Different Signaling Pathways Define Different Interferon-Stimulated Gene Expression during Mycobacteria Infection in Macrophages

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
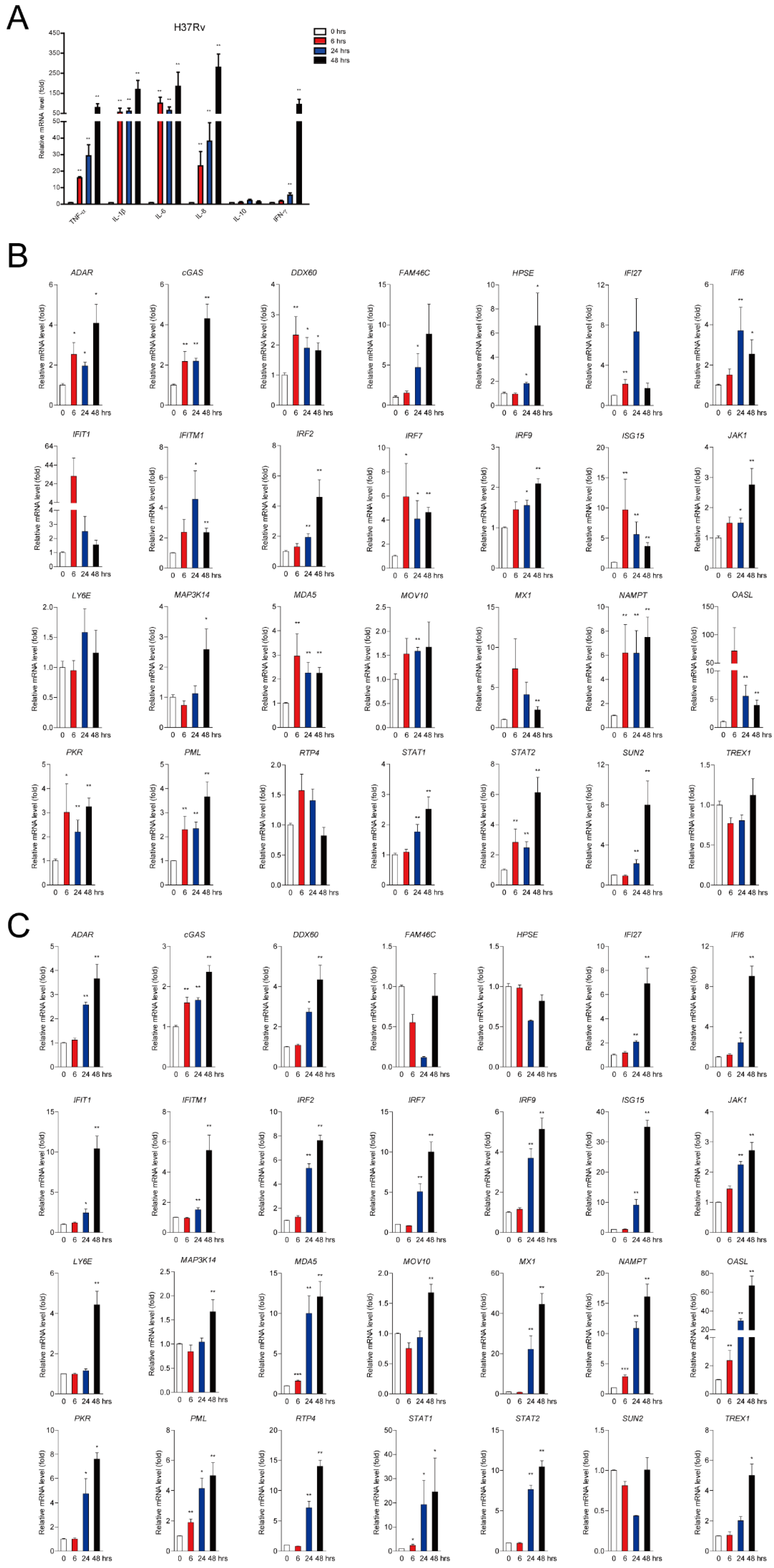
2.1. Mtb Infection Facilitates a Subset of ISG Production in Mϕs
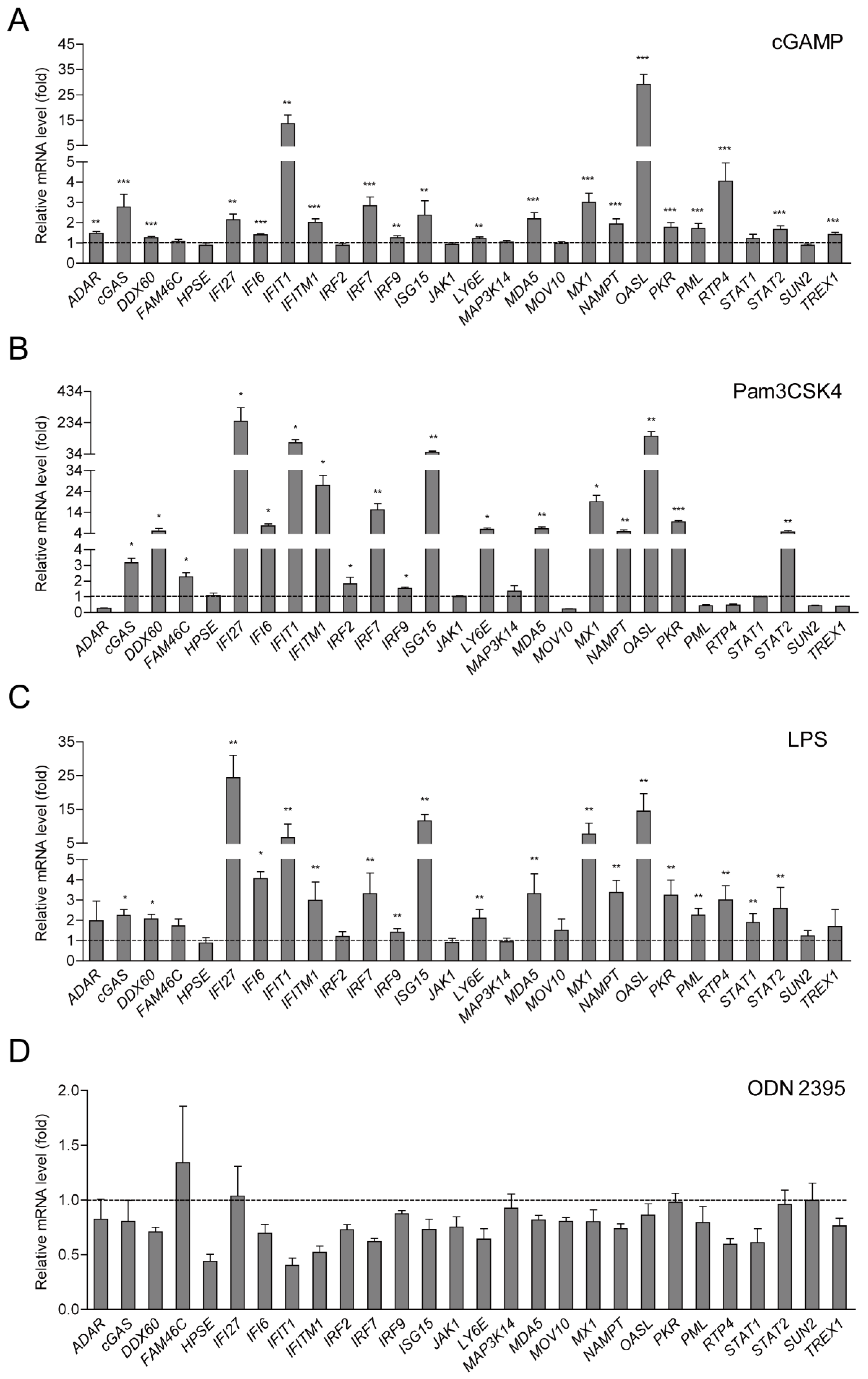
2.2. ISG Production is Dependent on cGAS-STING and TLR-2/4 Signaling Pathways
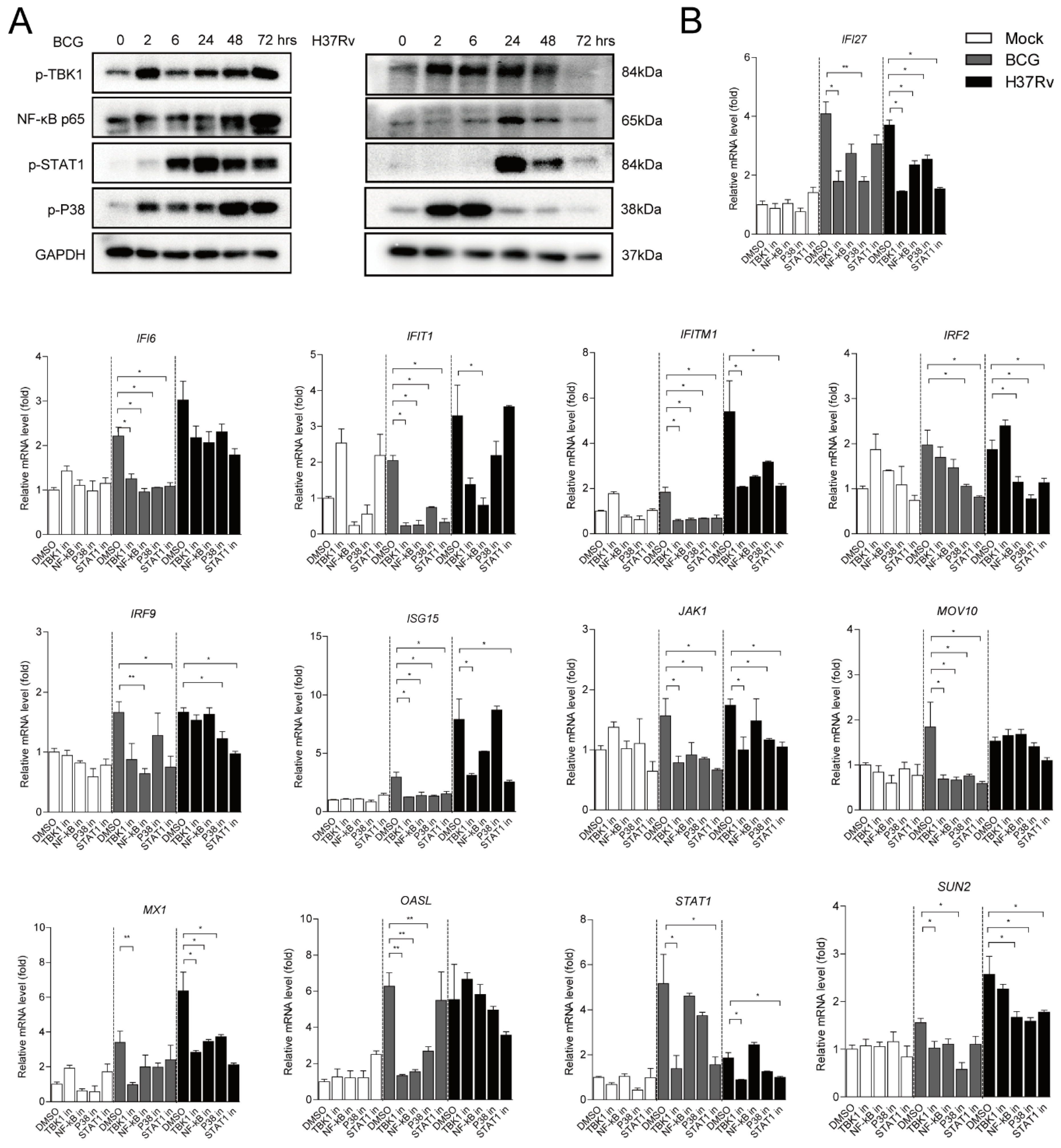
2.3. Different ISGs Are Diversely Induced by TBK1, NF-κB, MAPK and JAK-STAT Signaling Pathways
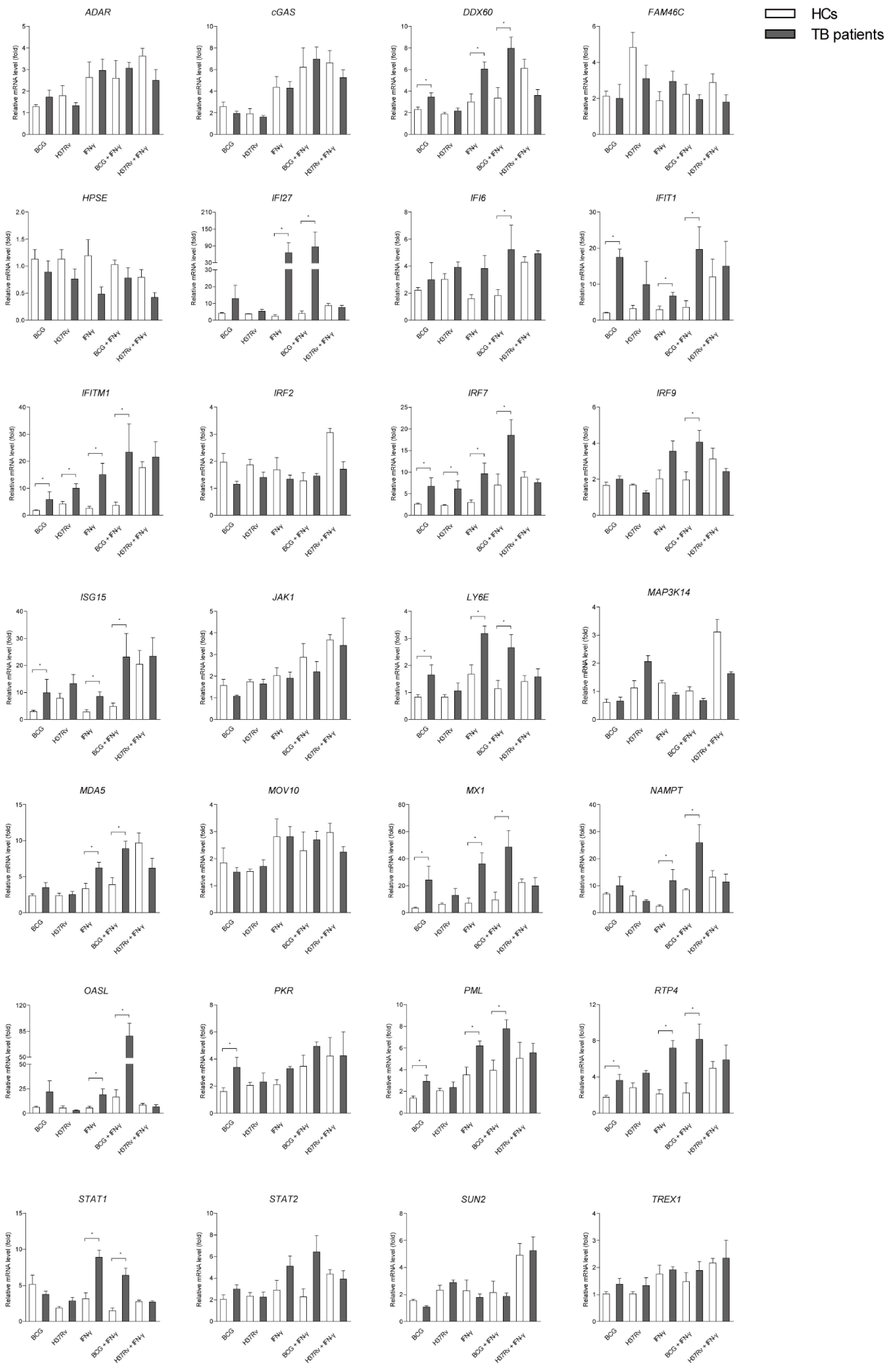
2.4. A Number of ISGs in Mϕs of TB Patients are More Susceptible to Mycobacterial Infection or/and IFN-γ Treatment than that of Healthy People
3. Discussion
4. Materials and Methods
4.1. Study of Populations/Participants
4.2. Cell Culture of hMDMs and THP-1-Mϕs and Mtb Infection
4.3. Reagents
4.4. RNA Extraction and Reverse Transcription
4.5. Real Time PCR Quantitation of Cytokine mRNA
4.6. Protein Preparation and Western Blot Analysis
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- WHO. Global Tuberculosis Report; World Health Organization: Geneva, Switzerland, 2017; pp. 1–147. [Google Scholar]
- Chin, K.L.; Anis, F.Z.; Sarmiento, M.E.; Norazmi, M.N.; Acosta, A. Role of interferons in the development of diagnostics, vaccines, and therapy for tuberculosis. J. Immunol. Res. 2017, 2017, 5212910. [Google Scholar] [CrossRef] [PubMed]
- Giosue, S.; Casarini, M.; Ameglio, F.; Zangrilli, P.; Palla, M.; Altieri, A.M.; Bisetti, A. Aerosolized interferon-alpha treatment in patients with multi-drug-resistant pulmonary tuberculosis. Eur. Cytokine Netw. 2000, 11, 99–104. [Google Scholar] [PubMed]
- Travar, M.; Petkovic, M.; Verhaz, A. Type I, II, and III interferons: Regulating immunity to Mycobacterium tuberculosis infection. Arch. Immunol. Ther. Exp. 2016, 64, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; deWeerd, N.A.; Stifter, S.A.; Liu, L.; Zhou, B.; Wang, W.; Zhou, Y.; Ying, B.; Hu, X.; Matthews, A.Y.; et al. A proline deletion in IFNAR1 impairs IFN-signaling and underlies increased resistance to tuberculosis in humans. Nat. Commun. 2018, 9, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira-Teixeira, L.; Sousa, J.; McNab, F.W.; Torrado, E.; Cardoso, F.; Machado, H.; Castro, F.; Cardoso, V.; Gaifem, J.; Wu, X.; et al. Type I IFN inhibits alternative macrophage activation during Mycobacterium tuberculosis infection and leads to enhanced protection in the absence of IFN-γ Signaling. J. Immunol. 2016, 197, 4714–4726. [Google Scholar] [CrossRef] [PubMed]
- Desvignes, L.; Wolf, A.J.; Ernst, J.D. Dynamic roles of type I and type II IFNs in early infection with Mycobacterium tuberculosis. J. Immunol. 2012, 188, 6205–6215. [Google Scholar] [CrossRef] [PubMed]
- Peddireddy, V.; Doddam, S.N.; Ahmed, N. Mycobacterial dormancy systems and host responses in tuberculosis. Front. Immunol. 2017, 8, 84. [Google Scholar] [CrossRef] [PubMed]
- Guirado, E.; Schlesinger, L.S.; Kaplan, G. Macrophages in tuberculosis: Friend or foe. Semin. Immunopathol. 2013, 35, 563–583. [Google Scholar] [CrossRef]
- Dorhoi, A.; Du Plessis, N. Monocytic myeloid-derived suppressor cells in chronic infections. Front. Immunol. 2017, 8, 1895. [Google Scholar] [CrossRef]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef] [Green Version]
- Schurz, H.; Daya, M.; Moller, M.; Hoal, E.G.; Salie, M. TLR1, 2, 4, 6 and 9 variants associated with tuberculosis susceptibility: A systematic review and META-analysis. PLoS ONE 2015, 10, e0139711. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Akhtar, S.; Jagannath, C.; Khan, A. Pattern recognition receptors and coordinated cellular pathways involved in tuberculosis immunopathogenesis: Emerging concepts and perspectives. Mol. Immunol. 2017, 87, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Watson, R.O.; Bell, S.L.; MacDuff, D.A.; Kimmey, J.M.; Diner, E.J.; Olivas, J.; Vance, R.E.; Stallings, C.L.; Virgin, H.W.; Cox, J.S. The cytosolic sensor cGAS detects Mycobacterium tuberculosis DNA to induce type I interferons and activate autophagy. Cell Host Microbe. 2015, 17, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Zuniga, J.; Torres-Garcia, D.; Santos-Mendoza, T.; Rodriguez-Reyna, T.S.; Granados, J.; Yunis, E.J. Cellular and humoral mechanisms involved in the control of tuberculosis. Clin. Dev. Immunol. 2012, 2012, 193923. [Google Scholar] [CrossRef] [PubMed]
- Jo, E.K.; Yang, C.S.; Choi, C.H.; Harding, C.V. Intracellular signalling cascades regulating innate immune responses to Mycobacteria: Branching out from Toll-like receptors. Cell Microbiol. 2007, 9, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.A.; Schimke, L.F.; Amaral, E.P.; Ishfaq, M.; Barbosa Bonfim, C.C.; Rahman, H.; Iqbal, A.; D’Imperio Lima, M.R.; Costa Carvalho, B.T.; Cabral-Marques, O.; et al. Interferon-γ reduces the proliferation of M. tuberculosis within macrophages from a patient with a novel hypomorphic NEMO mutation. Pediatr. Blood Cancer 2016, 63, 1863–1866. [Google Scholar] [CrossRef]
- Joosten, S.A.; Meijgaarden, K.E.V.; Arend, S.M.; Prins, C.; Oftung, F.; Korsvold, G.E.; Kik, S.V.; Arts, R.J.; Crevel, R.V.; Netea, M.G.; et al. Mycobacterial growth inhibition is associated with trained innate immunity. J. Clin. Investig. 2018. [Google Scholar] [CrossRef]
- Kaufmann, E.; Sanz, J.; Dunn, J.L.; Khan, N.; Mendonca, L.E.; Pacis, A.; Tzelepis, F.; Pernet, E.; Dumaine, A.; Grenier, J.C.; et al. BCG educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell 2018, 172, 176–190. [Google Scholar] [CrossRef]
- Ottenhoff, T.H.; Dass, R.H.; Yang, N.; Zhang, M.M.; Wong, H.E.; Sahiratmadja, E.; Khor, C.C.; Alisjahbana, B.; van Crevel, R.; Marzuki, S.; et al. Genome-wide expression profiling identifies type 1 interferon response pathways in active tuberculosis. PLoS ONE 2012, 7, e45839. [Google Scholar] [CrossRef]
- Donovan, M.L.; Schultz, T.E.; Duke, T.J.; Blumenthal, A. Type I interferons in the pathogenesis of tuberculosis: Molecular drivers and immunological consequences. Front. Immunol. 2017, 8, 1633. [Google Scholar] [CrossRef]
- Labzin, L.I.; Lauterbach, M.A.; Latz, E. Interferons and inflammasomes: Cooperation and counterregulation in disease. J. Allergy Clin. Immunol. 2016, 138, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, P.F.; Van Weyenbergh, J.; Delgobo, M.; Oliveira Patricio, D.; Ferguson, B.J.; Guabiraba, R.; Dierckx, T.; Menezes, S.M.; Bafica, A.; Mansur, D.S. ISG15-induced IL-10 is a novel anti-inflammatory myeloid axis disrupted during active tuberculosis. J. Immunol. 2018, 200, 1434–1442. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, X.; Pan, B.; Wang, H.; Shi, F.; Gan, W.; Yang, L.; Yin, X.; Xu, B.; Zhao, D. Expression pattern of interferon-inducible transcriptional genes in neutrophils during bovine tuberculosis infection. DNA Cell Biol. 2013, 32, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Remoli, M.E.; Giacomini, E.; Lutfalla, G.; Dondi, E.; Orefici, G.; Battistini, A.; Uze, G.; Pellegrini, S.; Coccia, E.M. Selective expression of type I IFN genes in human dendritic cells infected with Mycobacterium tuberculosis. J. Immunol. 2002, 169, 366–374. [Google Scholar] [CrossRef]
- Lavalett, L.; Rodriguez, H.; Ortega, H.; Sadee, W.; Schlesinger, L.S.; Barrera, L.F. Alveolar macrophages from tuberculosis patients display an altered inflammatory gene expression profile. Tuberculosis 2017, 107, 156–167. [Google Scholar] [CrossRef]
- Liu, C.H.; Liu, H.; Ge, B. Innate immunity in tuberculosis: Host defense vs pathogen evasion. Cell Mol. Immunol. 2017, 14, 963–975. [Google Scholar] [CrossRef]
- Dey, B.; Dey, R.J.; Cheung, L.S.; Pokkali, S.; Guo, H.; Lee, J.H.; Bishai, W.R. A bacterial cyclic dinucleotide activates the cytosolic surveillance pathway and mediates innate resistance to tuberculosis. Nat. Med. 2015, 21, 401–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, C.V.; Boom, W.H. Regulation of antigen presentation by Mycobacterium tuberculosis: A role for Toll-like receptors. Nat. Rev. Microbiol. 2010, 8, 296–307. [Google Scholar] [CrossRef]
- Pilli, M.; Arko-Mensah, J.; Ponpuak, M.; Roberts, E.; Master, S.; Mandell, M.A.; Dupont, N.; Ornatowski, W.; Jiang, S.; Bradfute, S.B.; et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 2012, 37, 223–234. [Google Scholar] [CrossRef]
- Liu, S.; Jia, H.; Hou, S.; Xin, T.; Guo, X.; Zhang, G.; Gao, X.; Li, M.; Zhu, W.; Zhu, H. Recombinant Mtb9.8 of Mycobacterium bovis stimulates TNF-α and IL-1β secretion by RAW264.7 macrophages through activation of NF-κB pathway via TLR2. Sci. Rep. 2018, 8, 1928. [Google Scholar] [CrossRef]
- Jia, H.; Liu, S.; Wu, J.; Hou, S.; Xin, T.; Guo, X.; Yuan, W.; Gao, X.; Zhang, G.; Li, M.; et al. Recombinant TB9.8 of Mycobacterium bovis triggers the production of IL-12 p40 and IL-6 in RAW264.7 macrophages via activation of the p38, ERK, and NF-κB signaling pathways. Inflammation 2015, 38, 1337–1346. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, X.; Yang, J.; Zhang, Z.; Zhang, L.; Zhu, B.; Lie, L.; Huang, Y.; Ma, R.; Zhou, C.; Hu, S.; et al. Different Signaling Pathways Define Different Interferon-Stimulated Gene Expression during Mycobacteria Infection in Macrophages. Int. J. Mol. Sci. 2019, 20, 663. https://doi.org/10.3390/ijms20030663
Zhou X, Yang J, Zhang Z, Zhang L, Zhu B, Lie L, Huang Y, Ma R, Zhou C, Hu S, et al. Different Signaling Pathways Define Different Interferon-Stimulated Gene Expression during Mycobacteria Infection in Macrophages. International Journal of Molecular Sciences. 2019; 20(3):663. https://doi.org/10.3390/ijms20030663
Chicago/Turabian StyleZhou, Xinying, Jiahui Yang, Zelin Zhang, Lijie Zhang, Bo Zhu, Linmiao Lie, Yubin Huang, Rui Ma, Chaoying Zhou, Shengfeng Hu, and et al. 2019. "Different Signaling Pathways Define Different Interferon-Stimulated Gene Expression during Mycobacteria Infection in Macrophages" International Journal of Molecular Sciences 20, no. 3: 663. https://doi.org/10.3390/ijms20030663