Crystal Structure of the Cyclostreptin-Tubulin Adduct: Implications for Tubulin Activation by Taxane-Site Ligands

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
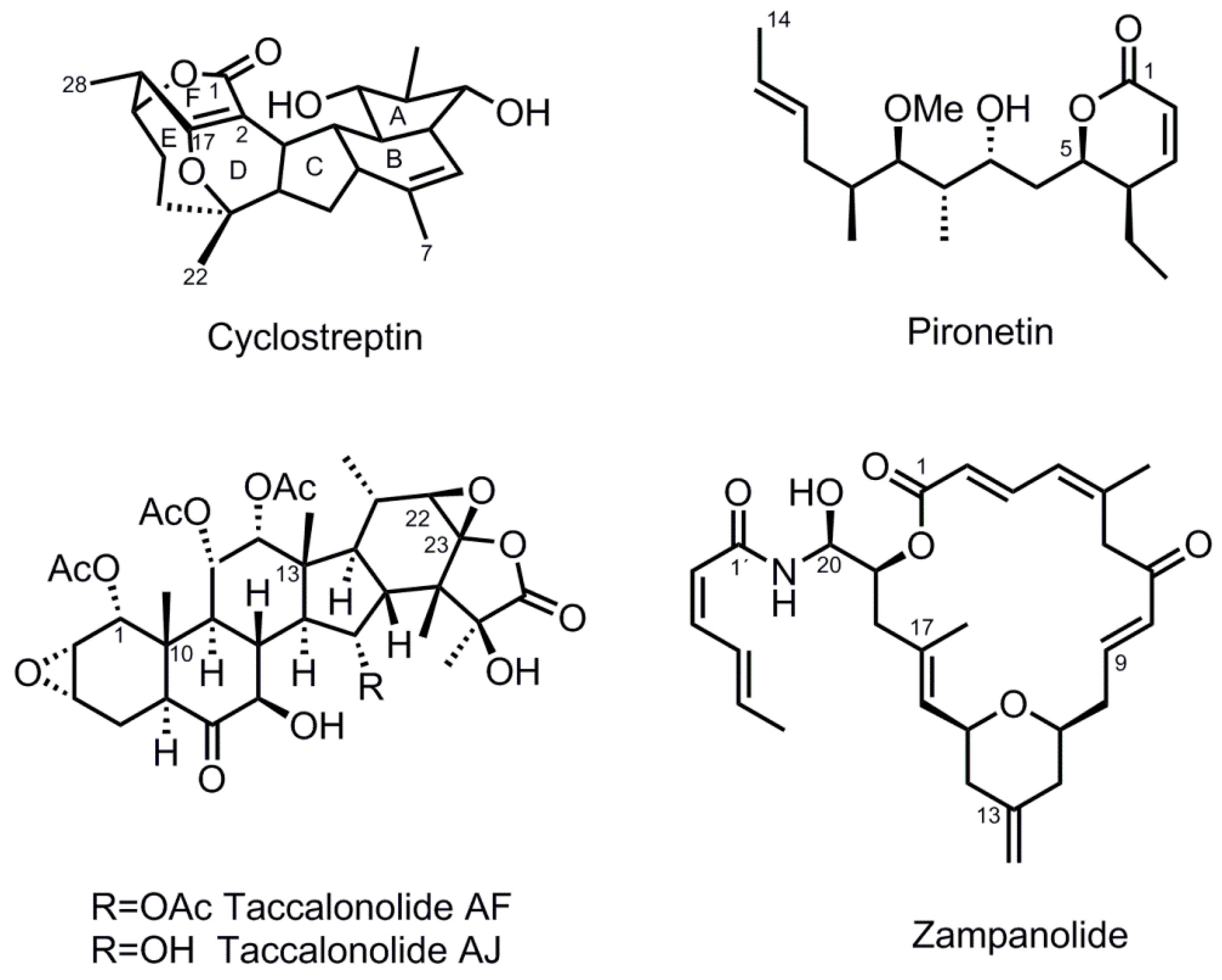
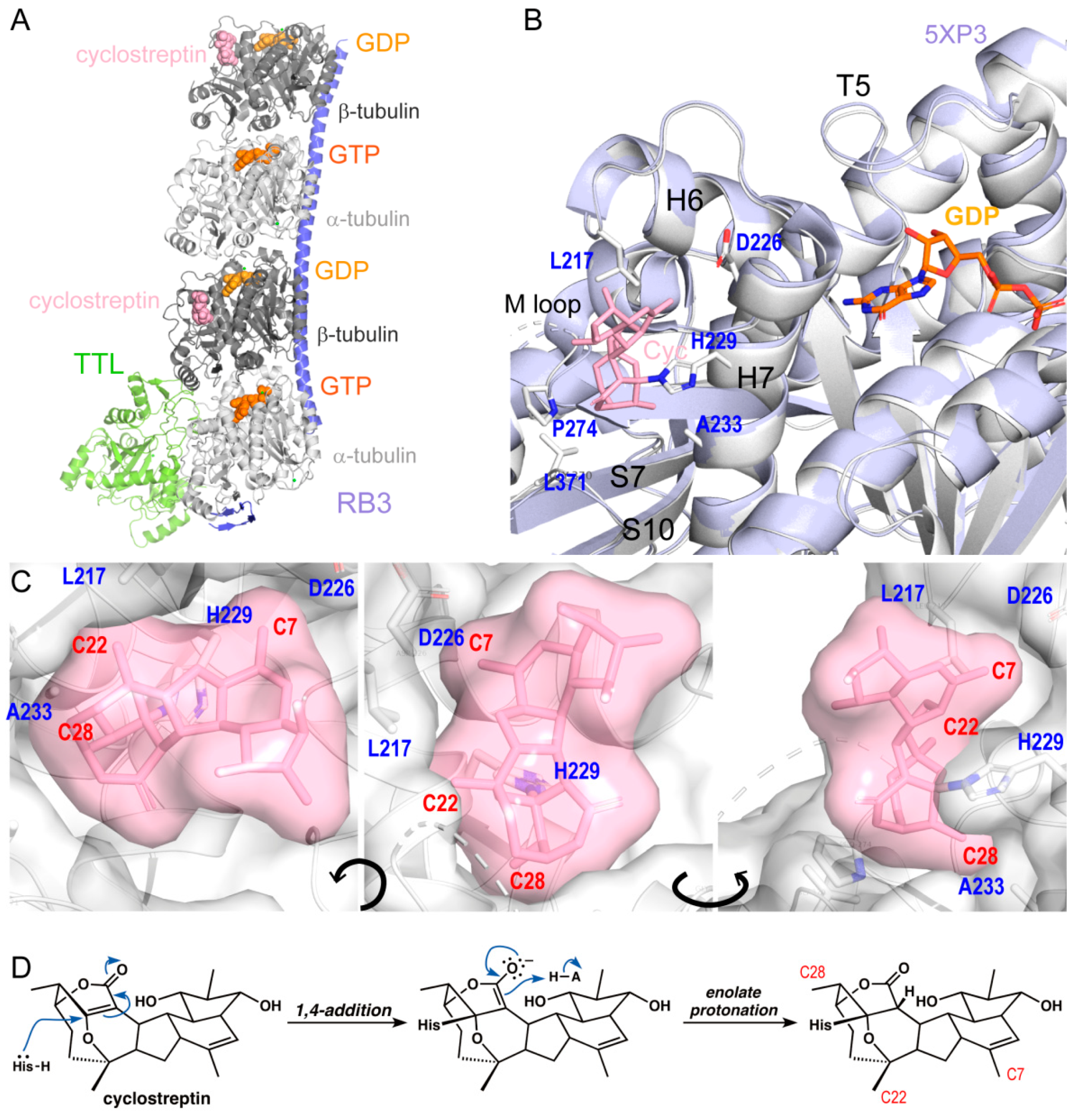
2.1. Cyclostreptin Covalently Binds to the Taxane-Site and Does Not Induce M-Loop Folding
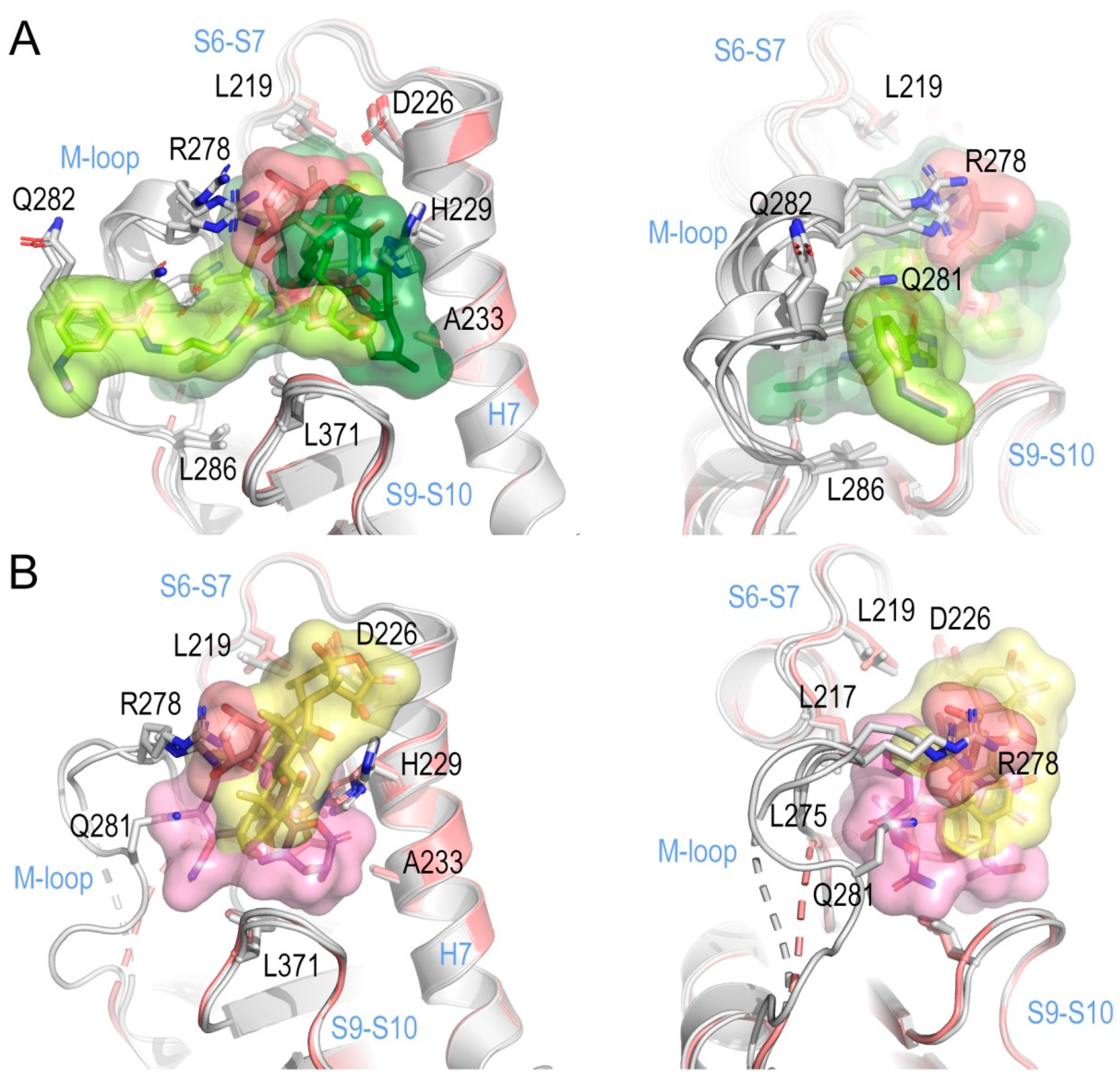
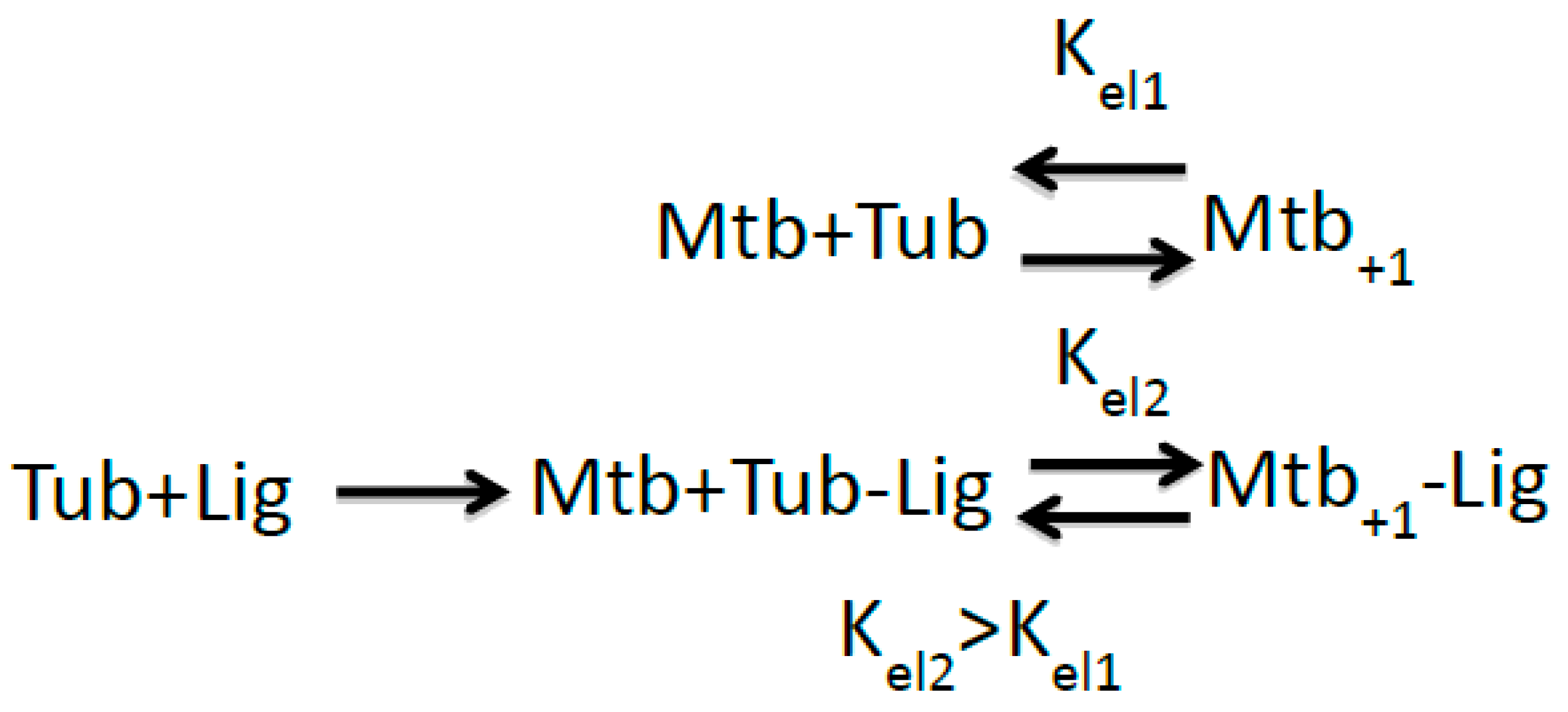
2.2. Implications of M-Loop Structuring by Taxane-Site Ligands
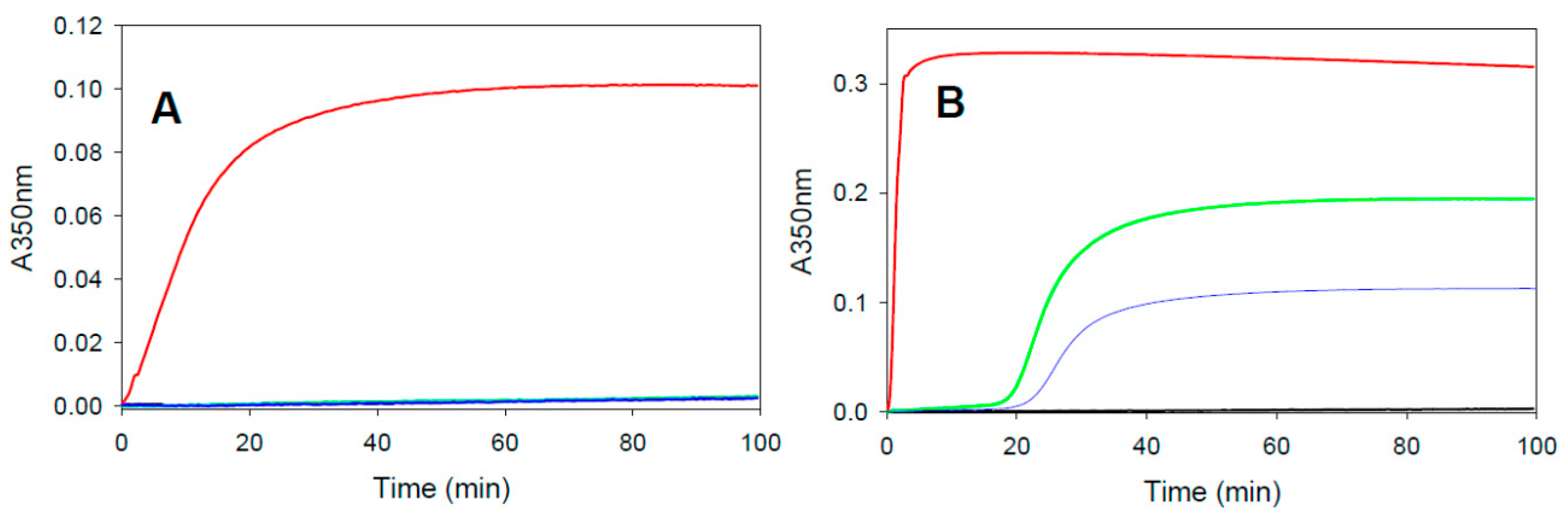
2.3. Assembly Promoting Activity of Covalent Taxane-Site Binding Agents
2.4. Potency of Covalent Binders in βIII-Tubulin Expressing Paclitaxel-Resistant Cells
3. Discussion
3.1. Cyclostreptin Interaction with βHis229 of the Taxane-Site Induces Tubulin Assembly Without Structuration of the M-Loop
3.2. Cytotoxicity of Tubulin Covalent Binders and Effect on Cancer Cell Resistance
4. Materials and Methods
4.1. Proteins and Chemicals
4.2. Cell Culture
4.3. Time Course of Binding of Cyclostreptin to Dimeric Tubulin
4.4. Tubulin Assembly in the Presence of the Drugs
4.5. Tubulin-Cyclostreptin Adduct Formation, Crystallization, Data Collection, and Structure Determination
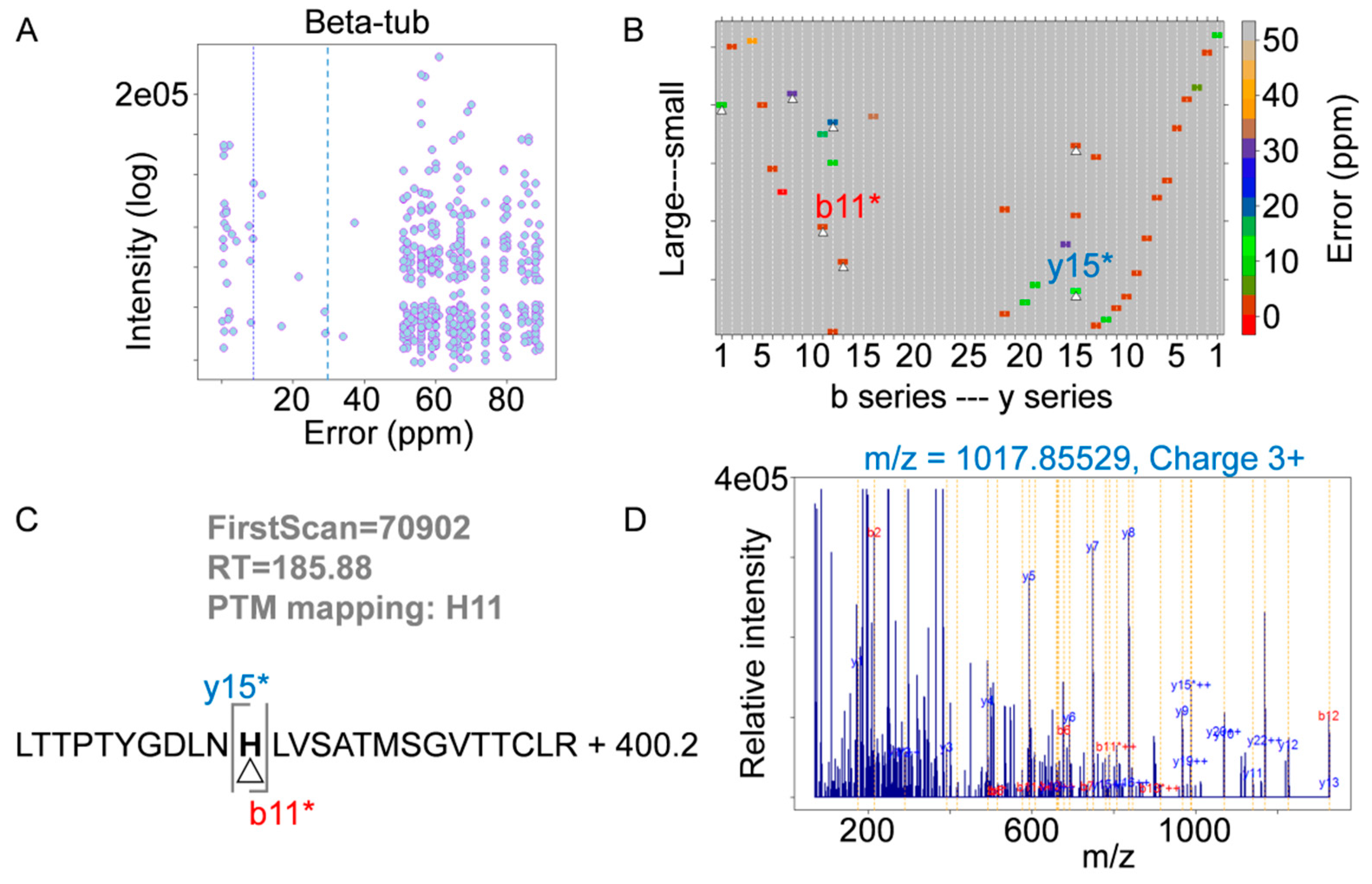
4.6. Mass Spectrometry Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Deposition
Abbreviations
| ADC | Antibody-drug conjugate |
| DMSO | Dimethylsulfoxide |
| DTT | Dithiothreitol |
| EDTA | thylene diamine tetraacetic acid |
| GDP | Guanosine diphosphate |
| GTP | Guanosine triphosphate |
| HCD | Higher-energy collisional dissociation |
| HPLC-MS | High performance liquid chromatography coupled to mass spectrometry |
| IC50 | Inhibitory concentration 50% |
| MSA | Microtubule stabilizing agent |
| RB3 | Rat brain 3 stathmin like domain |
| T2R | Complex of two α-β-tubulin heterodimers with a RB3 molecule |
| TTL | Tubulin tyrosin ligase |
References
- Dunphy, F.R.; Spitzer, G.; Buzdar, A.U.; Hortobagyi, G.N.; Horwitz, L.J.; Yau, J.C.; Spinolo, J.A.; Jagannath, S.; Holmes, F.; Wallerstein, R.O. Treatment of estrogen receptor-negative or hormonally refractory breast cancer with double high-dose chemotherapy intensification and bone marrow support. J. Clin. Oncol. 1990, 8, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Teply, B.A.; Wang, H.; Luber, B.; Sullivan, R.; Rifkind, I.; Bruns, A.; Spitz, A.; DeCarli, M.; Sinibaldi, V.; Pratz, C.F.; et al. Bipolar androgen therapy in men with metastatic castration-resistant prostate cancer after progression on enzalutamide: An open-label, phase 2, multicohort study. Lancet Oncol. 2018, 19, 76–86. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, H.; Viskov, C.; Garcia-Echeverria, C. Antibody-drug conjugates—A new wave of cancer drugs. Bioorg. Med. Chem. Lett. 2014, 24, 5357–5363. [Google Scholar] [CrossRef] [PubMed]
- Saville, M.W.; Lietzau, J.; Pluda, J.M.; Wilson, W.H.; Humphrey, R.W.; Feigel, E.; Steinberg, S.M.; Broder, S.; Yarchoan, R.; Odom, J.; et al. Treatment of hiv-associated kaposi’s sarcoma with paclitaxel. Lancet 1995, 346, 26–28. [Google Scholar] [CrossRef]
- Buey, R.M.; Calvo, E.; Barasoain, I.; Pineda, O.; Edler, M.C.; Matesanz, R.; Cerezo, G.; Vanderwal, C.D.; Day, B.W.; Sorensen, E.J.; et al. Cyclostreptin binds covalently to microtubule pores and lumenal taxoid binding sites. Nat. Chem. Biol. 2007, 3, 117–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Risinger, A.L.; Jackson, E.M.; Polin, L.A.; Helms, G.L.; LeBoeuf, D.A.; Joe, P.A.; Hopper-Borge, E.; Luduena, R.F.; Kruh, G.D.; Mooberry, S.L. The taccalonolides; microtubule stabilizers that circumvent clinically relevant taxane resistance mechanisms. Cancer Res. 2008, 68, 8881–8888. [Google Scholar] [CrossRef] [PubMed]
- Field, J.J.; Pera, B.; Calvo, E.; Canales, A.; Zurwerra, D.; Trigili, C.; Rodriguez-Salarichs, J.; Matesanz, R.; Kanakkanthara, A.; Wakefield, S.J.; et al. Zampanolide, a potent new microtubule-stabilizing agent, covalently reacts with the taxane luminal site in tubulin alpha,beta-heterodimers and microtubules. Chem. Biol. 2012, 19, 686–698. [Google Scholar] [CrossRef] [PubMed]
- Nogales, E.; Wolf, S.G.; Downing, K.H. Structure of the alpha beta tubulin dimer by electron crystallography. Nature 1998, 391, 199–203. [Google Scholar] [CrossRef]
- Prota, A.E.; Setter, J.; Waight, A.B.; Bargsten, K.; Murga, J.; Diaz, J.F.; Steinmetz, M.O. Pironetin binds covalently to αcys316 and perturbs a major loop and helix of α-tubulin to inhibit microtubule formation. J. Mol. Biol. 2016, 428, 2981–2988. [Google Scholar] [CrossRef]
- Yang, J.; Wang, Y.; Wang, T.; Jiang, J.; Botting, C.H.; Liu, H.; Chen, Q.; Yang, J.; Naismith, J.H.; Zhu, X.; et al. Pironetin reacts covalently with cysteine-316 of α-tubulin to destabilize microtubule. Nat. Commun. 2016, 7, 12103. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yu, Y.; Li, G.B.; Li, S.A.; Wu, C.; Gigant, B.; Qin, W.; Chen, H.; Wu, Y.; Chen, Q.; et al. Mechanism of microtubule stabilization by taccalonolide aj. Nat. Commun. 2017, 8, 15787. [Google Scholar] [CrossRef]
- Edler, M.C.; Buey, R.M.; Gussio, R.; Marcus, A.I.; Vanderwal, C.D.; Sorensen, E.J.; Diaz, J.F.; Giannakakou, P.; Hamel, E. Cyclostreptin (fr182877), an antitumor tubulin-polymerizing agent deficient in enhancing tubulin assembly despite its high affinity for the taxoid site. Biochemistry 2005, 44, 11525–11538. [Google Scholar] [CrossRef]
- Prota, A.E.; Bargsten, K.; Zurwerra, D.; Field, J.J.; Diaz, J.F.; Altmann, K.H.; Steinmetz, M.O. Molecular mechanism of action of microtubule-stabilizing anticancer agents. Science 2013, 339, 587–590. [Google Scholar] [CrossRef]
- Kellogg, E.H.; Hejab, N.M.A.; Howes, S.; Northcote, P.; Miller, J.H.; Diaz, J.F.; Downing, K.H.; Nogales, E. Insights into the distinct mechanisms of action of taxane and non-taxane microtubule stabilizers from cryo-em structures. J. Mol. Biol. 2017, 429, 633–646. [Google Scholar] [CrossRef]
- Trigili, C.; Barasoain, I.; Sánchez-Murcia, P.A.; Bargsten, K.; Redondo-Horcajo, M.; Nogales, A.; Gardner, N.M.; Meyer, A.; Naylor, G.J.; Gómez-Rubio, E.; et al. Structural determinants of the dictyostatin chemotype for tubulin binding affinity and antitumor activity against taxane- and epothilone-resistant cancer cells. ACS Omega 2016, 1, 1192–1204. [Google Scholar] [CrossRef]
- Prota, A.E.; Bargsten, K.; Redondo-Horcajo, M.; Smith, A.B., III; Yang, C.H.; McDaid, H.M.; Paterson, I.; Horwitz, S.B.; Fernando Diaz, J.; Steinmetz, M.O. Structural basis of microtubule stabilization by discodermolide. Chembiochem 2017, 18, 905–909. [Google Scholar] [CrossRef]
- Risinger, A.L.; Li, J.; Bennett, M.J.; Rohena, C.C.; Peng, J.; Schriemer, D.C.; Mooberry, S.L. Taccalonolide binding to tubulin imparts microtubule stability and potent in vivo activity. Cancer Res. 2013, 73, 6780–6792. [Google Scholar] [CrossRef] [Green Version]
- Calvo, E.; Barasoain, I.; Matesanz, R.; Pera, B.; Camafeita, E.; Pineda, O.; Hamel, E.; Vanderwal, C.D.; Andreu, J.M.; Lopez, J.A.; et al. Cyclostreptin derivatives specifically target cellular tubulin and further map the paclitaxel site. Biochemistry 2012, 51, 329–341. [Google Scholar] [CrossRef]
- Nawrotek, A.; Knossow, M.; Gigant, B. The determinants that govern microtubule assembly from the atomic structure of GTP-tubulin. J. Mol. Biol. 2011, 412, 35–42. [Google Scholar] [CrossRef]
- Nogales, E.; Whittaker, M.; Milligan, R.A.; Downing, K.H. High-resolution model of the microtubule. Cell 1999, 96, 79–88. [Google Scholar] [CrossRef]
- Canales, A.; Salarichs, J.R.; Trigili, C.; Nieto, L.; Coderch, C.; Andreu, J.M.; Paterson, I.; Jiménez-Barbero, J.; Díaz, J.F. Insights into the interaction of discodermolide and docetaxel with dimeric tubulin. Mapping the binding sites of microtubule-stabilizing agents using an integrated nmr and computational approach. ACS Chem. Biol. 2011, 6, 789–799. [Google Scholar] [CrossRef]
- Buey, R.M.; Barasoain, I.; Jackson, E.; Meyer, A.; Giannakakou, P.; Paterson, I.; Mooberry, S.; Andreu, J.M.; Díaz, J.F. Microtubule interactions with chemically diverse stabilizing agents: Thermodynamics of binding to the paclitaxel site predicts cytotoxicity. Chem. Biol. 2005, 12, 1269–1279. [Google Scholar] [CrossRef]
- Díaz, J.F.; Menéndez, M.; Andreu, J.M. Thermodynamics of ligand-induced assembly of tubulin. Biochemistry 1993, 32, 10067–10077. [Google Scholar] [CrossRef]
- Oosawa, F.; Asakura, S. Thermodynamics of the Polymerization of Protein; Academic Press: London, UK, 1975. [Google Scholar]
- Mozzetti, S.; Ferlini, C.; Concolino, P.; Filippetti, F.; Raspaglio, G.; Prislei, S.; Gallo, D.; Martinelli, E.; Ranelletti, F.O.; Ferrandina, G.; et al. Class iii beta-tubulin overexpression is a prominent mechanism of paclitaxel resistance in ovarian cancer patients. Clin. Cancer Res. 2005, 11, 298–305. [Google Scholar]
- Gan, P.; McCarroll, J.; Byrne, F.; Garner, J.; Kavallaris, M. Specific β-tubulin isotypes can functionally enhance or diminish epothilone b sensitivity in non-small cell lung cancer cells. PLoS ONE 2011, 6, e21717. [Google Scholar] [CrossRef]
- Kato, A.; Naiki-Ito, A.; Naitoh, I.; Hayashi, K.; Nakazawa, T.; Shimizu, S.; Nishi, Y.; Okumura, F.; Inoue, T.; Takada, H.; et al. The absence of class iii β-tubulin is predictive of a favorable response to nab-paclitaxel and gemcitabine in patients with unresectable pancreatic ductal adenocarcinoma. Hum. Pathol. 2018, 74, 92–98. [Google Scholar] [CrossRef]
- Buey, R.M.; Diaz, J.F.; Andreu, J.M.; O’Brate, A.; Giannakakou, P.; Nicolaou, K.C.; Sasmal, P.K.; Ritzen, A.; Namoto, K. Interaction of epothilone analogs with the paclitaxel binding site; relationship between binding affinity, microtubule stabilization, and cytotoxicity. Chem. Biol. 2004, 11, 225–236. [Google Scholar]
- Gupta, M.L., Jr.; Bode, C.J.; Georg, G.I.; Himes, R.H. Understanding tubulin-taxol interactions: Mutations that impart taxol binding to yeast tubulin. Proc. Natl. Acad. Sci. USA 2003, 100, 6394–6397. [Google Scholar] [CrossRef]
- Field, J.J.; Pera, B.; Gallego, J.E.; Calvo, E.; Rodriguez-Salarichs, J.; Saez-Calvo, G.; Zuwerra, D.; Jordi, M.; Andreu, J.M.; Prota, A.E.; et al. Zampanolide binding to tubulin indicates cross-talk of taxane site with colchicine and nucleotide sites. J. Nat. Prod. 2018, 81, 494–505. [Google Scholar] [CrossRef]
- Tan, B.; Piwnica-Worms, D.; Ratner, L. Multidrug resistance transporters and modulation. Curr. Opin. Oncol. 2000, 12, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, A.C.A.S.F.; Cloos, J.; Jansen, G.; Peters, G.J. How to overcome atp-binding cassette drug efflux transporter-mediated drug resistance? Cancer Drug Resist. 2018, 1, 6–29. [Google Scholar] [CrossRef]
- Kavallaris, M.; Burkhart, C.A.; Horwitz, S.B. Antisense oligonucleotides to class iii beta-tubulin sensitize drug-resistant cells to taxol. Br. J. Cancer 1999, 80, 1020–1025. [Google Scholar] [CrossRef] [PubMed]
- Kavallaris, M. Microtubules and resistance to tubulin-binding agents. Nat. Rev. Cancer 2010, 10, 194–204. [Google Scholar] [CrossRef]
- McCarroll, J.; Gan, P.; Liu, M.; Kavallaris, M. Betaiii-tubulin is a multifunctional protein involved in drug sensitivity and tumorigenesis in non-small cell lung cancer. Cancer Res. 2010, 70, 4995–5003. [Google Scholar] [CrossRef]
- Du, J.; Li, B.; Fang, Y.; Liu, Y.; Wang, Y.; Li, J.; Zhou, W.; Wang, X. Overexpression of class iii β-tubulin, sox2, and nuclear survivin is predictive of taxane resistance in patients with stage iii ovarian epithelial cancer. BMC Cancer 2015, 15, 536. [Google Scholar] [CrossRef]
- Marco, J.A.; Garcia-Pla, J.; Carda, M.; Murga, J.; Falomir, E.; Trigili, C.; Notararigo, S.; Diaz, J.F.; Barasoain, I. Design and synthesis of pironetin analogues with simplified structure and study of their interactions with microtubules. Eur. J. Med. Chem. 2011, 46, 1630–1637. [Google Scholar] [CrossRef] [Green Version]
- Cai, P.; Lu, P.; Sharom, F.J.; Fang, W.S. A semisynthetic taxane YG-3-46A effectively evades p-glycoprotein and B-III tubulin mediated tumor drug resistance in vitro. Cancer Lett. 2013, 341, 214–223. [Google Scholar] [CrossRef]
- St George, M.; Ayoub, A.T.; Banerjee, A.; Churchill, C.D.; Winter, P.; Klobukowski, M.; Cass, C.E.; Luduena, R.F.; Tuszynski, J.A.; Damaraju, S. Designing and testing of novel taxanes to probe the highly complex mechanisms by which taxanes bind to microtubules and cause cytotoxicity to cancer cells. PLoS ONE 2015, 10, e0129168. [Google Scholar] [CrossRef]
- Yeh, L.C.; Banerjee, A.; Prasad, V.; Tuszynski, J.A.; Weis, A.L.; Bakos, T.; Yeh, I.T.; Luduena, R.F.; Lee, J.C. Effect of CH-35, a novel anti-tumor colchicine analogue, on breast cancer cells overexpressing the betaiii isotype of tubulin. Investig. New Drugs 2016, 34, 129–137. [Google Scholar] [CrossRef]
- Raspaglio, G.; Filippetti, F.; Prislei, S.; Penci, R.; De Maria, I.; Cicchillitti, L.; Mozzetti, S.; Scambia, G.; Ferlini, C. Hypoxia induces class III β-tubulin gene expression by hif-1α binding to its 3′ flanking region. Gene 2008, 409, 100–108. [Google Scholar] [CrossRef]
- Cicchillitti, L.; Penci, R.; Di Michele, M.; Filippetti, F.; Rotilio, D.; Donati, M.B.; Scambia, G.; Ferlini, C. Proteomic characterization of cytoskeletal and mitochondrial class III β-tubulin. Mol. Cancer Ther. 2008, 7, 2070–2079. [Google Scholar] [CrossRef]
- Katsetos, C.D.; Dráberová, E.; Legido, A.; Dumontet, C.; Dráber, P. Tubulin targets in the pathobiology and therapy of glioblastoma multiforme. I. Class III β-tubulin. J. Cell. Physiol. 2009, 221, 505–513. [Google Scholar] [CrossRef]
- Kamath, K.; Wilson, L.; Cabral, F.; Jordan, M.A. ΒIII-tubulin induces paclitaxel resistance in association with reduced effects on microtubule dynamic instability. J. Biol. Chem. 2005, 280, 12902–12907. [Google Scholar] [CrossRef]
- Field, J.; Northcote, P.; Paterson, I.; Altmann, K.-H.; Díaz, J.; Miller, J. Zampanolide, a microtubule-stabilizing agent, is active in resistant cancer cells and inhibits cell migration. Int. J. Mol. Sci. 2017, 18, 971. [Google Scholar] [CrossRef]
- Diaz, J.F.; Strobe, R.; Engelborghs, Y.; Souto, A.A.; Andreu, J.M. Molecular recognition of taxol by microtubules. Kinetics and thermodynamics of binding of fluorescent taxol derivatives to an exposed site. J. Biol. Chem. 2000, 275, 26265–26276. [Google Scholar] [CrossRef]
- Diaz, J.F.; Barasoain, I.; Andreu, J.M. Fast kinetics of taxol binding to microtubules. Effects of solution variables and microtubule-associated proteins. J. Biol. Chem. 2003, 278, 8407–8419. [Google Scholar] [CrossRef]
- Diaz, J.F.; Barasoain, I.; Souto, A.A.; Amat-Guerri, F.; Andreu, J.M. Macromolecular accessibility of fluorescent taxoids bound at a paclitaxel binding site in the microtubule surface. J. Biol. Chem. 2005, 280, 3928–3937. [Google Scholar] [CrossRef]
- Diaz, J.F.; Buey, R.M. Characterizing ligand-microtubule binding by competition methods. Methods Mol. Med. 2007, 137, 245–260. [Google Scholar]
- Hari, M.; Yang, H.; Zeng, C.; Canizales, M.; Cabral, F. Expression of class III β-tubulin reduces microtubule assembly and confers resistance to paclitaxel. Cell Motil. Cytoskelet. 2003, 56, 45–56. [Google Scholar] [CrossRef]
- Díaz, J.F.; Andreu, J.M. Assembly of purified gdp-tubulin into microtubules induced by taxol and taxotere: Reversibility, ligand stoichiometry, and competition. Biochemistry 1993, 32, 2747–2755. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, R.B.; Gigant, B.; Curmi, P.A.; Jourdain, I.; Lachkar, S.; Sobel, A.; Knossow, M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 2004, 428, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Prota, A.E.; Magiera, M.M.; Kuijpers, M.; Bargsten, K.; Frey, D.; Wieser, M.; Jaussi, R.; Hoogenraad, C.C.; Kammerer, R.A.; Janke, C.; et al. Structural basis of tubulin tyrosination by tubulin tyrosine ligase. J. Cell. Biol. 2013, 200, 259–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanderwal, C.D.; Vosburg, D.A.; Weiler, S.; Sorensen, E.J. An enantioselective synthesis of Fr182877 provides a chemical rationalization of its structure and affords multigram quantities of its direct precursor. J. Am. Chem. Soc. 2003, 125, 5393–5407. [Google Scholar] [CrossRef]
- Joe, P.A.; Banerjee, A.; Luduena, R.F. The roles of cys124 and ser239 in the functional properties of human βIII tubulin. Cell. Motil. Cytoskelet. 2008, 65, 476–486. [Google Scholar] [CrossRef]
- Matesanz, R.; Trigili, C.; Rodriguez-Salarichs, J.; Zanardi, I.; Pera, B.; Nogales, A.; Fang, W.S.; Jimenez-Barbero, J.; Canales, A.; Barasoain, I.; et al. Taxanes with high potency inducing tubulin assembly overcome tumoural cell resistances. Bioorg. Med. Chem. 2014, 22, 5078–5090. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Rodriguez-Salarichs, J.; Zhao, Y.; Cai, P.; Estevez-Gallego, J.; Balaguer-Perez, F.; Redondo Horcajo, M.; Lucena-Agell, D.; Barasoain, I.; Diaz, J.F.; et al. Modification of C-seco taxoids through ring tethering and substituent replacement leading to effective agents against tumor drug resistance mediated by βIII-tubulin and P-glycoprotein (P-gp) overexpressions. Eur. J. Med. Chem. 2017, 137, 488–503. [Google Scholar] [CrossRef]
- Saez-Calvo, G.; Sharma, A.; Balaguer, F.A.; Barasoain, I.; Rodriguez-Salarichs, J.; Olieric, N.; Munoz-Hernandez, H.; Berbis, M.A.; Wendeborn, S.; Penalva, M.A.; et al. Triazolopyrimidines are microtubule-stabilizing agents that bind the vinca inhibitor site of tubulin. Cell. Chem. Biol. 2017, 24, 737–750.e6. [Google Scholar] [CrossRef]
- Prota, A.E.; Bargsten, K.; Northcote, P.T.; Marsh, M.; Altmann, K.H.; Miller, J.H.; Diaz, J.F.; Steinmetz, M.O. Structural basis of microtubule stabilization by laulimalide and peloruside A. Angew. Chem. Int. Ed. Engl. 2014, 53, 1621–1625. [Google Scholar] [CrossRef]
- Prota, A.E.; Danel, F.; Bachmann, F.; Bargsten, K.; Buey, R.M.; Pohlmann, J.; Reinelt, S.; Lane, H.; Steinmetz, M.O. The novel microtubule-destabilizing drug bal27862 binds to the colchicine site of tubulin with distinct effects on microtubule organization. J. Mol. Biol. 2014, 426, 1848–1860. [Google Scholar] [CrossRef]
- Kabsch, W. Xds. Acta Crystallogr. Sect. D 2010, 66, 125–132. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. Phenix: A comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Cogliati, S.; Calvo, E.; Loureiro, M.; Guaras, A.M.; Nieto-Arellano, R.; Garcia-Poyatos, C.; Ezkurdia, I.; Mercader, N.; Vázquez, J.; Enriquez, J.A. Mechanism of super-assembly of respiratory complexes III and IV. Nature 2016, 539, 579. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
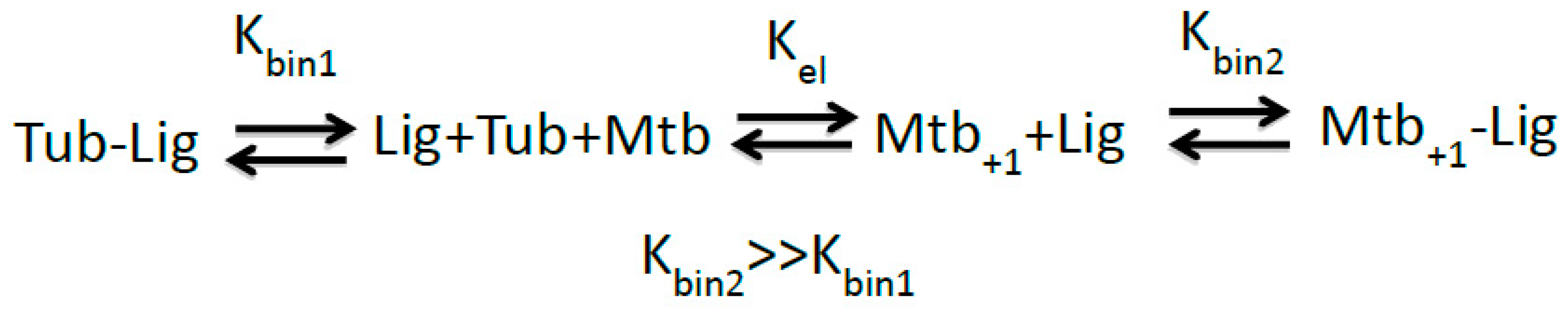{kind=link}
{kind=link}
| Compound | HeLa IC50 (nM) | HeLa βIII IC50 (nM) | R/S Ratio 2 |
|---|---|---|---|
| Zampanolide | 0.045 ± 0.007 | 0.22 ± 0.06 | 4.9 |
| Cyclostreptin | 19.3 ± 0.3 | 53.8 ± 4.3 | 2.8 |
| Taccalonolide AJ | 6.2 ± 0.3 | 9.6 ± 1.2 | 1.6 |
| Pironetin | 6.9 ± 1.6 | 3.9 ± 0.5 | 0.6 |
| Paclitaxel | 1.6 ± 0.3 | 25.7 ± 0.1 | 16.6 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balaguer, F.d.A.; Mühlethaler, T.; Estévez-Gallego, J.; Calvo, E.; Giménez-Abián, J.F.; Risinger, A.L.; Sorensen, E.J.; Vanderwal, C.D.; Altmann, K.-H.; Mooberry, S.L.; et al. Crystal Structure of the Cyclostreptin-Tubulin Adduct: Implications for Tubulin Activation by Taxane-Site Ligands. Int. J. Mol. Sci. 2019, 20, 1392. https://doi.org/10.3390/ijms20061392
Balaguer FdA, Mühlethaler T, Estévez-Gallego J, Calvo E, Giménez-Abián JF, Risinger AL, Sorensen EJ, Vanderwal CD, Altmann K-H, Mooberry SL, et al. Crystal Structure of the Cyclostreptin-Tubulin Adduct: Implications for Tubulin Activation by Taxane-Site Ligands. International Journal of Molecular Sciences. 2019; 20(6):1392. https://doi.org/10.3390/ijms20061392
Chicago/Turabian StyleBalaguer, Francisco de Asís, Tobias Mühlethaler, Juan Estévez-Gallego, Enrique Calvo, Juan Francisco Giménez-Abián, April L. Risinger, Erik J. Sorensen, Christopher D. Vanderwal, Karl-Heinz Altmann, Susan L. Mooberry, and et al. 2019. "Crystal Structure of the Cyclostreptin-Tubulin Adduct: Implications for Tubulin Activation by Taxane-Site Ligands" International Journal of Molecular Sciences 20, no. 6: 1392. https://doi.org/10.3390/ijms20061392