Essential Role of Visfatin in Lipopolysaccharide and Colon Ascendens Stent Peritonitis-Induced Acute Lung Injury

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
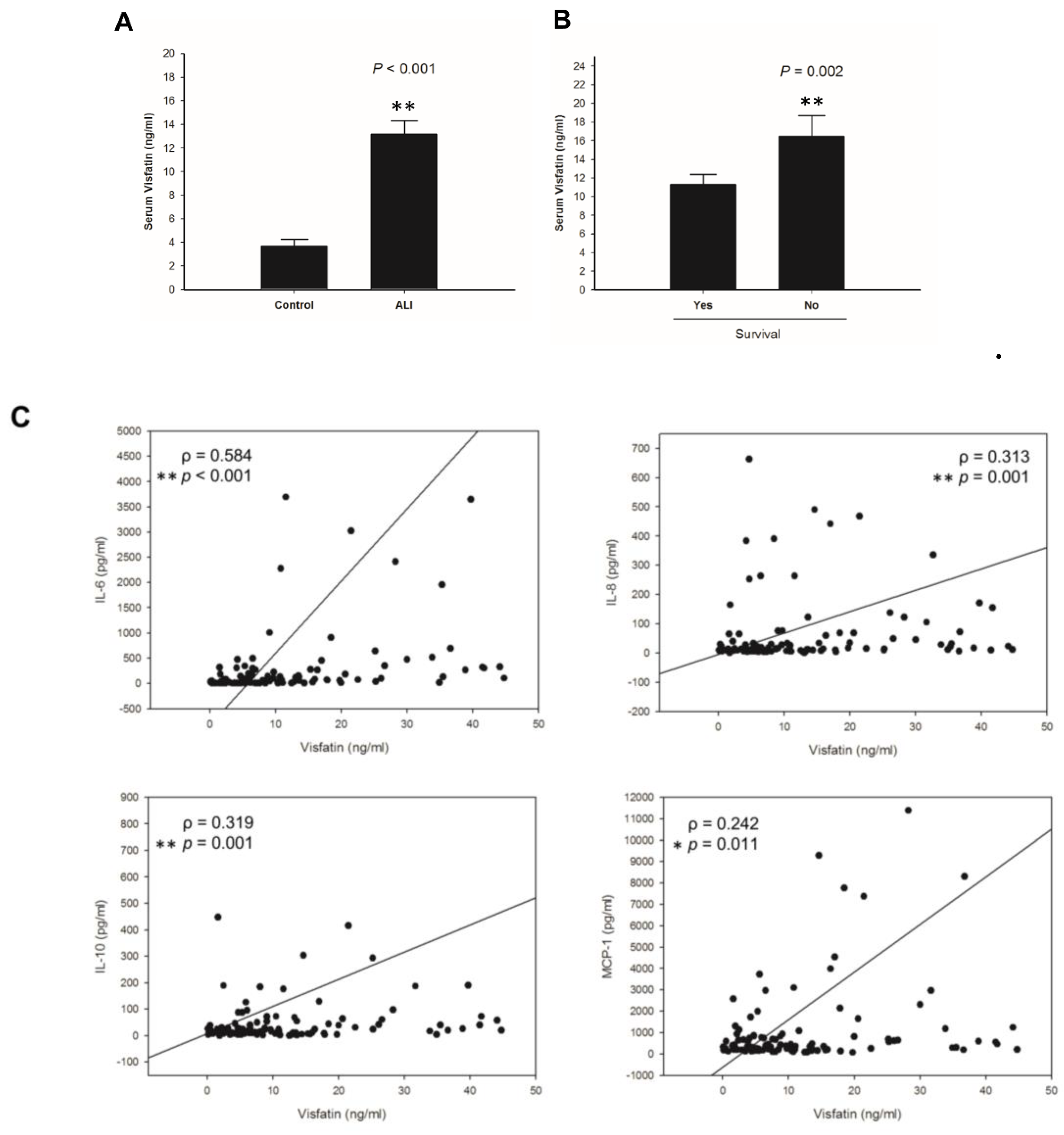
2.1. Visfatin Correlates with Poor Survival and Inflammatory Cytokines in ALI Patients
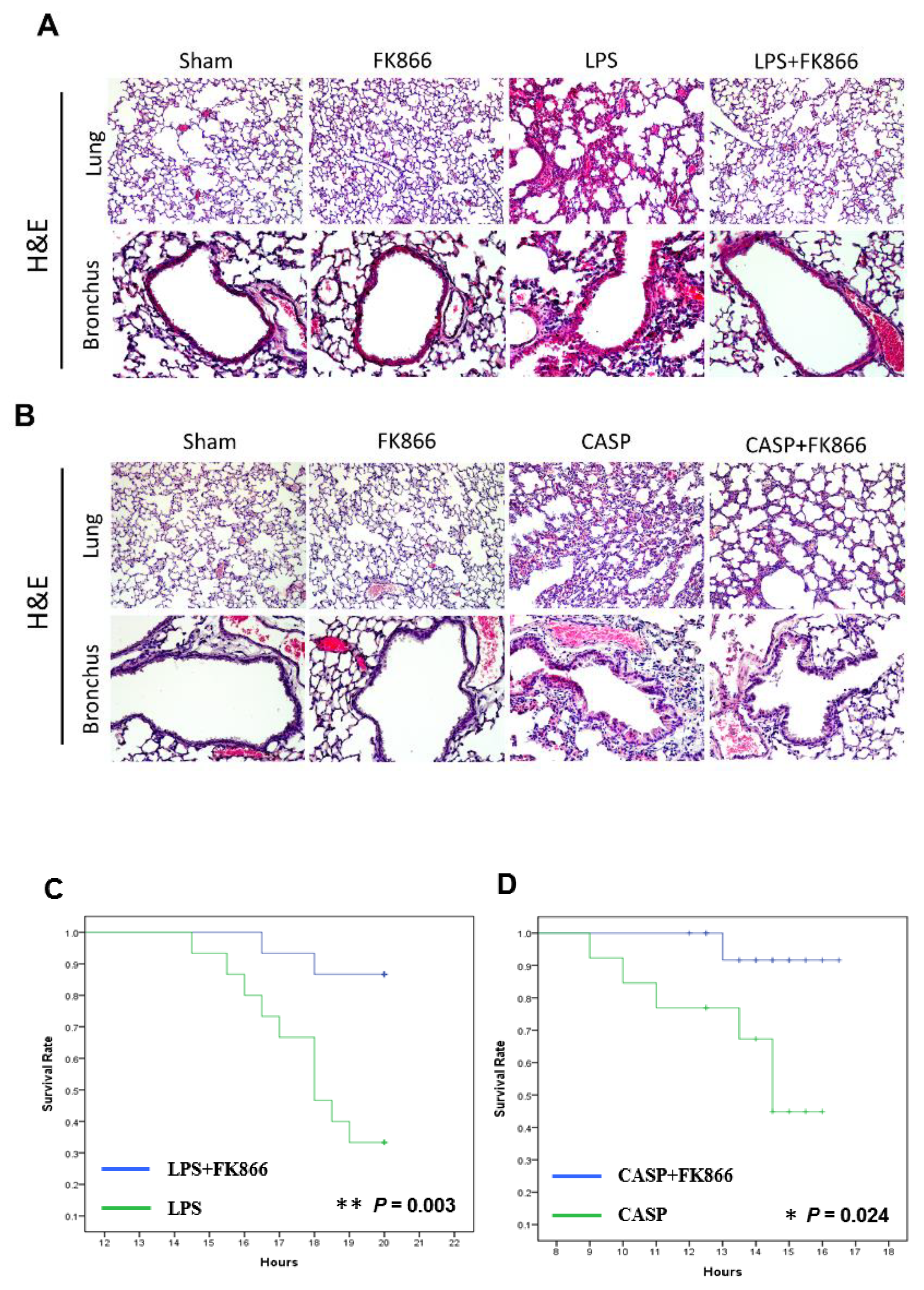
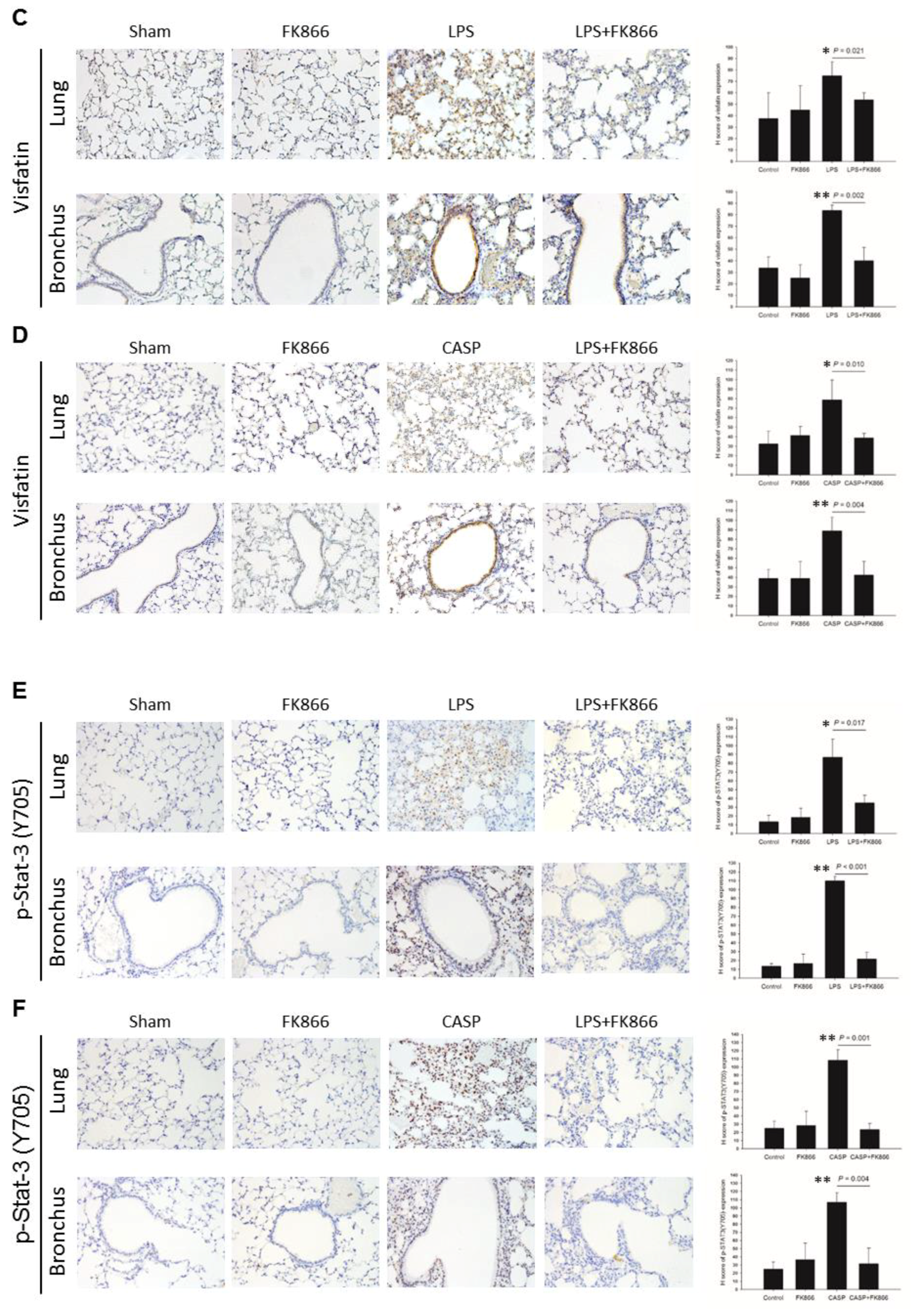
2.2. Visfatin Expression is Upregulated in Both LPS and CASP-Induced ALI Mice, While Visfatin Inhibitor FK866 Attenuated the ALI Phenotypes
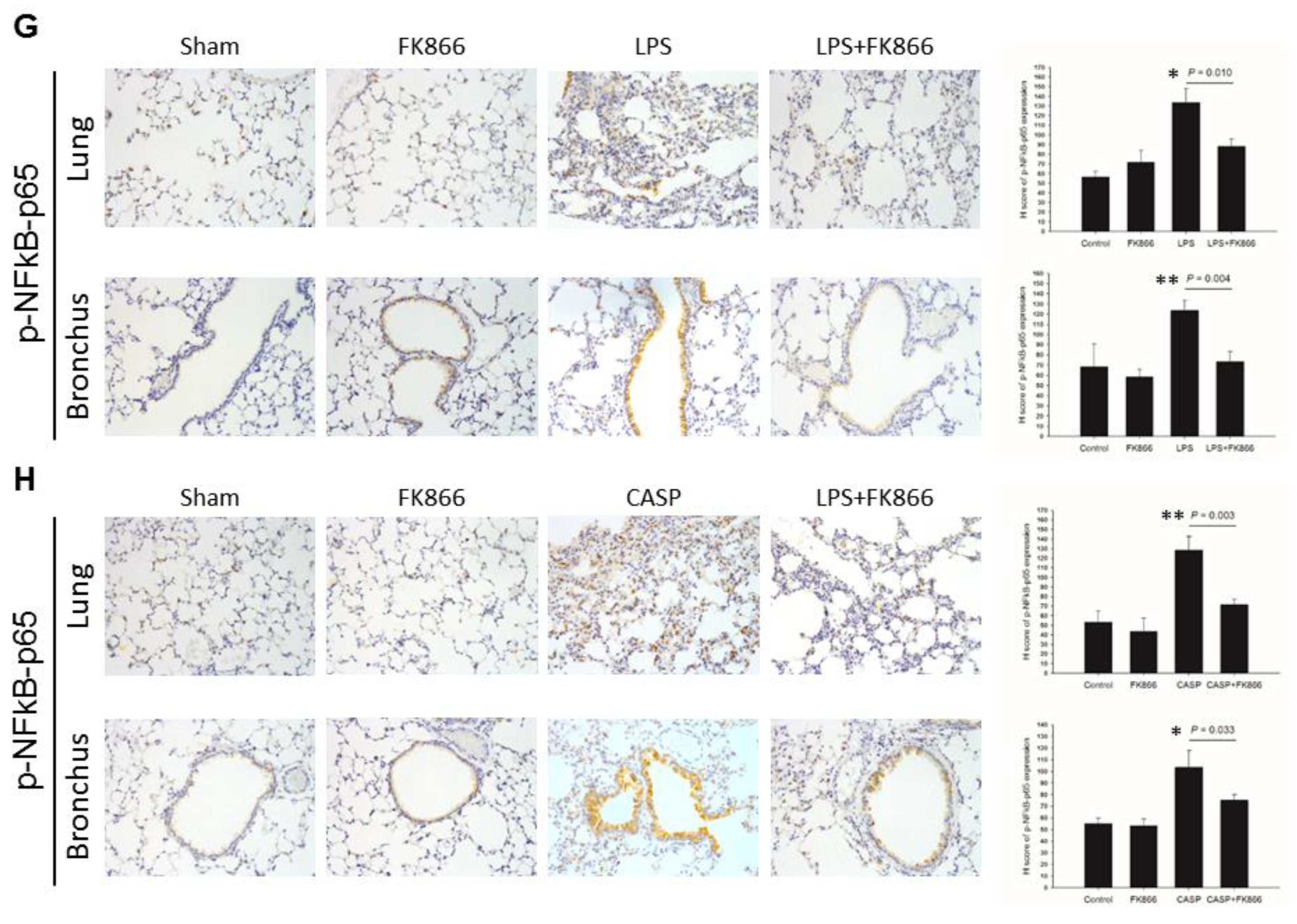
2.3. LPS-Upregulated Visfatin Expression in Human Bronchial Epithelial Cells Mediates the Activation of the STAT3/NF-κB Pathway
3. Discussion
4. Materials and Methods
4.1. Patient Enrollment
4.2. Reagents
4.3. Cell Culture
4.4. Animal Model of Abdominal Sepsis
4.5. Immunoblotting Analysis
4.6. Enzyme Immunoassay
4.7. Immunohistochemistry
4.8. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALI | Acute lung injury |
| H&E | Hematoxylin and eosin |
| LPS | Lipopolysaccharide |
| ARDS | Acute respiratory distress syndrome |
| PBEF | Pre-B-cell colony-enhancing factor |
| Nampt | Nicotinamide phosphoribosyltransferase |
| MCP-1 | Monocyte chemotactic protein-1 |
| IL-1β | Interleukin-1β |
| IL-6 | Interleukin-6 |
| TNF-α | Tumor necrosis factor-α |
| CASP | Colon ascendens stent peritonitis |
| TLR4 | Toll–like receptor 4 |
References
- Matthay, M.A.; Zimmerman, G.A. Acute lung injury and the acute respiratory distress syndrome: Four decades of inquiry into pathogenesis and rational management. Am. J. Respir. Cell Mol. Biol. 2005, 33, 319–327. [Google Scholar] [CrossRef]
- Mortelliti, M.P.; Manning, H.L. Acute respiratory distress syndrome. Am. Fam. Phys. 2002, 65, 1823–1830. [Google Scholar]
- Matthay, M.A.; Zimmerman, G.A.; Esmon, C.; Bhattacharya, J.; Coller, B.; Doerschuk, C.M.; Floros, J.; Gimbrone, M.A., Jr.; Hoffman, E.; Hubmayr, R.D.; et al. Future research directions in acute lung injury: Summary of a National Heart, Lung, and Blood Institute working group. Am. J. Respir. Crit. Care Med. 2003, 167, 1027–1035. [Google Scholar] [CrossRef]
- Rubenfeld, G.D.; Caldwell, E.; Peabody, E.; Weaver, J.; Martin, D.P.; Neff, M.; Stern, E.J.; Hudson, L.D. Incidence and outcomes of acute lung injury. New Engl. J. Med. 2005, 353, 1685–1693. [Google Scholar] [CrossRef]
- Goss, C.H.; Brower, R.G.; Hudson, L.D.; Rubenfeld, G.D.; Network, A. Incidence of acute lung injury in the United States. Crit. Care Med. 2003, 31, 1607–1611. [Google Scholar] [CrossRef]
- Vasudevan, A.; Lodha, R.; Kabra, S.K. Acute lung injury and acute respiratory distress syndrome. Indian J. Pediatrics 2004, 71, 743–750. [Google Scholar]
- Ware, L.B.; Matthay, M.A. The acute respiratory distress syndrome. New Engl. J. Med. 2000, 342, 1334–1349. [Google Scholar] [CrossRef]
- Ware, L.B. Pathophysiology of acute lung injury and the acute respiratory distress syndrome. Semin. Respir. Crit. Care Med. 2006, 27, 337–349. [Google Scholar] [CrossRef]
- Luh, S.P.; Chiang, C.H. Acute lung injury/acute respiratory distress syndrome (ALI/ARDS): The mechanism, present strategies and future perspectives of therapies. J. Zhejiang Univ. Sci. B 2007, 8, 60–69. [Google Scholar] [CrossRef]
- Johnson, E.R.; Matthay, M.A. Acute lung injury: Epidemiology, pathogenesis, and treatment. J. Aerosol Med. Pulm. Drug Deliv. 2010, 23, 243–252. [Google Scholar] [CrossRef]
- Calfee, C.S.; Matthay, M.A. Nonventilatory treatments for acute lung injury and ARDS. Chest 2007, 131, 913–920. [Google Scholar] [CrossRef]
- Diaz, J.V.; Brower, R.; Calfee, C.S.; Matthay, M.A. Therapeutic strategies for severe acute lung injury. Crit. Care Med. 2010, 38, 1644–1650. [Google Scholar] [CrossRef]
- Barrera, G.; Landoni, V.; Martire-Greco, D.; Chiarella, P.; Meiss, R.; Gomez, S.A.; Alves-Rosa, F.; Rearte, B.; Isturiz, M.; Palermo, M.S.; et al. Model of polymicrobial peritonitis that induces the proinflammatory and immunosuppressive phases of sepsis. Infect. Immun. 2011, 79, 1280–1288. [Google Scholar] [CrossRef]
- Feterowski, C.; Emmanuilidis, K.; Miethke, T.; Gerauer, K.; Rump, M.; Ulm, K.; Holzmann, B.; Weighardt, H. Effects of functional Toll-like receptor-4 mutations on the immune response to human and experimental sepsis. Immunology 2003, 109, 426–431. [Google Scholar] [Green Version]
- Parsons, P.E. Mediators and mechanisms of acute lung injury. Clin. Chest Med. 2000, 21, 467–476. [Google Scholar]
- Bhatia, M.; Moochhala, S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J. Pathol. 2004, 202, 145–156. [Google Scholar] [CrossRef]
- Herold, S.; Gabrielli, N.M.; Vadasz, I. Novel concepts of acute lung injury and alveolar-capillary barrier dysfunction. Am. J. Physiology. Lung Cell. Mol. Physiol. 2013, 305, L665–L681. [Google Scholar] [CrossRef]
- Samal, B.; Sun, Y.; Stearns, G.; Xie, C.; Suggs, S.; McNiece, I. Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol. Cell. Biol. 1994, 14, 1431–1437. [Google Scholar]
- Li, H.; Liu, P.; Cepeda, J.; Fang, D.; Easley, R.B.; Simon, B.A.; Zhang, L.Q.; Ye, S.Q. Augmentation of Pulmonary Epithelial Cell IL-8 Expression and Permeability by Pre-B-cell Colony Enhancing Factor. J. Inflamm. 2008, 5, 15. [Google Scholar] [CrossRef]
- Kitani, T.; Okuno, S.; Fujisawa, H. Growth phase-dependent changes in the subcellular localization of pre-B-cell colony-enhancing factor. FEBS Lett. 2003, 544, 74–78. [Google Scholar]
- Jia, S.H.; Li, Y.; Parodo, J.; Kapus, A.; Fan, L.; Rotstein, O.D.; Marshall, J.C. Pre-B cell colony-enhancing factor inhibits neutrophil apoptosis in experimental inflammation and clinical sepsis. J. Clin. Investig. 2004, 113, 1318–1327. [Google Scholar] [CrossRef]
- Sun, Z.; Lei, H.; Zhang, Z. Pre-B cell colony enhancing factor (PBEF), a cytokine with multiple physiological functions. Cytokine Growth Factor Rev. 2013, 24, 433–442. [Google Scholar] [CrossRef]
- Sonoli, S.S.; Shivprasad, S.; Prasad, C.V.; Patil, A.B.; Desai, P.B.; Somannavar, M.S. Visfatin—A review. Eur. Rev. Med Pharmacol. Sci. 2011, 15, 9–14. [Google Scholar]
- Revollo, J.R.; Grimm, A.A.; Imai, S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J. Biol. Chem. 2004, 279, 50754–50763. [Google Scholar] [CrossRef]
- Rongvaux, A.; Shea, R.J.; Mulks, M.H.; Gigot, D.; Urbain, J.; Leo, O.; Andris, F. Pre-B-cell colony-enhancing factor, whose expression is up-regulated in activated lymphocytes, is a nicotinamide phosphoribosyltransferase, a cytosolic enzyme involved in NAD biosynthesis. Eur. J. Immunol. 2002, 32, 3225–3234. [Google Scholar] [CrossRef]
- Ognjanovic, S.; Bryant-Greenwood, G.D. Pre-B-cell colony-enhancing factor, a novel cytokine of human fetal membranes. Am. J. Obstet. Gynecol. 2002, 187, 1051–1058. [Google Scholar]
- Fukuhara, A.; Matsuda, M.; Nishizawa, M.; Segawa, K.; Tanaka, M.; Kishimoto, K.; Matsuki, Y.; Murakami, M.; Ichisaka, T.; Murakami, H.; et al. Visfatin: A protein secreted by visceral fat that mimics the effects of insulin. Science 2005, 307, 426–430. [Google Scholar] [CrossRef]
- Moschen, A.R.; Kaser, A.; Enrich, B.; Mosheimer, B.; Theurl, M.; Niederegger, H.; Tilg, H. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J. Immunol. 2007, 178, 1748–1758. [Google Scholar]
- Ye, S.Q.; Zhang, L.Q.; Adyshev, D.; Usatyuk, P.V.; Garcia, A.N.; Lavoie, T.L.; Verin, A.D.; Natarajan, V.; Garcia, J.G. Pre-B-cell-colony-enhancing factor is critically involved in thrombin-induced lung endothelial cell barrier dysregulation. Microvasc. Res. 2005, 70, 142–151. [Google Scholar] [CrossRef]
- Nowell, M.A.; Richards, P.J.; Fielding, C.A.; Ognjanovic, S.; Topley, N.; Williams, A.S.; Bryant-Greenwood, G.; Jones, S.A. Regulation of pre-B cell colony-enhancing factor by STAT-3-dependent interleukin-6 trans-signaling: Implications in the pathogenesis of rheumatoid arthritis. Arthritis Rheum. 2006, 54, 2084–2095. [Google Scholar] [CrossRef]
- Dahl, T.B.; Yndestad, A.; Skjelland, M.; Oie, E.; Dahl, A.; Michelsen, A.; Damas, J.K.; Tunheim, S.H.; Ueland, T.; Smith, C.; et al. Increased expression of visfatin in macrophages of human unstable carotid and coronary atherosclerosis: Possible role in inflammation and plaque destabilization. Circulation 2007, 115, 972–980. [Google Scholar] [CrossRef]
- Hong, S.B.; Huang, Y.; Moreno-Vinasco, L.; Sammani, S.; Moitra, J.; Barnard, J.W.; Ma, S.F.; Mirzapoiazova, T.; Evenoski, C.; Reeves, R.R.; et al. Essential role of pre-B-cell colony enhancing factor in ventilator-induced lung injury. Am. J. Respir. Crit. Care Med. 2008, 178, 605–617. [Google Scholar] [CrossRef]
- Ye, S.Q.; Simon, B.A.; Maloney, J.P.; Zambelli-Weiner, A.; Gao, L.; Grant, A.; Easley, R.B.; McVerry, B.J.; Tuder, R.M.; Standiford, T.; et al. Pre-B-cell colony-enhancing factor as a potential novel biomarker in acute lung injury. Am. J. Respir. Crit. Care Med. 2005, 171, 361–370. [Google Scholar] [CrossRef]
- Manicone, A.M. Role of the pulmonary epithelium and inflammatory signals in acute lung injury. Expert Rev. Clin. Immunol. 2009, 5, 63–75. [Google Scholar] [CrossRef]
- Wu, M.H.; Tsai, C.H.; Huang, Y.L.; Fong, Y.C.; Tang, C.H. Visfatin Promotes IL-6 and TNF-alpha Production in Human Synovial Fibroblasts by Repressing miR-199a-5p through ERK, p38 and JNK Signaling Pathways. Int. J. Mol. Sci. 2018, 19, 190. [Google Scholar] [CrossRef]
- Lustig, M.K.; Bac, V.H.; Pavlovic, D.; Maier, S.; Grundling, M.; Grisk, O.; Wendt, M.; Heidecke, C.D.; Lehmann, C. Colon ascendens stent peritonitis—A model of sepsis adopted to the rat: Physiological, microcirculatory and laboratory changes. Shock 2007, 28, 59–64. [Google Scholar] [CrossRef]
- Traeger, T.; Koerner, P.; Kessler, W.; Cziupka, K.; Diedrich, S.; Busemann, A.; Heidecke, C.D.; Maier, S. Colon ascendens stent peritonitis (CASP)—A standardized model for polymicrobial abdominal sepsis. J. Vis. Exp. JoVE 2010. [Google Scholar] [CrossRef]
- Matsuda, A.; Yang, W.L.; Jacob, A.; Aziz, M.; Matsuo, S.; Matsutani, T.; Uchida, E.; Wang, P. FK866, a visfatin inhibitor, protects against acute lung injury after intestinal ischemia-reperfusion in mice via NF-kappaB pathway. Ann. Surg. 2014, 259, 1007–1017. [Google Scholar] [CrossRef]
- Schuster, S.; Penke, M.; Gorski, T.; Gebhardt, R.; Weiss, T.S.; Kiess, W.; Garten, A. FK866-induced NAMPT inhibition activates AMPK and downregulates mTOR signaling in hepatocarcinoma cells. Biochem. Biophys. Res. Commun. 2015, 458, 334–340. [Google Scholar] [CrossRef]
- Kim, J.Y.; Bae, Y.H.; Bae, M.K.; Kim, S.R.; Park, H.J.; Wee, H.J.; Bae, S.K. Visfatin through STAT3 activation enhances IL-6 expression that promotes endothelial angiogenesis. Biochim. Biophys. Acta 2009, 1793, 1759–1767. [Google Scholar] [CrossRef] [Green Version]
- Bi, T.Q.; Che, X.M. Nampt/PBEF/visfatin and cancer. Cancer Biol. Ther. 2010, 10, 119–125. [Google Scholar] [CrossRef] [Green Version]
- Hung, A.C.; Lo, S.; Hou, M.F.; Lee, Y.C.; Tsai, C.H.; Chen, Y.Y.; Liu, W.; Su, Y.H.; Lo, Y.H.; Wang, C.H.; et al. Extracellular Visfatin-Promoted Malignant Behavior in Breast Cancer Is Mediated Through c-Abl and STAT3 Activation. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 4478–4490. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhang, Y.; Dorweiler, B.; Cui, D.; Wang, T.; Woo, C.W.; Brunkan, C.S.; Wolberger, C.; Imai, S.; Tabas, I. Extracellular Nampt promotes macrophage survival via a nonenzymatic interleukin-6/STAT3 signaling mechanism. J. Biol. Chem. 2008, 283, 34833–34843. [Google Scholar] [CrossRef]
- Oita, R.C.; Ferdinando, D.; Wilson, S.; Bunce, C.; Mazzatti, D.J. Visfatin induces oxidative stress in differentiated C2C12 myotubes in an Akt- and MAPK-independent, NFkB-dependent manner. Pflug. Arch. Eur. J. Physiol. 2010, 459, 619–630. [Google Scholar] [CrossRef]
- Camp, S.M.; Ceco, E.; Evenoski, C.L.; Danilov, S.M.; Zhou, T.; Chiang, E.T.; Moreno-Vinasco, L.; Mapes, B.; Zhao, J.; Gursoy, G.; et al. Unique Toll-Like Receptor 4 Activation by NAMPT/PBEF Induces NFkappaB Signaling and Inflammatory Lung Injury. Sci. Rep. 2015, 5, 13135. [Google Scholar] [CrossRef]
- Lee, M.W., Jr.; Sevryugina, Y.V.; Khan, A.; Ye, S.Q. Carboranes increase the potency of small molecule inhibitors of nicotinamide phosphoribosyltranferase. J. Med. Chem. 2012, 55, 7290–7294. [Google Scholar] [CrossRef]
- Huang, P.; Lee, M.W., Jr.; Sadrerafi, K.; Heruth, D.P.; Zhang, L.Q.; Maulik, D.; Ye, S.Q. MC-PPEA as a new and more potent inhibitor of CLP-induced sepsis and pulmonary inflammation than FK866. Drug Des. Dev. Ther. 2017, 11, 629–641. [Google Scholar] [CrossRef] [Green Version]
- Seymour, C.W.; Liu, V.X.; Iwashyna, T.J.; Brunkhorst, F.M.; Rea, T.D.; Scherag, A.; Rubenfeld, G.; Kahn, J.M.; Shankar-Hari, M.; Singer, M.; et al. Assessment of Clinical Criteria for Sepsis: For the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 762–774. [Google Scholar] [CrossRef]
- Rubenfeld, G.D.; Herridge, M.S. Epidemiology and outcomes of acute lung injury. Chest 2007, 131, 554–562. [Google Scholar] [CrossRef]
- Zhang, L.; Tian, Y.; Yang, J.; Li, J.; Tang, H.; Wang, Y. Colon Ascendens Stent Peritonitis (CASP) Induces Excessive Inflammation and Systemic Metabolic Dysfunction in a Septic Rat Model. J. Proteome Res. 2018, 17, 680–688. [Google Scholar] [CrossRef]
- Debaugnies, F.; Mahadeb, B.; Ferster, A.; Meuleman, N.; Rozen, L.; Demulder, A.; Corazza, F. Performances of the H-Score for Diagnosis of Hemophagocytic Lymphohistiocytosis in Adult and Pediatric Patients. Am. J. Clin. Pathol. 2016, 145, 862–870. [Google Scholar] [CrossRef] [Green Version]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.-C.; Lin, C.-Y.; Chen, Y.-H.; Chiu, W.-C.; Wang, Y.-Y.; Hsu, C.; Hu, S.C.-S.; Su, Y.-H.; Yuan, S.-S.F. Essential Role of Visfatin in Lipopolysaccharide and Colon Ascendens Stent Peritonitis-Induced Acute Lung Injury. Int. J. Mol. Sci. 2019, 20, 1678. https://doi.org/10.3390/ijms20071678
Lee Y-C, Lin C-Y, Chen Y-H, Chiu W-C, Wang Y-Y, Hsu C, Hu SC-S, Su Y-H, Yuan S-SF. Essential Role of Visfatin in Lipopolysaccharide and Colon Ascendens Stent Peritonitis-Induced Acute Lung Injury. International Journal of Molecular Sciences. 2019; 20(7):1678. https://doi.org/10.3390/ijms20071678
Chicago/Turabian StyleLee, Yi-Chen, Chun-Yu Lin, Yen-Hsu Chen, Wen-Chin Chiu, Yen-Yun Wang, Chin Hsu, Stephen Chu-Sung Hu, Yu-Han Su, and Shyng-Shiou F. Yuan. 2019. "Essential Role of Visfatin in Lipopolysaccharide and Colon Ascendens Stent Peritonitis-Induced Acute Lung Injury" International Journal of Molecular Sciences 20, no. 7: 1678. https://doi.org/10.3390/ijms20071678