Depletion of Gram-Positive Bacteria Impacts Hepatic Biological Functions During the Light Phase

 , ,
, ,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
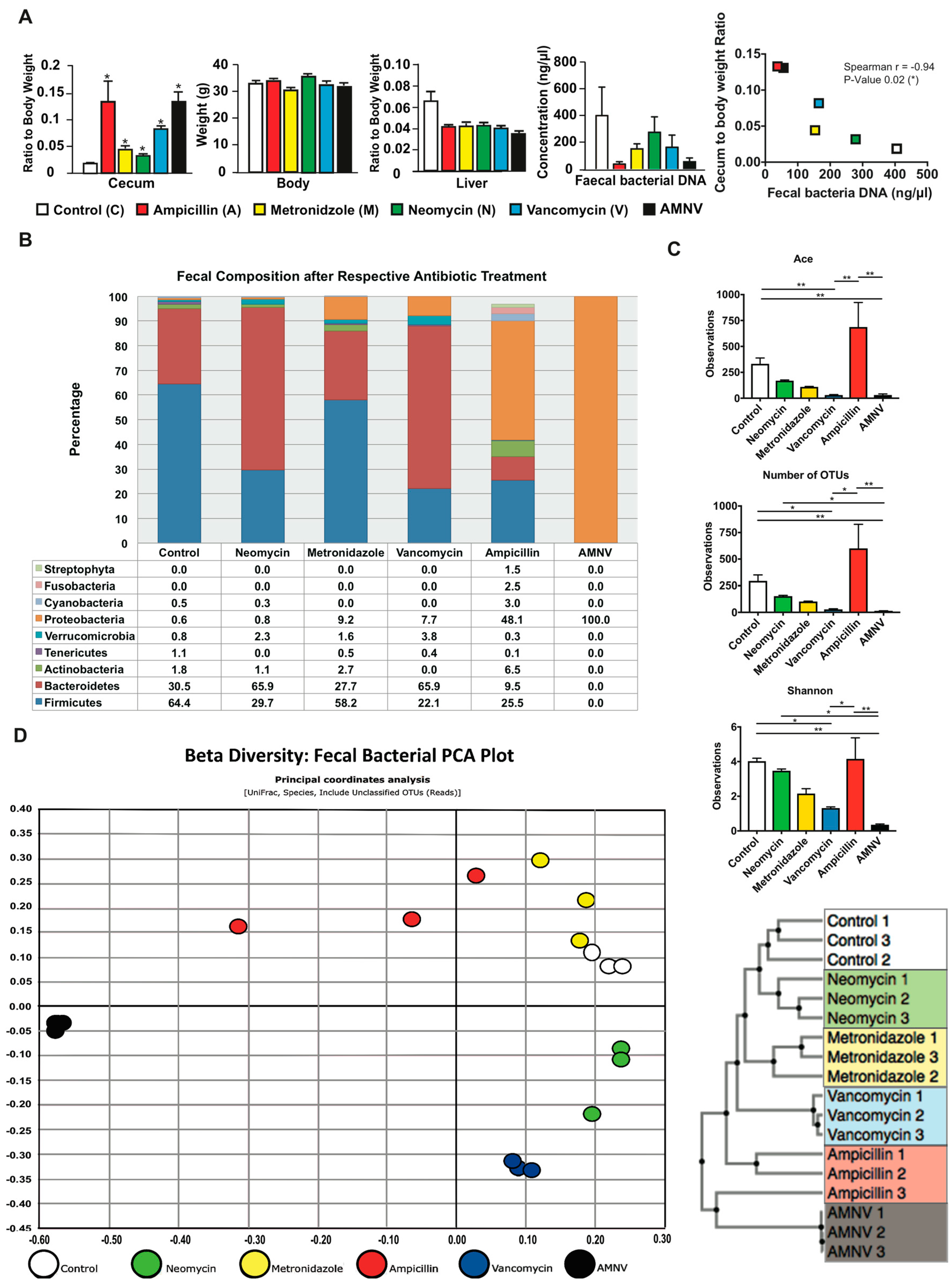
2.1. Faecal Microbiota Differs between Mice with Different Antibiotic Treatments
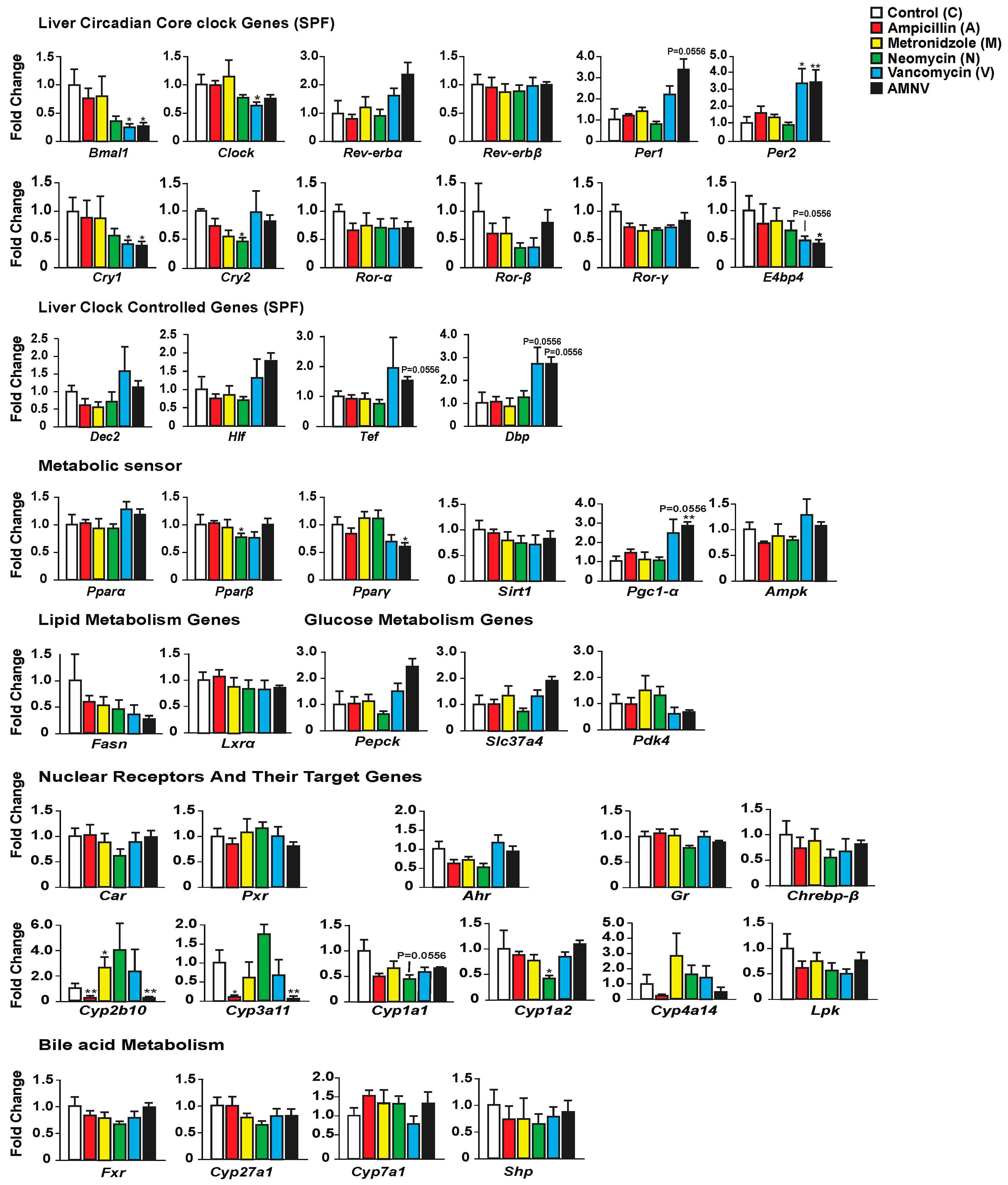
2.2. Effect of Antibiotic Treatments on Hepatic Clock, Metabolism and Detoxification Gene Expression
2.3. Antibiotic-Associated Daily Rhythm Disruption during Light Phase in SPF Mice is Mediated by the Gut Microbiota
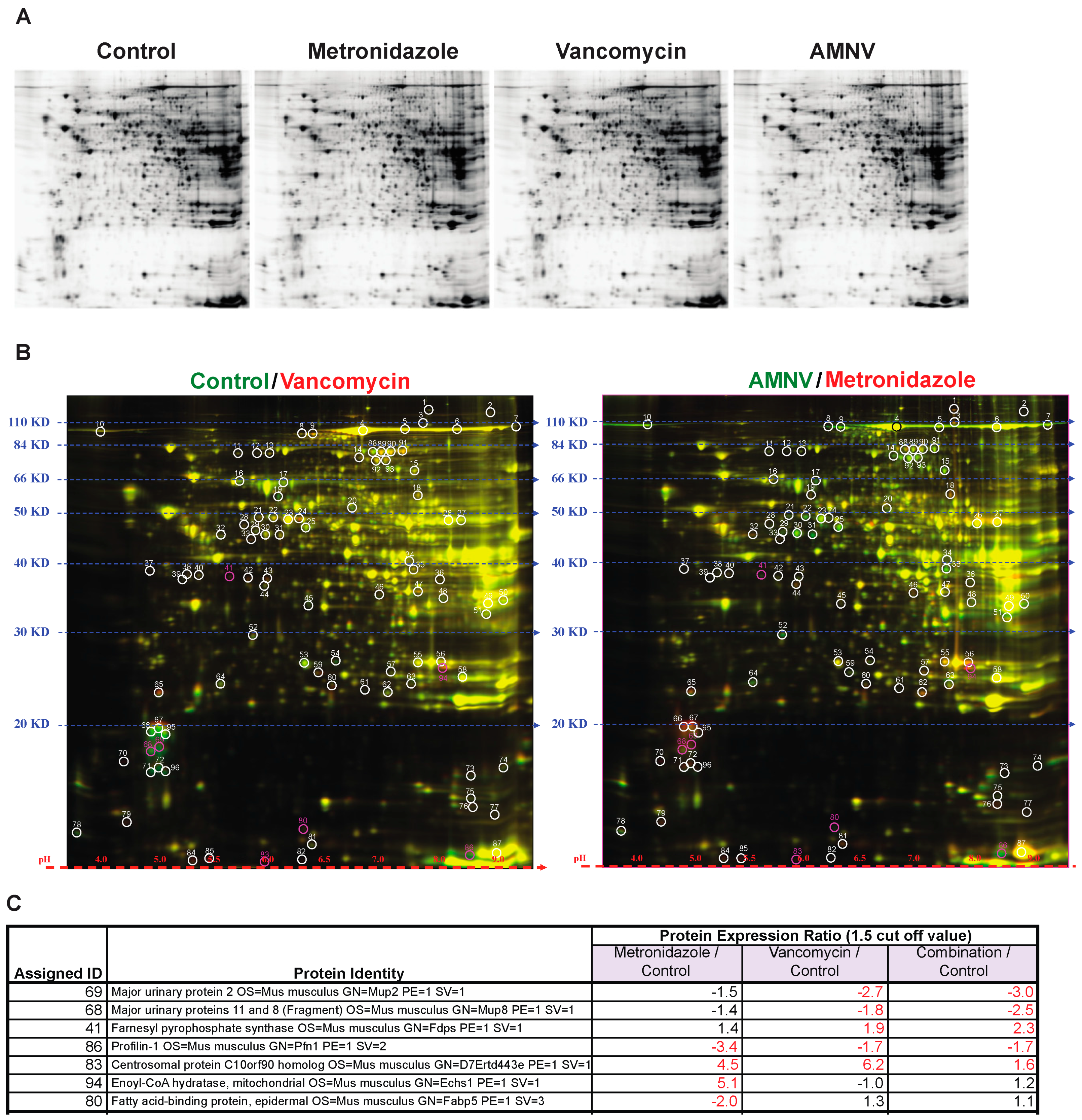
2.4. Antibiotics-Induced Differential Changes in the Post-Transcriptional Regulation of Circadian and Metabolic Genes
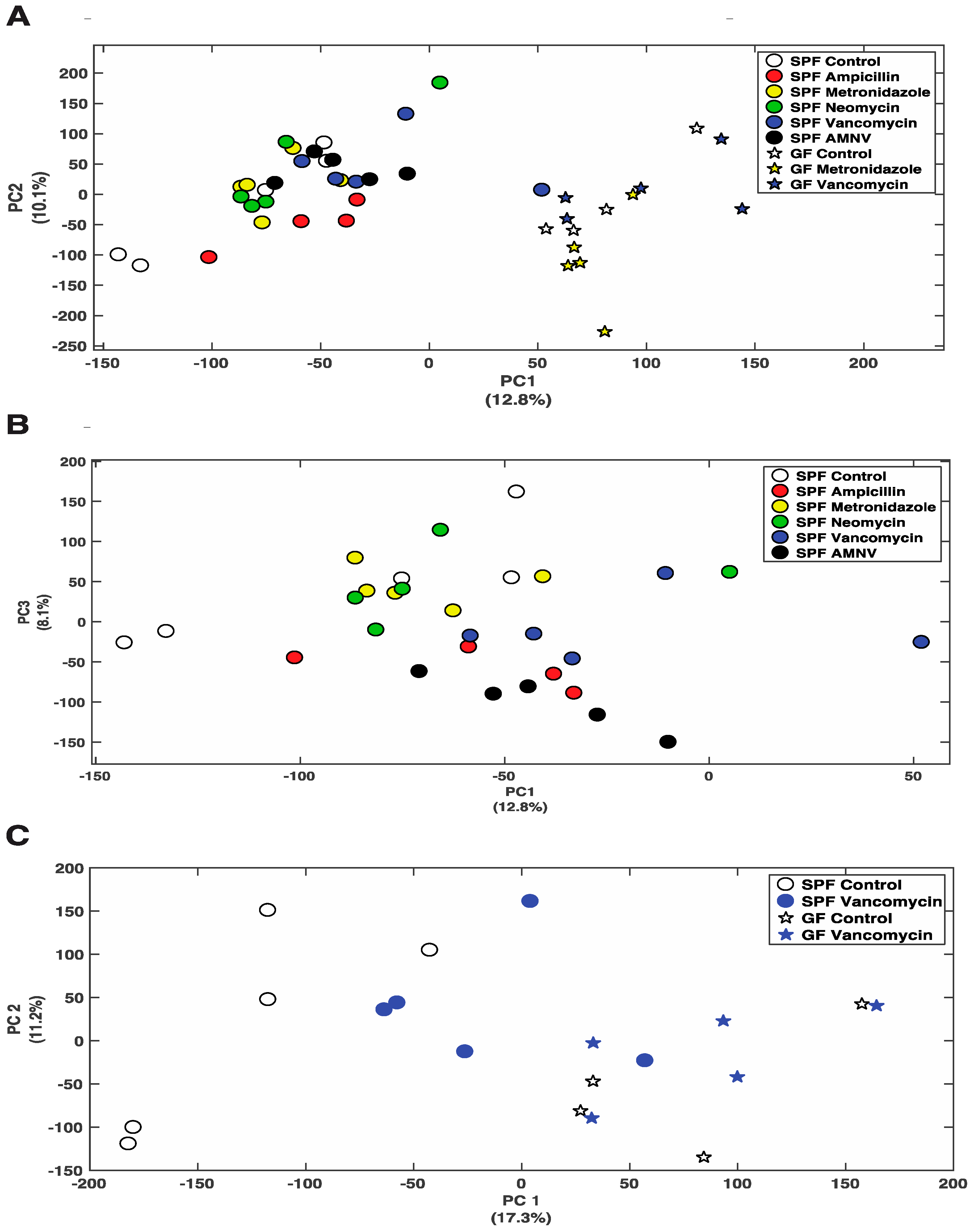
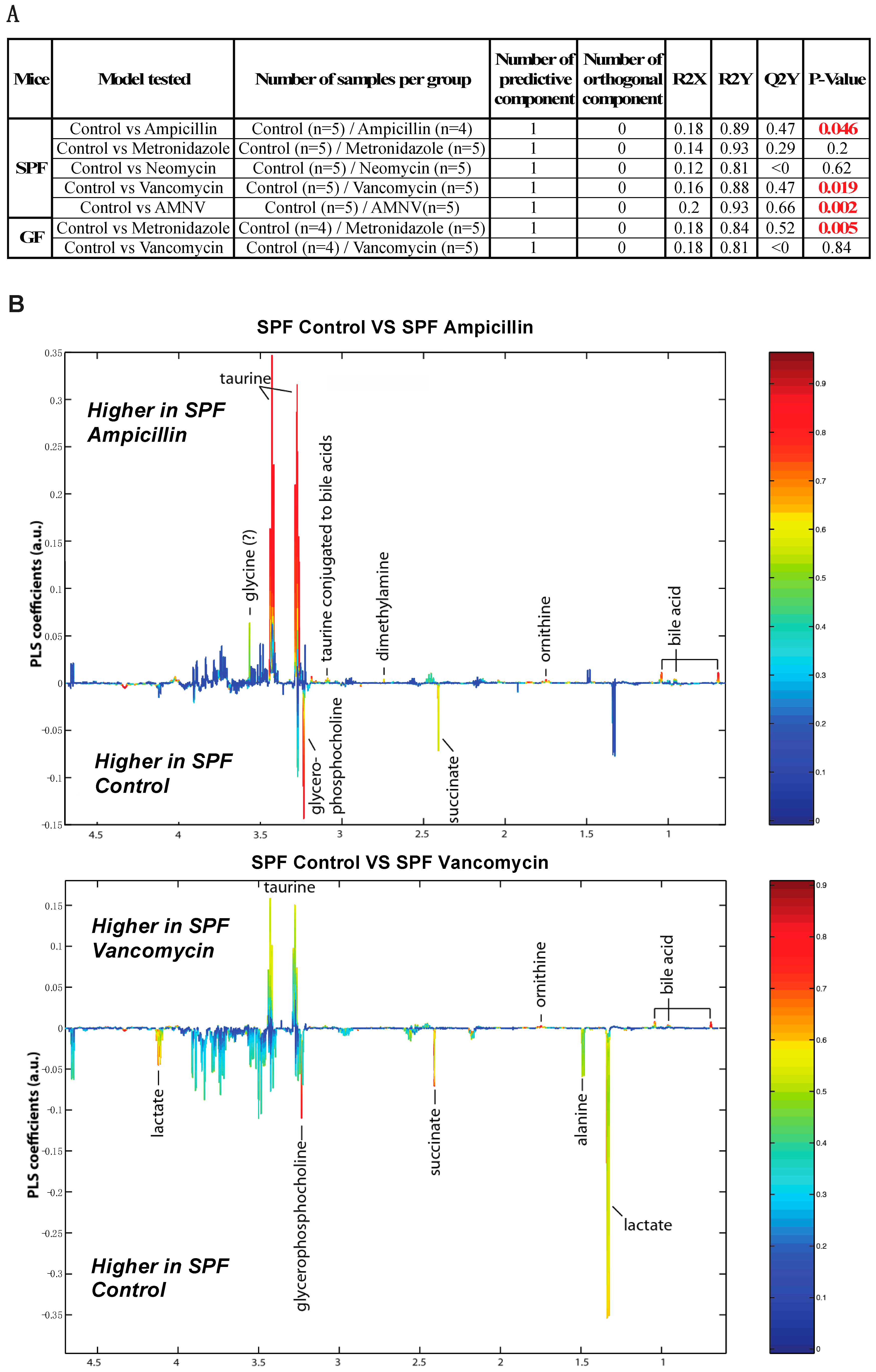
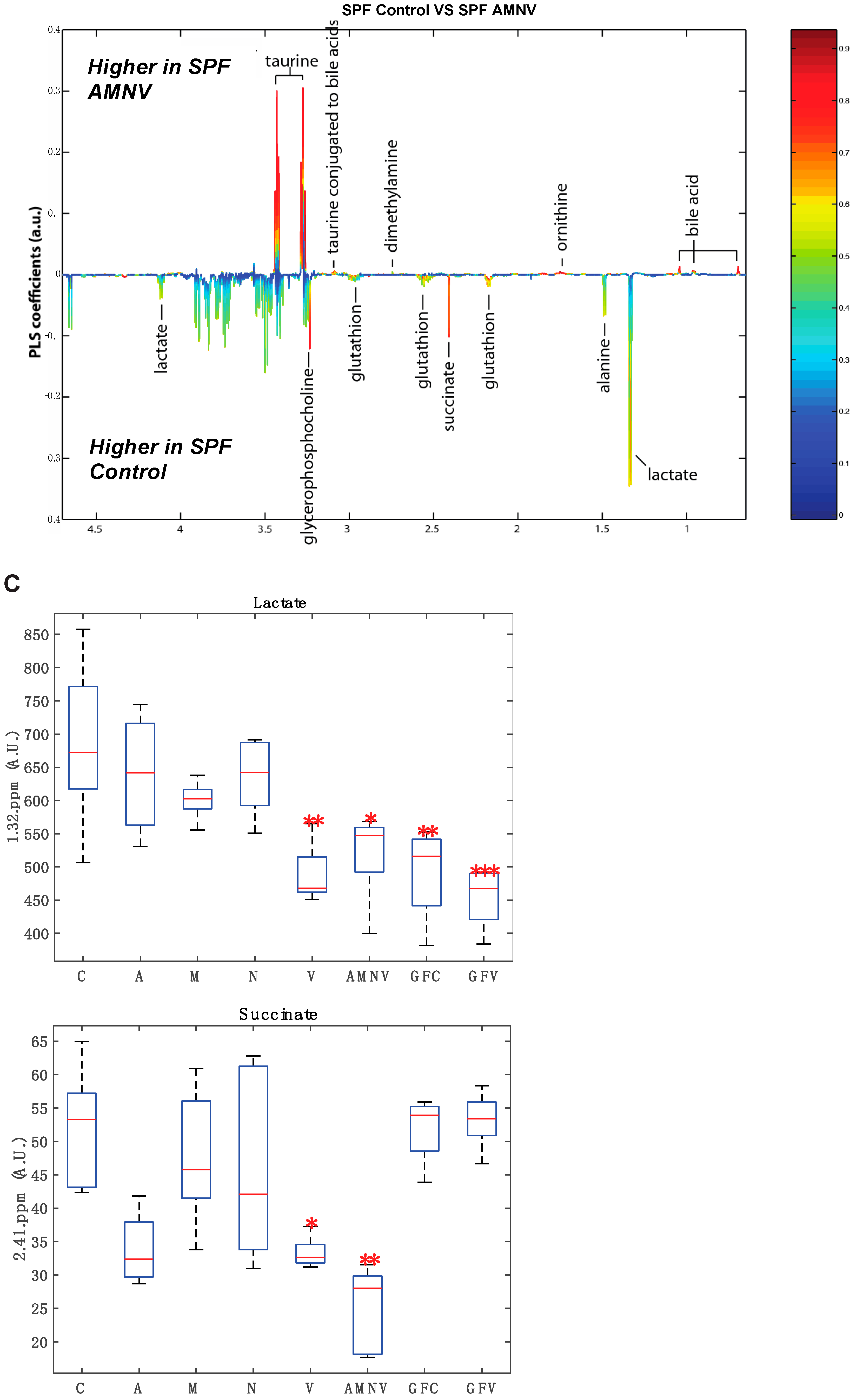
2.5. Antibiotics Cause Changes in Liver Metabolite Profile
3. Discussion
3.1. Selective Reduction in Microbiota by Antibiotics
3.2. Vancomycin Treatment-Induced Liver Clock Perturbation at ZT4–6 is due to Dysbiosis
3.3. Microbiota Protects against Metronidazole-Induced Circadian Perturbation
3.4. Visceral Adiposity Observed upon Neomycin Treatment
3.5. Regulation of Liver Regeneration by the Gut Microbiota
4. Materials and Methods
4.1. Animal Care and Experimental Procedures
4.2. Animals
4.3. Antibiotic Treatments
4.4. Faecal DNA Analysis
4.5. Quantitative Reverse Transcriptase Polymerase Chain Reaction (RT-qPCR)
4.6. Statistical Analysis
4.7. D-DIGE
4.8. Protein Identification by Mass Spectrometry
4.9. H-NMR Based Metabolomics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2-D DIGE | 2-Dimensional Fluorescence Difference Gel Electrophoresis |
| ACE | Abundance-based Coverage Estimator |
| AHR | Aryl Hydrocarbon Receptor |
| AMPK | AMP-activated Protein Kinase |
| BMAL1 | Brain and Muscle Arnt-like Protein-1 |
| CAR | Constitutive Androstane Receptor |
| CHAPS | 3-[(3-Cholamidopropyl)Dimethylammonio]-1-Propanesulfonate Hydrate |
| CHREBP | Carbohydrate-Responsive Element-Binding Protein |
| CLOCK | Circadian Locomotor Output Cycles Kaput |
| CRY | Cryptochrome |
| COSY | homonuclear Correlation Spectroscopy |
| CYPs | Cytochrome P450s |
| DBP | Albumin Site D-Binding Protein |
| DEC2 | Differentiated Embryonic Chondrocyte 2 |
| E4BP4 | Nuclear factor, Interleukin 3 Regulated |
| ECHS | Enoyl-CoA Hydratase |
| FABP5 | Fatty Acid Binding Protein 5 |
| FASN | Fatty Acid Synthase |
| FATS | Fragile-site Associated Tumour Suppressor |
| FPPS | Farnesyl Pyrophosphate Synthase |
| FXR | Farnesoid X Receptor |
| GAPDH | Glyceraldehyde-3-Phosphate Dehydrogenase |
| GF | Germ-Free |
| GR | Glucocorticoid Receptor |
| HLF | Hepatic Leukemia Factor |
| HSQC | Heteronuclear Single Quantum Correlation Spectroscopy |
| IACUC | Institutional Animal Care and Use Committee |
| IEF | Isoelectric Focusing |
| IPG | Immobilized pH Gradient |
| LPK | Liver Pyruvate Kinase |
| LXRα | Liver X Receptor Alpha |
| MALDI-TOF MS | Matrix-Assisted Laser Desorption Ionization-Time of Flight Mass Spectrometry |
| MTP | Microbiome Taxonomic Profiling |
| MUPs | Major Urinary Proteins |
| NMR | Nuclear Magnetic Resonance |
| O-PLS-DA | Orthogonal Projections to Latent Structures Discriminant Analysis |
| OTUs | Operational Taxonomic Units |
| PCA | Principal Component Analysis |
| PCoA | Principal Coordinate Analysis |
| PDK4 | Pyruvate Dehydrogenase Kinase 4 |
| PER | Period |
| PEPCK | Phosphoenolpyruvate Carboxykinase |
| PFN1 | Profilin-1 |
| PGC-1α | PPARγ-Coactivator-1 Alpha |
| PPARs | Peroxisome Proliferator-Activated Receptors |
| PXR | Pregnane X Receptor (NR1I2) |
| q-PCR | Quantitative Polymerase Chain Reaction |
| RT-qPCR | Quantitative reverse transcriptase polymerase chain reaction |
| RORs | Retinoid-related Orphan Receptors (NR1F) |
| REV-ERB | Rev-Erb receptors (NR1D) |
| SDS | Sodium Dodecyl Sulfate |
| SDS-PAGE | Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis |
| SEMC | Singhealth Experimental Medicine Centre |
| SHP | Small heterodimer partner |
| SIRT1 | Sirtuin 1 |
| SLC | Solute Carrier Transporter |
| SPF | Specific-pathogen Free |
| TEF | Thyrotroph embryonic factor |
| Tris-HCl | Tris-hydrochloride |
| TOCSY | Total Correlation Spectroscopy |
| TSP | Trimethylsilylpropanoic acid |
| UPGMA | Unweighted Pair Group Method with Arithmetic Mean |
| ZT | Zeitgeber Time |
References
- Margulis, L.; Fester, R. Symbiosis as a Source of Evolutionary Innovation; Margulis, L., Fester, R., Eds.; MIT Press: Cambridge, MA, USA; London, UK, 1991. [Google Scholar]
- Schnorr, S.L.; Sankaranarayanan, K.; Lewis, C.M., Jr.; Warinner, C. Insights into Human Evolution from Ancient and Contemporary Microbiome Studies. Curr. Opin. Genet. Dev. 2016, 41, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A Human Gut Microbial Gene Catalogue Established by Metagenomic Sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the Gut Microbiota in Disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [CrossRef] [PubMed]
- Eckel-Mahan, K.; Sassone-Corsi, P. Metabolism and the Circadian Clock Converge. Physiol. Rev. 2013, 93, 107–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flint, H.J.; Bayer, E.A.; Rincon, M.T.; Lamed, R.; White, B.A. Polysaccharide Utilization by Gut Bacteria: Potential for New Insights from Genomic Analysis. Nat. Rev. Microbiol. 2008, 6, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.A.; Velazquez, K.T.; Herbert, K.M. Influence of High-Fat-Diet on Gut Microbiota: A Driving Force for Chronic Disease Risk. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 515–520. [Google Scholar] [CrossRef]
- Sherman, H.; Genzer, Y.; Cohen, R.; Chapnik, N.; Madar, Z.; Froy, O. Timed High-Fat Diet Resets Circadian Metabolism and Prevents Obesity. FASEB J. 2012, 3493–3502. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-Gut Microbiota Metabolic Interactions. Science 2012, 108, 1262–1268. [Google Scholar] [CrossRef]
- Tremaroli, V.; Bäckhed, F. Functional Interactions between the Gut Microbiota and Host Metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef]
- Damiola, F.; Minh, N.L.; Preitner, N.; Fleury-olela, F. Restricted Feeding Uncouples Circadian Oscillators in Peripheral Tissues from the Central Pacemaker in the Suprachiasmatic Nucleus. Genes Dev. 2000, 14, 2950–2961. [Google Scholar] [CrossRef]
- Stokkan, K.; Yamazaki, S.; Tei, H.; Sakaki, Y.; Menaker, M. Entrainment of the Circadian Clock in the Liver by Feeding. Science 2001, 291, 490–493. [Google Scholar] [CrossRef] [PubMed]
- Antunes, L.C.; Levandovski, R.; Dantas, G.; Caumo, W.; Hidalgo, M.P. Obesity and Shift Work: Chronobiological Aspects. Nutr. Res. Rev. 2010, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Duszka, K.; Wahli, W. Enteric microbiota–gut–brain axis from the perspective of nuclear receptors. Int. J. Mol. Sci. 2018, 19, 2210. [Google Scholar] [CrossRef] [PubMed]
- Mukherji, A.; Kobiita, A.; Ye, T.; Chambon, P. Homeostasis in Intestinal Epithelium Is Orchestrated by the Circadian Clock and Microbiota Cues Transduced by TLRs. Cell 2013, 153, 812–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reikvam, D.H.; Erofeev, A.; Sandvik, A.; Grcic, V.; Jahnsen, F.L.; Gaustad, P.; McCoy, K.D.; Macpherson, A.J.; Meza-Zepeda, L.A.; Johansen, F.E. Depletion of Murine Intestinal Microbiota: Effects on Gut Mucosa and Epithelial Gene Expression. PLoS ONE 2011, 6, e17996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montagner, A.; Korecka, A.; Polizzi, A.; Lippi, Y.; Blum, Y.; Canlet, C.; Tremblay-Franco, M.; Gautier-Stein, A.; Burcelin, R.; Yen, Y.-C.; et al. Hepatic Circadian Clock Oscillators and Nuclear Receptors Integrate Microbiome-Derived Signals. Sci. Rep. 2016, 6, 20127. [Google Scholar] [CrossRef] [PubMed]
- Russell, W.S.; Rene, D.; Costello, R. Association of Germfree Mice with Bacteria. J. Exp. Med. 1965, 122, 77–83. [Google Scholar]
- Claus, S.P.; Ellero, S.L.; Berger, B.; Krause, L.; Bruttin, A.; Molina, J.; Paris, A.; Want, E.J.; De Waziers, I.; Cloarec, O.; et al. Colonization-Induced Host-Gut Microbial Metabolic Interaction. mBio 2011, 2, 1–8. [Google Scholar] [CrossRef]
- Ley, R.E.; Backhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity Alters Gut Microbial Ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef]
- Ubeda, C.; Taur, Y.; Jenq, R.R.; Equinda, M.J.; Son, T.; Samstein, M.; Viale, A.; Socci, N.D.; Van Den Brink, M.R.M.; Kamboj, M.; et al. Vancomycin-Resistant Enterococcus Domination of Intestinal Microbiota Is Enabled by Antibiotic Treatment in Mice and Precedes Bloodstream Invasion in Humans. J. Clin. Investig. 2010, 120. [Google Scholar] [CrossRef]
- Wallace, B.D.; Redinbo, M.R. Xenobiotic-Sensing Nuclear Receptors Involved in Drug Metabolism: A Structural Perspective. Drug Metab Rev. 2016, 45, 79–100. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Jiang, J.C.; Guo, L.; Hu, S.J. Establishment of Transgenic Mice Carrying the Gene for Farnesyl Pyrophosphate Synthase. Biotechnol. Lett. 2011, 33, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Gnocchi, D.; Pedrelli, M.; Hurt-Camejo, E.; Parini, P. Lipids around the Clock: Focus on Circadian Rhythms and Lipid Metabolism. Biology 2015, 4, 104–132. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kuang, Z.; Yu, X.; Ruhn, K.A.; Kubo, M.; Hooper, L.V. The Intestinal Microbiota Regulates Body Composition through NFIL3 and the Circadian Clock. Science 2017, 916, 912–916. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An Obesity-Associated Gut Microbiome with Increased Capacity for Energy Harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Korpela, K.; Salonen, A.; Virta, L.J.; Kekkonen, R.A.; Forslund, K.; Bork, P.; de Vos, W.M. Intestinal Microbiome Is Related to Lifetime Antibiotic Use in Finnish Pre-School Children. Nat. Commun. 2016, 7, 10410. [Google Scholar] [CrossRef] [PubMed]
- Grasa, L.; Abecia, L.; Forcén, R.; Castro, M.; de Jalón, J.A.G.; Latorre, E.; Alcalde, A.I.; Murillo, M.D. Antibiotic-Induced Depletion of Murine Microbiota Induces Mild Inflammation and Changes in Toll-Like Receptor Patterns and Intestinal Motility. Microb. Ecol. 2015, 70, 835–848. [Google Scholar] [CrossRef]
- Leone, V.; Gibbons, S.M.; Martinez, K.; Hutchison, A.L.; Edmond, Y.; Cham, C.M.; Pierre, J.F.; Heneghan, A.F.; Hubert, N.; Zale, E.; et al. Effects of Diurnal Variation of Gut Microbes and High Fat Feeding on Host Circadian Clock Function and Metabolism Vanessa. Cell Host Microbe 2015, 17, 681–689. [Google Scholar] [CrossRef]
- Montagner, A.; Polizzi, A.; Fouché, E.; Ducheix, S.; Lippi, Y.; Lasserre, F.; Barquissau, V.; Régnier, M.; Lukowicz, C.; Benhamed, F.; et al. Liver PPARα Is Crucial for Whole-Body Fatty Acid Homeostasis and Is Protective against NAFLD. Gut 2016, 65, 1202–1214. [Google Scholar] [CrossRef]
- Rooks, M.G.; Veiga, P.; Wardwell-Scott, L.H.; Tickle, T.; Segata, N.; Michaud, M.; Gallini, C.A.; Beal, C.; van Hylckama-Vlieg, J.E.; Ballal, S.A.; et al. Gut Microbiome Composition and Function in Experimental Colitis during Active Disease and Treatment-Induced Remission. ISME J. 2014, 8, 1403–1417. [Google Scholar] [CrossRef]
- Ellekilde, M.; Selfjord, E.; Larsen, C.S.; Jakesevic, M.; Rune, I.; Tranberg, B.; Vogensen, F.K.; Nielsen, D.S.; Bahl, M.I.; Licht, T.R.; et al. Transfer of Gut Microbiota from Lean and Obese Mice to Antibiotic-Treated Mice. Sci. Rep. 2014, 4, 5922. [Google Scholar] [CrossRef] [PubMed]
- Shin, N.; Whon, T.W.; Bae, J. Proteobacteria: Microbial Signature of Dysbiosis in Gut Microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Thaiss, C.A.; Levy, M.; Korem, T.; Dohnalová, L.; Shapiro, H.; Jaitin, D.A.; David, E.; Winter, D.R.; Gury-Benari, M.; Tatirovsky, E.; et al. Microbiota Diurnal Rhythmicity Programs Host Transcriptome Oscillations. Cell 2016, 167. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.Y.; Sun, W.W.; Ma, Y.; Zhu, H.; Yang, P.; Wei, H.; Zeng, B.H.; Zhang, Q.; Liu, Y.; Li, W.X.; et al. Microbiota Prevents Cholesterol Loss from the Body by Regulating Host Gene Expression in Mice. Sci. Rep. 2015, 5, 10512. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, S.; Liu, T.; Borjigin, J.; Lin, J.D. Transcriptional Coactivator PGC-1α Integrates the Mammalian Clock and Energy Metabolism. Nature 2007, 447, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Tung, Y.T.; Chen, Y.J.; Chuang, H.L.; Huang, W.C.; Lo, C.T.; Liao, C.C.; Huang, C.C. Characterization of the Serum and Liver Proteomes in Gut-Microbiota-Lacking Mice. Int. J. Med. Sci. 2017, 14, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Proia, P.; Di Liegro, C.M.; Schiera, G.; Fricano, A.; Di Liegro, I. Lactate as a Metabolite and a Regulator in the Central Nervous System. Int. J. Mol. Sci. 2016, 17, 1450. [Google Scholar] [CrossRef]
- Tretter, L.; Patocs, A.; Chinopoulos, C. Succinate, an Intermediate in Metabolism, Signal Transduction, ROS, Hypoxia, and Tumorigenesis. Biochim. Biophys. Acta Bioenerg. 2016, 1857, 1086–1101. [Google Scholar] [CrossRef]
- Sharrow, S.D.; Vaughn, J.L.; Žídek, L.; Novotny, M.V.; Stone, M.J.; Zídek, L.; Novotny, M.V.; Stone, M.J. Pheromone Binding by Polymorphic Mouse Major Urinary Proteins. Protein Sci. 2002, 11, 2247–2256. [Google Scholar] [CrossRef]
- Roberts, S.A.; Simpson, D.M.; Armstrong, S.D.; Davidson, A.J.; Robertson, D.H.; Mclean, L.; Beynon, R.J.; Hurst, J.L. Darcin: A Male Pheromone That Stimulates Female Memory and Sexual Attraction to an Individual Male’s Odour. BMC Biol. 2010, 464, 75. [Google Scholar] [CrossRef]
- Zhou, Y.; Rui, L. Major Urinary Protein Regulation of Chemical Communication and Nutrient Metabolism. Vitam. Horm. 2010, 100, 151–163. [Google Scholar] [CrossRef]
- Wu, Y.; Dong, Y.; Atefi, M.; Liu, Y.; Elshimali, Y.; Vadgama, J.V. Lactate, a Neglected Factor for Diabetes and Cancer Interaction. Mediat. Inflamm. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- DeBruyne, J.P.; Weaver, D.R.; Dallmann, R. The Hepatic Circadian Clock Modulates Xenobiotic Metabolism in Mice. J. Biol. Rhythms 2014, 29, 277–287. [Google Scholar] [CrossRef] [Green Version]
- Pascussi, J.M.; Gerbal-Chaloin, S.; Drocourt, L.; Maurel, P.; Vilarem, M.J. The Expression of CYP2B6, CYP2C9 and CYP3A4 Genes: A Tangle of Networks of Nuclear and Steroid Receptors. Biochim. Biophys. Acta Gen. Subj. 2003, 1619, 243–253. [Google Scholar] [CrossRef]
- Lembergers, T.; Staels, B.; Saladin, R.; Desvergne, B.; Auwerx, J.; Wahli, W. Regulation of the Peroxisome Proliferator-Activated Receptor Gene by Glucocorticoids. J. Biol. Chem. 1994, 269, 24527–24530. [Google Scholar]
- Rando, G.; Tan, C.K.; Khaled, N.; Montagner, A.; Leuenberger, N.; Bertrand-Michel, J.; Paramalingam, E.; Guillou, H.; Wahli, W. Glucocorticoid Receptor-PPARα Axis in Fetal Mouse Liver Prepares Neonates for Milk Lipid Catabolism. eLife 2016, 5, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Selwyn, F.P.; Cui, J.Y.; Klaassen, C.D. Special Section on Drug Metabolism and the Microbiome RNA-Seq Quantification of Hepatic Drug Processing Genes in Germ-Free Mice. Drug Metab. Dispos. 2015, 43, 1572–1580. [Google Scholar] [CrossRef] [PubMed]
- Alomar, M.J. Factors Affecting the Development of Adverse Drug Reactions (Review Article). Saudi Pharm. J. 2014, 22, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Duncan, S.H.; Lobley, G.E.; Holtrop, G.; Ince, J.; Johnstone, A.M.; Louis, P.; Flint, H.J. Human Colonic Microbiota Associated with Diet, Obesity and Weight Loss. Int. J. Obes. (Lond.) 2008, 1720–1724. [Google Scholar] [CrossRef]
- Chakraborti, C.K. New-Found Link between Microbiota and Obesity. World J. Gastrointest. Pathophysiol. 2015, 6, 110–119. [Google Scholar] [CrossRef]
- Després, J.P. Body Fat Distribution and Risk of Cardiovascular Disease: An Update. Circulation 2012, 126, 1301–1313. [Google Scholar] [CrossRef] [PubMed]
- Serino, M.; Luche, E.; Gres, S.; Baylac, A.; Bergé, M.; Cenac, C.; Waget, A.; Klopp, P.; Iacovoni, J.; Klopp, C.; et al. Metabolic Adaptation to a High-Fat Diet Is Associated with a Change in the Gut Microbiota. Gut 2012, 61, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Bae, Y.H.; Roy, P. Molecular Insights on Context-Specific Role of Profilin-1 in Cell Migration. Cell Adhes. Migr. 2012, 6, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Keiji, H.; Yamashiro, K.; Hiranuma, K.; Shibata, K.; Higashi, K.; Morita, T.; Hirano, H. Increase of Hepatic MRNAs of Profilin, Actin and Extracellular Matrix Proteins After Carbon Tetrachloride Treatment and Partial Hepatectomy in Rats. Bichem. Biophys. Res. Commun. 1994, 198, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Qiu, L.; Ma, K.; Zhang, X.; Zhao, Y.; Zhang, J.; Li, X.; Hao, X.; Li, Z. FATS is an E2-Independent Ubiquitin Ligase That Stabilizes P53 and Promotes Its Activation in Response to DNA Damage. Oncogene 2014, 33, 5424–5433. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Q.; Mao, J.-H.; Weise, A.; Mrasek, K.; Fan, X.; Zhang, X.; Liehr, T.; Lu, K.H.; Balmain, A.; et al. An HDAC1-Binding Domain within FATS Bridges P21 Turnover to Radiation-Induced Tumorigenesis. Oncogene 2010, 29, 2659–2671. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-X.; Keane, R.; Sheng, L.; Wan, Y.-J.Y. Implications of Microbiota and Bile Acid in Liver Injury and Regeneration. J. Hepatol. 2016, 63, 1502–1510. [Google Scholar] [CrossRef] [PubMed]
- Sinturel, F.; Gerber, A.; Mauvoisin, D.; Wang, J.; Gatfield, D.; Stubblefield, J.J.; Green, C.B.; Gachon, F.; Schibler, U. Diurnal Oscillations in Liver Mass and Cell Size Accompany Ribosome Assembly Cycles. Cell 2017, 169, 651–663.E14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manickam, R.; Oh, H.; Tan, C.; Paramalingam, E.; Wahli, W. Metronidazole Causes Skeletal Muscle Atrophy and Modulates Muscle Chronometabolism. Int. J. Mol. Sci. 2018, 19, 2418. [Google Scholar] [CrossRef]
- Mao, H.; Feng, X.Z.; Guang, S.H. Treating Liver Cancer with Antibiotics? Acta Pharmacol. Sin. 2013, 34, 989–990. [Google Scholar] [CrossRef]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-Induced Gut Microbial Metabolite Promotes Liver Cancer through Senescence Secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef]
- Aryal, R.P.; Kwak, P.B.; Tamayo, A.G.; Gebert, M.; Chiu, P.-L.; Walz, T.; Weitz, C.J. Macromolecular Assemblies of the Mammalian Circadian Clock. Mol. Cell 2017, 67, 770–782.e6. [Google Scholar] [CrossRef] [PubMed]
- Morgun, A.; Dzutsev, A.; Dong, X.; Greer, R.L.; Sexton, D.J.; Ravel, J.; Schuster, M.; Hsiao, W.; Matzinger, P.; Shulzhenko, N. Uncovering Effects of Antibiotics on the Host and Microbiota Using Transkingdom Gene Networks. Gut 2015, 64, 732–743. [Google Scholar] [CrossRef] [PubMed]
- Rakoff-Nahoum, S.; Paglino, J.; Eslami-Varzaneh, F.; Edberg, S.; Medzhitov, R. Recognition of Commensal Microflora by Toll-like Receptors Is Required for Intestinal Homeostasis. Cell 2004, 118, 229–241. [Google Scholar] [CrossRef]
- Croswell, A.; Amir, E.; Teggatz, P.; Barman, M.; Salzman, N.H. Prolonged Impact of Antibiotics on Intestinal Microbial Ecology and Susceptibility to Enteric Salmonella Infection. Infect. Immun. 2009, 77, 2741–2753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, J.; Kim, K.Y.; Lee, J.-H.; Choi, Y. The Analysis of Oral Microbial Communities of Wild-Type and Toll-like Receptor 2-Deficient Mice Using a 454 GS FLX Titanium Pyrosequencer. BMC Microbiol. 2010, 10, 101. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A Taxonomically United Database of 16S RRNA Gene Sequences and Whole-Genome Assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef]
- Beckonert, O.; Keun, H.C.; Ebbels, T.M.D.; Bundy, J.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. Metabolic Profiling, Metabolomic and Metabonomic Procedures for NMR Spectroscopy of Urine, Plasma, Serum and Tissue Extracts. Nat. Protoc. 2007, 2, 2692–2703. [Google Scholar] [CrossRef]
- Dieterle, F.; Ross, A.; Schlotterbeck, G.; Senn, H. Probabilistic Quotient Normalization as Robust Method to Account for Dilution of Complex Biological Mixtures. Application In 1H NMR Metabonomics. Anal. Chem. 2006, 78, 4281–4290. [Google Scholar] [CrossRef]
- Veselkov, K.A.; Lindon, J.C.; Ebbels, T.M.D.; Crockford, D.; Volynkin, V.V.; Holmes, E.; Davies, D.B.; Nicholson, J.K. Recursive Segment-Wise Peak Alignment of Biological H NMR Spectra for Improved Metabolic Biomarker Recovery Recursive Segment-Wise Peak Alignment of Biological 1 H NMR Spectra for Improved Metabolic Biomarker Recovery. Anal. Chem 2009, 81, 56–66. [Google Scholar] [CrossRef]
- Cloarec, O.; Dumas, M.E.; Craig, A.; Barton, R.H.; Trygg, J.; Hudson, J.; Blancher, C.; Gauguier, D.; Lindon, J.C.; Holmes, E.; et al. Statistical Total Correlation Spectroscopy: An Exploratory Approach for Latent Biomarker Identification from Metabolic 1H NMR Data Sets. Anal. Chem. 2005, 77, 1282–1289. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, H.Y.P.; Ellero-Simatos, S.; Manickam, R.; Tan, N.S.; Guillou, H.; Wahli, W. Depletion of Gram-Positive Bacteria Impacts Hepatic Biological Functions During the Light Phase. Int. J. Mol. Sci. 2019, 20, 812. https://doi.org/10.3390/ijms20040812
Oh HYP, Ellero-Simatos S, Manickam R, Tan NS, Guillou H, Wahli W. Depletion of Gram-Positive Bacteria Impacts Hepatic Biological Functions During the Light Phase. International Journal of Molecular Sciences. 2019; 20(4):812. https://doi.org/10.3390/ijms20040812
Chicago/Turabian StyleOh, Hui Yun Penny, Sandrine Ellero-Simatos, Ravikumar Manickam, Nguan Soon Tan, Hervé Guillou, and Walter Wahli. 2019. "Depletion of Gram-Positive Bacteria Impacts Hepatic Biological Functions During the Light Phase" International Journal of Molecular Sciences 20, no. 4: 812. https://doi.org/10.3390/ijms20040812