197, two sites critical for its proteasomal degradation. Mol. Cell. Biol. 2005, 25, 854–864. [Google Scholar] [CrossRef]
- Jurek, A.; Amagasaki, K.; Gembarska, A.; Heldin, C.H.; Lennartsson, J. Negative and positive regulation of MAPK phosphatase 3 controls platelet-derived growth factor-induced Erk activation. J. Biol. Chem. 2009, 284, 4626–4634. [Google Scholar] [CrossRef]
- Bermudez, O.; Marchetti, S.; Pages, G.; Gimond, C. Post-translational regulation of the ERK phosphatase DUSP6/MKP3 by the mTOR pathway. Oncogene 2008, 27, 3685–3691. [Google Scholar] [CrossRef]
- Castelli, M.; Camps, M.; Gillieron, C.; Leroy, D.; Arkinstall, S.; Rommel, C.; Nichols, A. MAP kinase phosphatase 3 (MKP3) interacts with and is phosphorylated by protein kinase CK2alpha. J. Biol. Chem. 2004, 279, 44731–44739. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, R.J.; Delavaine, L.; Cejudo-Marin, R.; Stewart, G.; Staples, C.J.; Didmon, M.P.; Trinidad, A.G.; Alonso, A.; Pulido, R.; Keyse, S.M. Phosphorylation of the kinase interaction motif in mitogen-activated protein (MAP) kinase phosphatase-4 mediates cross-talk between protein kinase A and MAP kinase signaling pathways. J. Biol. Chem. 2011, 286, 38018–38026. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Bao, C.; Padalko, E.; Lowenstein, C.J. Acetylation of mitogen-activated protein kinase phosphatase-1 inhibits Toll-like receptor signaling. J. Exp. Med. 2008, 205, 1491–1503. [Google Scholar] [CrossRef] [PubMed]
- Cejudo-Marin, R.; Tarrega, C.; Nunes-Xavier, C.E.; Pulido, R. Caspase-3 cleavage of DUSP6/MKP3 at the interdomain region generates active MKP3 fragments that regulate ERK1/2 subcellular localization and function. J. Mol. Biol. 2012, 420, 128–138. [Google Scholar] [CrossRef]
- Muda, M.; Boschert, U.; Dickinson, R.; Martinou, J.C.; Martinou, I.; Camps, M.; Schlegel, W.; Arkinstall, S. MKP-3, a novel cytosolic protein-tyrosine phosphatase that exemplifies a new class of mitogen-activated protein kinase phosphatase. J. Biol. Chem. 1996, 271, 4319–4326. [Google Scholar] [CrossRef] [PubMed]
- Misra-Press, A.; Rim, C.S.; Yao, H.; Roberson, M.S.; Stork, P.J. A novel mitogen-activated protein kinase phosphatase. Structure, expression, and regulation. J. Biol. Chem. 1995, 270, 14587–14596. [Google Scholar] [CrossRef] [PubMed]
- Waetzig, V.; Herdegen, T. The concerted signaling of ERK1/2 and JNKs is essential for PC12 cell neuritogenesis and converges at the level of target proteins. Mol. Cell. Neurosci. 2003, 24, 238–249. [Google Scholar] [CrossRef]
- Peinado-Ramon, P.; Wallen, A.; Hallbook, F. MAP kinase phosphatase-1 mRNA is expressed in embryonic sympathetic neurons and is upregulated after NGF stimulation. Brain Res. Mol. Brain Res. 1998, 56, 256–267. [Google Scholar] [CrossRef]
- Reffas, S.; Schlegel, W. Compartment-specific regulation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) mitogen-activated protein kinases (MAPKs) by ERK-dependent and non-ERK-dependent inductions of MAPK phosphatase (MKP)-3 and MKP-1 in differentiating P19 cells. Biochem. J. 2000, 352, 701–708. [Google Scholar]
- Jeanneteau, F.; Deinhardt, K.; Miyoshi, G.; Bennett, A.M.; Chao, M.V. The MAP kinase phosphatase MKP-1 regulates BDNF-induced axon branching. Nat. Neurosci. 2010, 13, 1373–1379. [Google Scholar] [CrossRef]
- Collins, L.M.; O’Keeffe, G.W.; Long-Smith, C.M.; Wyatt, S.L.; Sullivan, A.M.; Toulouse, A.; Nolan, Y.M. Mitogen-activated protein kinase phosphatase (MKP)-1 as a neuroprotective agent: Promotion of the morphological development of midbrain dopaminergic neurons. Neuromol. Med. 2013, 15, 435–446. [Google Scholar] [CrossRef]
- Dickinson, R.J.; Eblaghie, M.C.; Keyse, S.M.; Morriss-Kay, G.M. Expression of the ERK-specific MAP kinase phosphatase PYST1/MKP3 in mouse embryos during morphogenesis and early organogenesis. Mech. Dev. 2002, 113, 193–196. [Google Scholar] [CrossRef]
- Eblaghie, M.C.; Lunn, J.S.; Dickinson, R.J.; Munsterberg, A.E.; Sanz-Ezquerro, J.J.; Farrell, E.R.; Mathers, J.; Keyse, S.M.; Storey, K.; Tickle, C. Negative feedback regulation of FGF signaling levels by Pyst1/MKP3 in chick embryos. Curr. Biol. 2003, 13, 1009–1018. [Google Scholar] [CrossRef]
- Echevarria, D.; Martinez, S.; Marques, S.; Lucas-Teixeira, V.; Belo, J.A. Mkp3 is a negative feedback modulator of Fgf8 signaling in the mammalian isthmic organizer. Dev. Biol. 2005, 277, 114–128. [Google Scholar] [CrossRef]
- Smith, T.G.; Sweetman, D.; Patterson, M.; Keyse, S.M.; Munsterberg, A. Feedback interactions between MKP3 and ERK MAP kinase control scleraxis expression and the specification of rib progenitors in the developing chick somite. Development 2005, 132, 1305–1314. [Google Scholar] [CrossRef]
- Kawakami, Y.; Rodriguez-Leon, J.; Koth, C.M.; Buscher, D.; Itoh, T.; Raya, A.; Ng, J.K.; Esteban, C.R.; Takahashi, S.; Henrique, D.; et al. MKP3 mediates the cellular response to FGF8 signalling in the vertebrate limb. Nat. Cell. Biol. 2003, 5, 513–519. [Google Scholar] [CrossRef]
- Smith, T.G.; Karlsson, M.; Lunn, J.S.; Eblaghie, M.C.; Keenan, I.D.; Farrell, E.R.; Tickle, C.; Storey, K.G.; Keyse, S.M. Negative feedback predominates over cross-regulation to control ERK MAPK activity in response to FGF signalling in embryos. FEBS Lett. 2006, 580, 4242–4245. [Google Scholar] [CrossRef]
- Tsang, M.; Maegawa, S.; Kiang, A.; Habas, R.; Weinberg, E.; Dawid, I.B. A role for MKP3 in axial patterning of the zebrafish embryo. Development 2004, 131, 2769–2779. [Google Scholar] [CrossRef]
- Molina, G.; Vogt, A.; Bakan, A.; Dai, W.; Queiroz de Oliveira, P.; Znosko, W.; Smithgall, T.E.; Bahar, I.; Lazo, J.S.; Day, B.W.; et al. Zebrafish chemical screening reveals an inhibitor of Dusp6 that expands cardiac cell lineages. Nat. Chem. Biol. 2009, 5, 680–687. [Google Scholar] [CrossRef]
- Jeanneteau, F.; Deinhardt, K. Fine-tuning MAPK signaling in the brain: The role of MKP-1. Commun. Integr. Biol. 2011, 4, 281–283. [Google Scholar] [CrossRef]
- Stanciu, M.; Wang, Y.; Kentor, R.; Burke, N.; Watkins, S.; Kress, G.; Reynolds, I.; Klann, E.; Angiolieri, M.R.; Johnson, J.W.; et al. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J. Biol. Chem. 2000, 275, 12200–12206. [Google Scholar] [CrossRef]
- Choi, B.H.; Hur, E.M.; Lee, J.H.; Jun, D.J.; Kim, K.T. Protein kinase Cdelta-mediated proteasomal degradation of MAP kinase phosphatase-1 contributes to glutamate-induced neuronal cell death. J. Cell. Sci. 2006, 119, 1329–1340. [Google Scholar] [CrossRef]
- Luo, Y.; DeFranco, D.B. Opposing roles for ERK1/2 in neuronal oxidative toxicity: Distinct mechanisms of ERK1/2 action at early versus late phases of oxidative stress. J. Biol. Chem. 2006, 281, 16436–16442. [Google Scholar] [CrossRef]
- Huang, X.; Liao, W.; Huang, Y.; Jiang, M.; Chen, J.; Wang, M.; Lin, H.; Guan, S.; Liu, J. Neuroprotective effect of dual specificity phosphatase 6 against glutamate-induced cytotoxicity in mouse hippocampal neurons. Biomed. Pharmacother. 2017, 91, 385–392. [Google Scholar] [CrossRef]
- Levinthal, D.J.; Defranco, D.B. Reversible oxidation of ERK-directed protein phosphatases drives oxidative toxicity in neurons. J. Biol. Chem. 2005, 280, 5875–5883. [Google Scholar] [CrossRef]
- Xu, Q.; Konta, T.; Nakayama, K.; Furusu, A.; Moreno-Manzano, V.; Lucio-Cazana, J.; Ishikawa, Y.; Fine, L.G.; Yao, J.; Kitamura, M. Cellular defense against H2O2-induced apoptosis via MAP kinase-MKP-1 pathway. Free Radic. Biol. Med. 2004, 36, 985–993. [Google Scholar] [CrossRef]
- Mendell, A.L.; MacLusky, N.J. The testosterone metabolite 3alpha-androstanediol inhibits oxidative stress-induced ERK phosphorylation and neurotoxicity in SH-SY5Y cells through an MKP3/DUSP6-dependent mechanism. Neurosci. Lett. 2019, 696, 60–66. [Google Scholar] [CrossRef]
- Kim, G.S.; Choi, Y.K.; Song, S.S.; Kim, W.K.; Han, B.H. MKP-1 contributes to oxidative stress-induced apoptosis via inactivation of ERK1/2 in SH-SY5Y cells. Biochem. Biophys. Res. Commun. 2005, 338, 1732–1738. [Google Scholar] [CrossRef]
- Domercq, M.; Alberdi, E.; Sanchez-Gomez, M.V.; Ariz, U.; Perez-Samartin, A.; Matute, C. Dual-specific phosphatase-6 (Dusp6) and ERK mediate AMPA receptor-induced oligodendrocyte death. J. Biol. Chem. 2011, 286, 11825–11836. [Google Scholar] [CrossRef]
- Hetman, M.; Vashishta, A.; Rempala, G. Neurotoxic mechanisms of DNA damage: Focus on transcriptional inhibition. J. Neurochem. 2010, 114, 1537–1549. [Google Scholar] [CrossRef]
- Gozdz, A.; Habas, A.; Jaworski, J.; Zielinska, M.; Albrecht, J.; Chlystun, M.; Jalili, A.; Hetman, M. Role of N-methyl-D-aspartate receptors in the neuroprotective activation of extracellular signal-regulated kinase 1/2 by cisplatin. J. Biol. Chem. 2003, 278, 43663–43671. [Google Scholar] [CrossRef]
- Gozdz, A.; Vashishta, A.; Kalita, K.; Szatmari, E.; Zheng, J.J.; Tamiya, S.; Delamere, N.A.; Hetman, M. Cisplatin-mediated activation of extracellular signal-regulated kinases 1/2 (ERK1/2) by inhibition of ERK1/2 phosphatases. J. Neurochem. 2008, 106, 2056–2067. [Google Scholar] [CrossRef]
- Sperlagh, B.; Illes, P. P2X7 receptor: An emerging target in central nervous system diseases. Trends Pharmacol. Sci. 2014, 35, 537–547. [Google Scholar] [CrossRef]
- Carrasquero, L.M.; Delicado, E.G.; Bustillo, D.; Gutierrez-Martin, Y.; Artalejo, A.R.; Miras-Portugal, M.T. P2X7 and P2Y13 purinergic receptors mediate intracellular calcium responses to BzATP in rat cerebellar astrocytes. J. Neurochem. 2009, 110, 879–889. [Google Scholar] [CrossRef]
- Diaz-Hernandez, M.; del Puerto, A.; Diaz-Hernandez, J.I.; Diez-Zaera, M.; Lucas, J.J.; Garrido, J.J.; Miras-Portugal, M.T. Inhibition of the ATP-gated P2X7 receptor promotes axonal growth and branching in cultured hippocampal neurons. J. Cell. Sci. 2008, 121, 3717–3728. [Google Scholar] [CrossRef]
- Neary, J.T.; Kang, Y. Signaling from P2 nucleotide receptors to protein kinase cascades induced by CNS injury: Implications for reactive gliosis and neurodegeneration. Mol. Neurobiol. 2005, 31, 95–103. [Google Scholar] [CrossRef]
- Ortega, F.; Perez-Sen, R.; Delicado, E.G.; Miras-Portugal, M.T. P2X7 nucleotide receptor is coupled to GSK-3 inhibition and neuroprotection in cerebellar granule neurons. Neurotox. Res. 2009, 15, 193–204. [Google Scholar] [CrossRef]
- Ortega, F.; Perez-Sen, R.; Delicado, E.G.; Teresa Miras-Portugal, M. ERK1/2 activation is involved in the neuroprotective action of P2Y13 and P2X7 receptors against glutamate excitotoxicity in cerebellar granule neurons. Neuropharmacology 2011, 61, 1210–1221. [Google Scholar] [CrossRef]
- Carrasquero, L.M.; Delicado, E.G.; Sanchez-Ruiloba, L.; Iglesias, T.; Miras-Portugal, M.T. Mechanisms of protein kinase D activation in response to P2Y(2) and P2X7 receptors in primary astrocytes. Glia 2010, 58, 984–995. [Google Scholar] [CrossRef]
- Queipo, M.J.; Gil-Redondo, J.C.; Morente, V.; Ortega, F.; Miras-Portugal, M.T.; Delicado, E.G.; Perez-Sen, R. P2X7 Nucleotide and EGF Receptors Exert Dual Modulation of the Dual-Specificity Phosphatase 6 (MKP-3) in Granule Neurons and Astrocytes, Contributing to Negative Feedback on ERK Signaling. Front. Mol. Neurosci. 2017, 10, 448. [Google Scholar] [CrossRef]
- Morente, V.; Perez-Sen, R.; Ortega, F.; Huerta-Cepas, J.; Delicado, E.G.; Miras-Portugal, M.T. Neuroprotection elicited by P2Y13 receptors against genotoxic stress by inducing DUSP2 expression and MAPK signaling recovery. Biochim. Biophys. Acta 2014, 1843, 1886–1898. [Google Scholar] [CrossRef]
- Kristiansen, M.; Hughes, R.; Patel, P.; Jacques, T.S.; Clark, A.R.; Ham, J. Mkp1 is a c-Jun target gene that antagonizes JNK-dependent apoptosis in sympathetic neurons. J. Neurosci. 2010, 30, 10820–10832. [Google Scholar] [CrossRef]
- Li, B.; Yi, P.; Zhang, B.; Xu, C.; Liu, Q.; Pi, Z.; Xu, X.; Chevet, E.; Liu, J. Differences in endoplasmic reticulum stress signalling kinetics determine cell survival outcome through activation of MKP-1. Cell Signal. 2011, 23, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Cadalbert, L.; Sloss, C.M.; Cameron, P.; Plevin, R. Conditional expression of MAP kinase phosphatase-2 protects against genotoxic stress-induced apoptosis by binding and selective dephosphorylation of nuclear activated c-jun N-terminal kinase. Cell Signal. 2005, 17, 1254–1264. [Google Scholar] [CrossRef]
- Kawahara, N.; Wang, Y.; Mukasa, A.; Furuya, K.; Shimizu, T.; Hamakubo, T.; Aburatani, H.; Kodama, T.; Kirino, T. Genome-wide gene expression analysis for induced ischemic tolerance and delayed neuronal death following transient global ischemia in rats. J. Cereb. Blood Flow Metab. 2004, 24, 212–223. [Google Scholar] [CrossRef]
- Dreixler, J.C.; Bratton, A.; Du, E.; Shaikh, A.R.; Savoie, B.; Alexander, M.; Marcet, M.M.; Roth, S. Mitogen-activated protein kinase phosphatase-1 (MKP-1) in retinal ischemic preconditioning. Exp. Eye Res. 2011, 93, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Koga, S.; Kojima, S.; Kishimoto, T.; Kuwabara, S.; Yamaguchi, A. Over-expression of map kinase phosphatase-1 (MKP-1) suppresses neuronal death through regulating JNK signaling in hypoxia/re-oxygenation. Brain Res. 2012, 1436, 137–146. [Google Scholar] [CrossRef]
- Liu, L.; Doran, S.; Xu, Y.; Manwani, B.; Ritzel, R.; Benashski, S.; McCullough, L.; Li, J. Inhibition of mitogen-activated protein kinase phosphatase-1 (MKP-1) increases experimental stroke injury. Exp. Neurol. 2014, 261, 404–411. [Google Scholar] [CrossRef]
- Ndong, C.; Landry, R.P.; DeLeo, J.A.; Romero-Sandoval, E.A. Mitogen activated protein kinase phosphatase-1 prevents the development of tactile sensitivity in a rodent model of neuropathic pain. Mol. Pain 2012, 8, 34. [Google Scholar] [CrossRef]
- Saha, M.; Skopelja, S.; Martinez, E.; Alvarez, D.L.; Liponis, B.S.; Romero-Sandoval, E.A. Spinal mitogen-activated protein kinase phosphatase-3 (MKP-3) is necessary for the normal resolution of mechanical allodynia in a mouse model of acute postoperative pain. J. Neurosci. 2013, 33, 17182–17187. [Google Scholar] [CrossRef] [PubMed]
- Skopelja-Gardner, S.; Saha, M.; Alvarado-Vazquez, P.A.; Liponis, B.S.; Martinez, E.; Romero-Sandoval, E.A. Mitogen-activated protein kinase phosphatase-3 (MKP-3) in the surgical wound is necessary for the resolution of postoperative pain in mice. J. Pain Res. 2017, 10, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Romero-Sandoval, E.A.; Horvath, R.; Landry, R.P.; DeLeo, J.A. Cannabinoid receptor type 2 activation induces a microglial anti-inflammatory phenotype and reduces migration via MKP induction and ERK dephosphorylation. Mol. Pain 2009, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Eljaschewitsch, E.; Witting, A.; Mawrin, C.; Lee, T.; Schmidt, P.M.; Wolf, S.; Hoertnagl, H.; Raine, C.S.; Schneider-Stock, R.; Nitsch, R.; et al. The endocannabinoid anandamide protects neurons during CNS inflammation by induction of MKP-1 in microglial cells. Neuron 2006, 49, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, G.; Chatterjee, N. Endocannabinoids alleviate proinflammatory conditions by modulating innate immune response in muller glia during inflammation. Glia 2012, 60, 1629–1645. [Google Scholar] [CrossRef]
- Landry, R.P.; Martinez, E.; DeLeo, J.A.; Romero-Sandoval, E.A. Spinal cannabinoid receptor type 2 agonist reduces mechanical allodynia and induces mitogen-activated protein kinase phosphatases in a rat model of neuropathic pain. J. Pain 2012, 13, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Bhore, N.; Wang, B.J.; Chen, Y.W.; Liao, Y.F. Critical Roles of Dual-Specificity Phosphatases in Neuronal Proteostasis and Neurological Diseases. Int. J. Mol. Sci. 2017, 18, 1963. [Google Scholar] [CrossRef] [PubMed]
- Colucci-D’Amato, L.; Perrone-Capano, C.; di Porzio, U. Chronic activation of ERK and neurodegenerative diseases. Bioessays 2003, 25, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Lee, H.G.; Raina, A.K.; Perry, G.; Smith, M.A. The role of mitogen-activated protein kinase pathways in Alzheimer’s disease. Neurosignals 2002, 11, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Reijonen, S.; Kukkonen, J.P.; Hyrskyluoto, A.; Kivinen, J.; Kairisalo, M.; Takei, N.; Lindholm, D.; Korhonen, L. Downregulation of NF-kappaB signaling by mutant huntingtin proteins induces oxidative stress and cell death. Cell. Mol. Life Sci. 2010, 67, 1929–1941. [Google Scholar] [CrossRef]
- Gianfriddo, M.; Melani, A.; Turchi, D.; Giovannini, M.G.; Pedata, F. Adenosine and glutamate extracellular concentrations and mitogen-activated protein kinases in the striatum of Huntington transgenic mice. Selective antagonism of adenosine A2A receptors reduces transmitter outflow. Neurobiol. Dis. 2004, 17, 77–88. [Google Scholar] [CrossRef]
- Banzhaf-Strathmann, J.; Benito, E.; May, S.; Arzberger, T.; Tahirovic, S.; Kretzschmar, H.; Fischer, A.; Edbauer, D. MicroRNA-125b induces tau hyperphosphorylation and cognitive deficits in Alzheimer’s disease. EMBO J. 2014, 33, 1667–1680. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Zheng, Y.; Fang, W.; Liao, S.; Xiong, Y.; Li, Y.; Xiao, S.; Zhang, X.; Liu, J. Dual Specificity Phosphatase 6 Protects Neural Stem Cells from beta-Amyloid-Induced Cytotoxicity through ERK1/2 Inactivation. Biomolecules 2018, 8, 181. [Google Scholar] [CrossRef]
- Jeanneteau, F.; Chao, M.V. Are BDNF and glucocorticoid activities calibrated? Neuroscience 2013, 239, 173–195. [Google Scholar] [CrossRef] [PubMed]
- Menke, A.; Arloth, J.; Putz, B.; Weber, P.; Klengel, T.; Mehta, D.; Gonik, M.; Rex-Haffner, M.; Rubel, J.; Uhr, M.; et al. Dexamethasone stimulated gene expression in peripheral blood is a sensitive marker for glucocorticoid receptor resistance in depressed patients. Neuropsychopharmacology 2012, 37, 1455–1464. [Google Scholar] [CrossRef]
- Arango-Lievano, M.; Peguet, C.; Catteau, M.; Parmentier, M.L.; Wu, S.; Chao, M.V.; Ginsberg, S.D.; Jeanneteau, F. Deletion of Neurotrophin Signaling through the Glucocorticoid Receptor Pathway Causes Tau Neuropathology. Sci. Rep. 2016, 6, 37231. [Google Scholar] [CrossRef]
- Luthi-Carter, R.; Hanson, S.A.; Strand, A.D.; Bergstrom, D.A.; Chun, W.; Peters, N.L.; Woods, A.M.; Chan, E.Y.; Kooperberg, C.; Krainc, D.; et al. Dysregulation of gene expression in the R6/2 model of polyglutamine disease: Parallel changes in muscle and brain. Hum. Mol. Genet. 2002, 11, 1911–1926. [Google Scholar] [CrossRef]
- Taylor, D.M.; Moser, R.; Regulier, E.; Breuillaud, L.; Dixon, M.; Beesen, A.A.; Elliston, L.; Silva Santos Mde, F.; Kim, J.; Jones, L.; et al. MAP kinase phosphatase 1 (MKP-1/DUSP1) is neuroprotective in Huntington’s disease via additive effects of JNK and p38 inhibition. J. Neurosci. 2013, 33, 2313–2325. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Kubilus, J.K.; Lee, J.; Ryu, H.; Beesen, A.; Zucker, B.; Smith, K.; Kowall, N.W.; Ratan, R.R.; Luthi-Carter, R.; et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J. Neurosci. 2003, 23, 9418–9427. [Google Scholar] [CrossRef]
- Collins, L.M.; Downer, E.J.; Toulouse, A.; Nolan, Y.M. Mitogen-Activated Protein Kinase Phosphatase (MKP)-1 in Nervous System Development and Disease. Mol. Neurobiol. 2015, 51, 1158–1167. [Google Scholar] [CrossRef]
- Heiman, M.; Heilbut, A.; Francardo, V.; Kulicke, R.; Fenster, R.J.; Kolaczyk, E.D.; Mesirov, J.P.; Surmeier, D.J.; Cenci, M.A.; Greengard, P. Molecular adaptations of striatal spiny projection neurons during levodopa-induced dyskinesia. Proc. Natl. Acad. Sci. USA 2014, 111, 4578–4583. [Google Scholar] [CrossRef]
- Brehm, N.; Bez, F.; Carlsson, T.; Kern, B.; Gispert, S.; Auburger, G.; Cenci, M.A. A Genetic Mouse Model of Parkinson’s Disease Shows Involuntary Movements and Increased Postsynaptic Sensitivity to Apomorphine. Mol. Neurobiol. 2015, 52, 1152–1164. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Ahn, Y.M.; Joo, E.J.; Chang, J.S.; Kim, Y.S. The association of DUSP6 gene with schizophrenia and bipolar disorder: Its possible role in the development of bipolar disorder. Mol. Psychiatry 2006, 11, 425–426. [Google Scholar] [CrossRef] [PubMed]
- Toyota, T.; Watanabe, A.; Shibuya, H.; Nankai, M.; Hattori, E.; Yamada, K.; Kurumaji, A.; Karkera, J.D.; Detera-Wadleigh, S.D.; Yoshikawa, T. Association study on the DUSP6 gene, an affective disorder candidate gene on 12q23, performed by using fluorescence resonance energy transfer-based melting curve analysis on the LightCycler. Mol. Psychiatry 2000, 5, 489–494. [Google Scholar] [CrossRef]
- Kim, S.H.; Shin, S.Y.; Lee, K.Y.; Joo, E.J.; Song, J.Y.; Ahn, Y.M.; Lee, Y.H.; Kim, Y.S. The genetic association of DUSP6 with bipolar disorder and its effect on ERK activity. Prog. Neuropsychopharmacol. Biol. Psychiatry 2012, 37, 41–49. [Google Scholar] [CrossRef]
- Kalkman, H.O. Potential opposite roles of the extracellular signal-regulated kinase (ERK) pathway in autism spectrum and bipolar disorders. Neurosci. Biobehav. Rev. 2012, 36, 2206–2213. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.J.; Wei, H.; Landgraf, D.; Le Roux, M.J.; Welsh, D.K. Disinhibition of the extracellular-signal-regulated kinase restores the amplification of circadian rhythms by lithium in cells from bipolar disorder patients. Eur. Neuropsychopharmacol. 2016, 26, 1310–1319. [Google Scholar] [CrossRef] [PubMed]
- Duric, V.; Banasr, M.; Licznerski, P.; Schmidt, H.D.; Stockmeier, C.A.; Simen, A.A.; Newton, S.S.; Duman, R.S. A negative regulator of MAP kinase causes depressive behavior. Nat. Med. 2010, 16, 1328–1332. [Google Scholar] [CrossRef]
- Wang, J.Q.; Mao, L. The ERK Pathway: Molecular Mechanisms and Treatment of Depression. Mol. Neurobiol. 2019. [Google Scholar] [CrossRef]
- Akbarian, S.; Davis, R.J. Keep the ‘phospho’ on MAPK, be happy. Nat. Med. 2010, 16, 1187–1188. [Google Scholar] [CrossRef] [PubMed]
- Vogt, A.; Tamewitz, A.; Skoko, J.; Sikorski, R.P.; Giuliano, K.A.; Lazo, J.S. The benzo[c]phenanthridine alkaloid, sanguinarine, is a selective, cell-active inhibitor of mitogen-activated protein kinase phosphatase-1. J. Biol. Chem. 2005, 280, 19078–19086. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, H.; Zhang, R.; Wang, H.; Peng, Z.; Sun, R.; Tan, Q. Microinjection of sanguinarine into the ventrolateral orbital cortex inhibits Mkp-1 and exerts an antidepressant-like effect in rats. Neurosci. Lett. 2012, 506, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The epidemiology of glioma in adults: A “state of the science” review. Neuro-Oncology 2014, 16, 896–913. [Google Scholar] [CrossRef]
- Prabhakar, S.; Asuthkar, S.; Lee, W.; Chigurupati, S.; Zakharian, E.; Tsung, A.J.; Velpula, K.K. Targeting DUSPs in glioblastomas—wielding a double-edged sword? Cell. Biol. Int. 2014, 38, 145–153. [Google Scholar] [CrossRef]
- Dedobbeleer, M.; Willems, E.; Freeman, S.; Lombard, A.; Goffart, N.; Rogister, B. Phosphatases and solid tumors: Focus on glioblastoma initiation, progression and recurrences. Biochem. J. 2017, 474, 2903–2924. [Google Scholar] [CrossRef] [PubMed]
- Mills, B.N.; Albert, G.P.; Halterman, M.W. Expression Profiling of the MAP Kinase Phosphatase Family Reveals a Role for DUSP1 in the Glioblastoma Stem Cell Niche. Cancer Microenviron. 2017, 10, 57–68. [Google Scholar] [CrossRef]
- Laderoute, K.R.; Mendonca, H.L.; Calaoagan, J.M.; Knapp, A.M.; Giaccia, A.J.; Stork, P.J. Mitogen-activated protein kinase phosphatase-1 (MKP-1) expression is induced by low oxygen conditions found in solid tumor microenvironments. A candidate MKP for the inactivation of hypoxia-inducible stress-activated protein kinase/c-Jun N-terminal protein kinase activity. J. Biol. Chem. 1999, 274, 12890–12897. [Google Scholar] [PubMed]
- Arrizabalaga, O.; Moreno-Cugnon, L.; Auzmendi-Iriarte, J.; Aldaz, P.; Ibanez de Caceres, I.; Garros-Regulez, L.; Moncho-Amor, V.; Torres-Bayona, S.; Pernia, O.; Pintado-Berninches, L.; et al. High expression of MKP1/DUSP1 counteracts glioma stem cell activity and mediates HDAC inhibitor response. Oncogenesis 2017, 6, 401. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.M.; Jan, H.J.; Lee, C.C.; Tao, H.Y.; Shih, Y.L.; Wei, H.W.; Lee, H.M. Dexamethasone reduced invasiveness of human malignant glioblastoma cells through a MAPK phosphatase-1 (MKP-1) dependent mechanism. Eur. J. Pharmacol. 2008, 593, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jan, H.J.; Lee, C.C.; Lin, Y.M.; Lai, J.H.; Wei, H.W.; Lee, H.M. Rosiglitazone reduces cell invasiveness by inducing MKP-1 in human U87MG glioma cells. Cancer Lett. 2009, 277, 141–148. [Google Scholar] [CrossRef]
- Ahmad, M.K.; Abdollah, N.A.; Shafie, N.H.; Yusof, N.M.; Razak, S.R.A. Dual-specificity phosphatase 6 (DUSP6): A review of its molecular characteristics and clinical relevance in cancer. Cancer Biol. Med. 2018, 15, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Messina, S.; Frati, L.; Leonetti, C.; Zuchegna, C.; Di Zazzo, E.; Calogero, A.; Porcellini, A. Dual-specificity phosphatase DUSP6 has tumor-promoting properties in human glioblastomas. Oncogene 2011, 30, 3813–3820. [Google Scholar] [CrossRef] [PubMed]
- Ortega, F.; Perez-Sen, R.; Morente, V.; Delicado, E.G.; Miras-Portugal, M.T. P2X7, NMDA and BDNF receptors converge on GSK3 phosphorylation and cooperate to promote survival in cerebellar granule neurons. Cell. Mol. Life Sci. 2010, 67, 1723–1733. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Sen, R.; Queipo, M.J.; Gil-Redondo, J.C.; Ortega, F.; Gómez-Villafuertes, R.; Miras-Portugal, M.T.; Delicado, E.G. Dual-Specificity Phosphatase Regulation in Neurons and Glial Cells. Int. J. Mol. Sci. 2019, 20, 1999. https://doi.org/10.3390/ijms20081999
Pérez-Sen R, Queipo MJ, Gil-Redondo JC, Ortega F, Gómez-Villafuertes R, Miras-Portugal MT, Delicado EG. Dual-Specificity Phosphatase Regulation in Neurons and Glial Cells. International Journal of Molecular Sciences. 2019; 20(8):1999. https://doi.org/10.3390/ijms20081999
Chicago/Turabian StylePérez-Sen, Raquel, María José Queipo, Juan Carlos Gil-Redondo, Felipe Ortega, Rosa Gómez-Villafuertes, María Teresa Miras-Portugal, and Esmerilda G. Delicado. 2019. "Dual-Specificity Phosphatase Regulation in Neurons and Glial Cells" International Journal of Molecular Sciences 20, no. 8: 1999. https://doi.org/10.3390/ijms20081999