Class I Phosphoinositide 3-Kinase PIK3CA/p110α and PIK3CB/p110β Isoforms in Endometrial Cancer


Abstract
:
1. The Phosphoinositide 3-Kinase Pathway
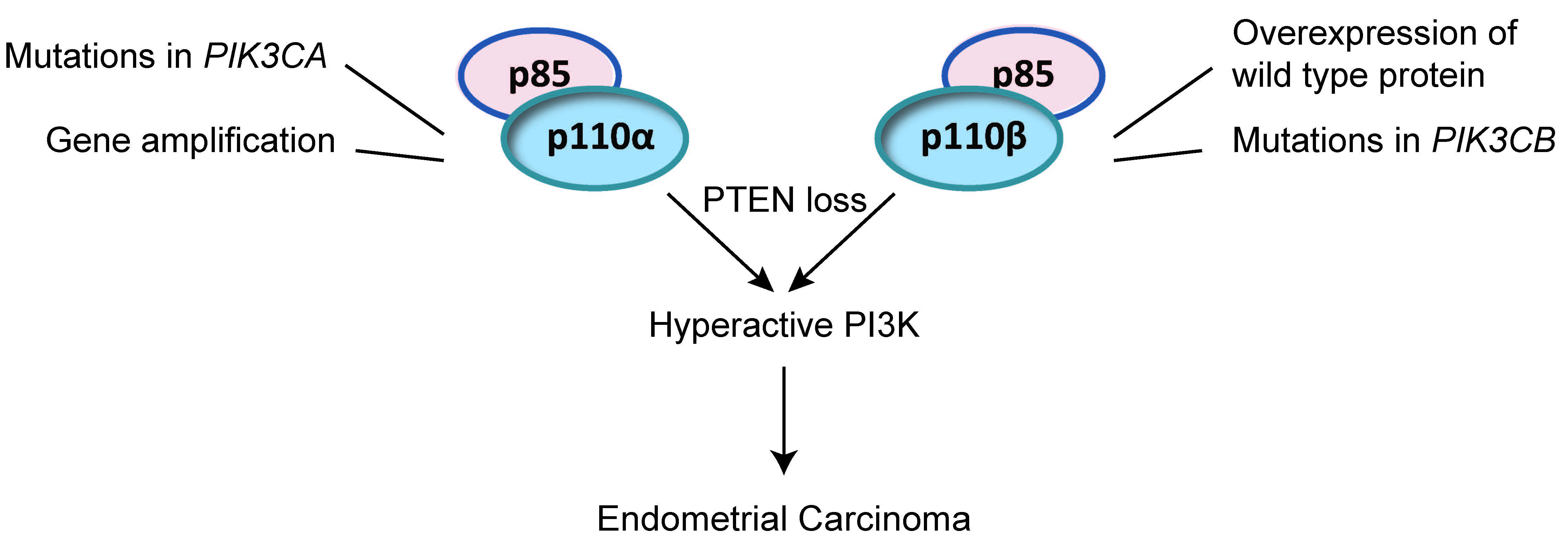
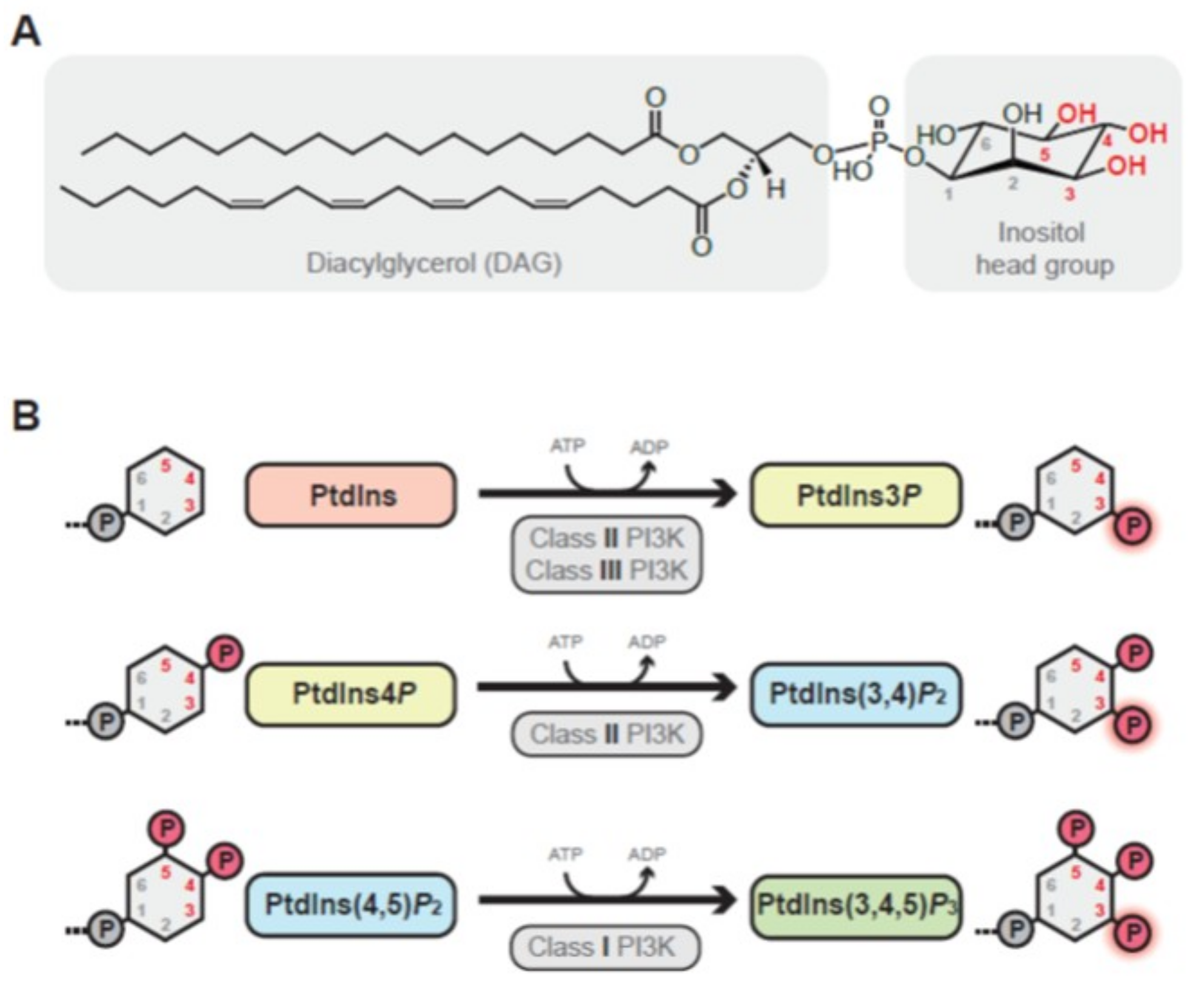
1.1. Class I PI3K Enzymes
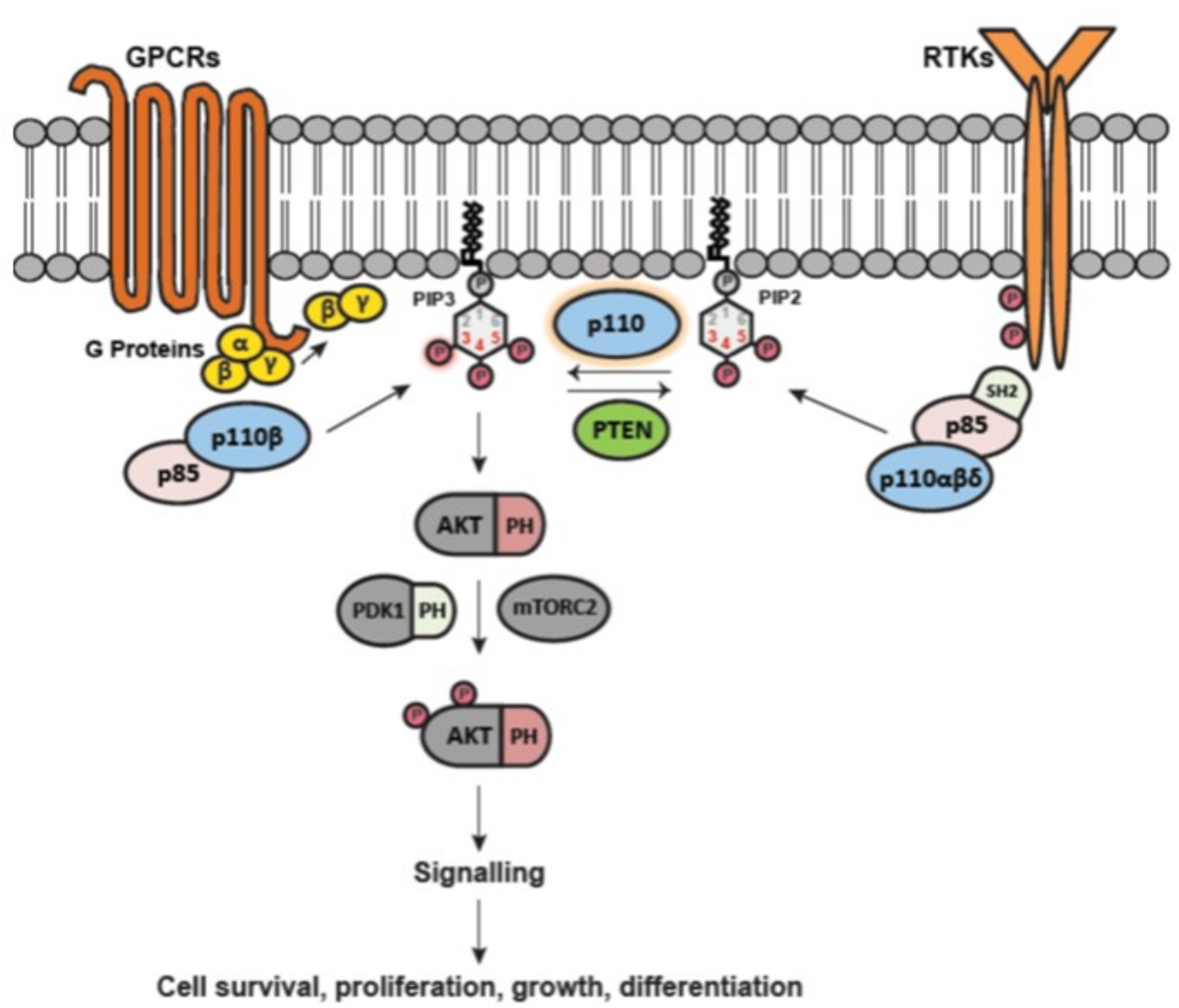
1.2. Activation of Class I PI3Ks and the PI3K Pathway
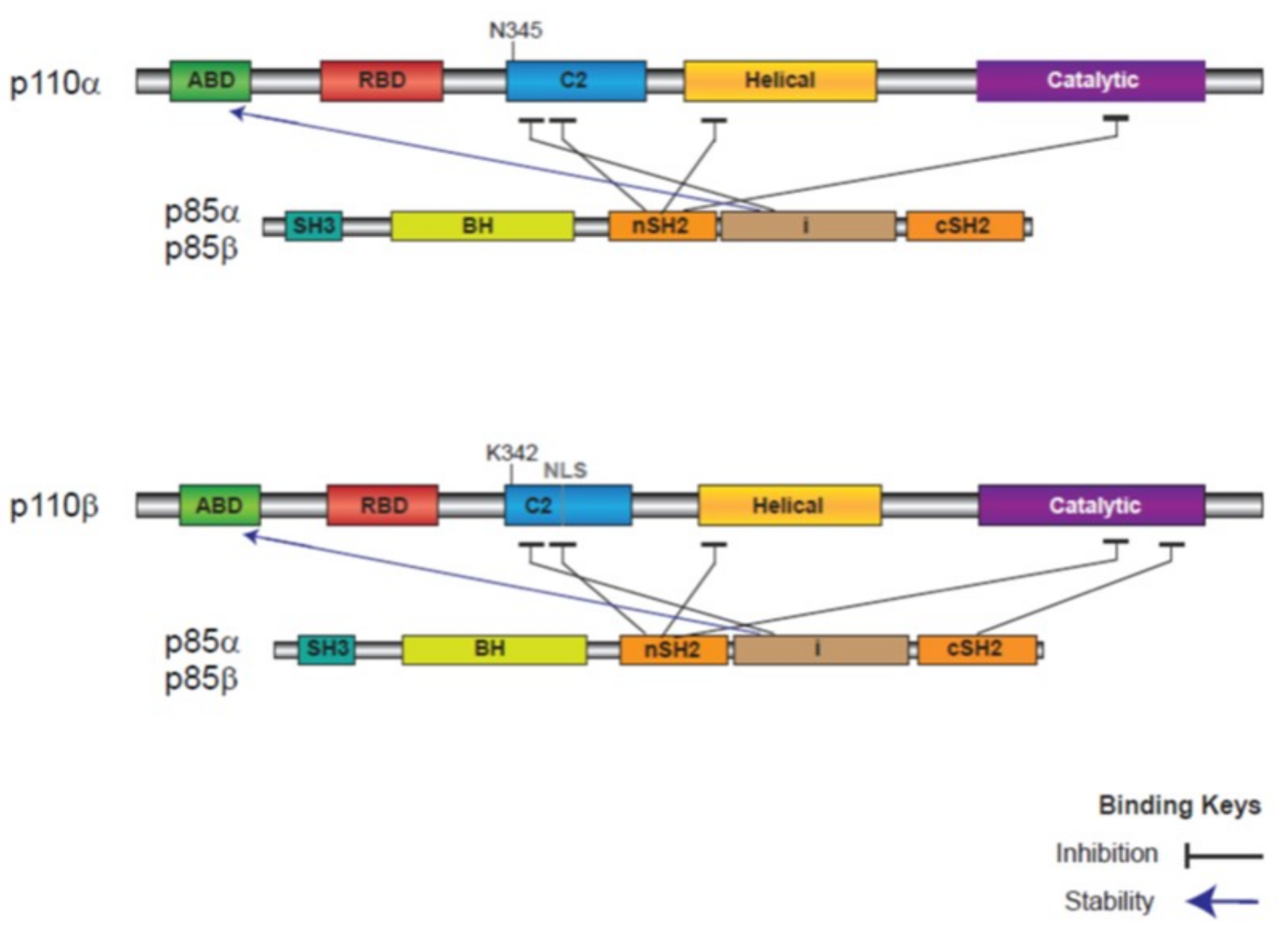
1.3. Properties of PI3K p110α and p110β and Mode of Activation in Cancer
2. Alteration of the PI3K Pathway in Endometrial Cancer
2.1. Endometrial Cancer
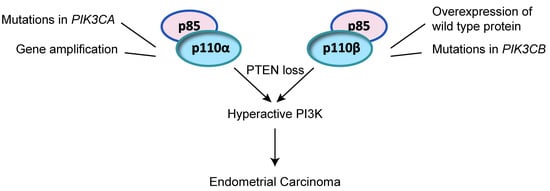
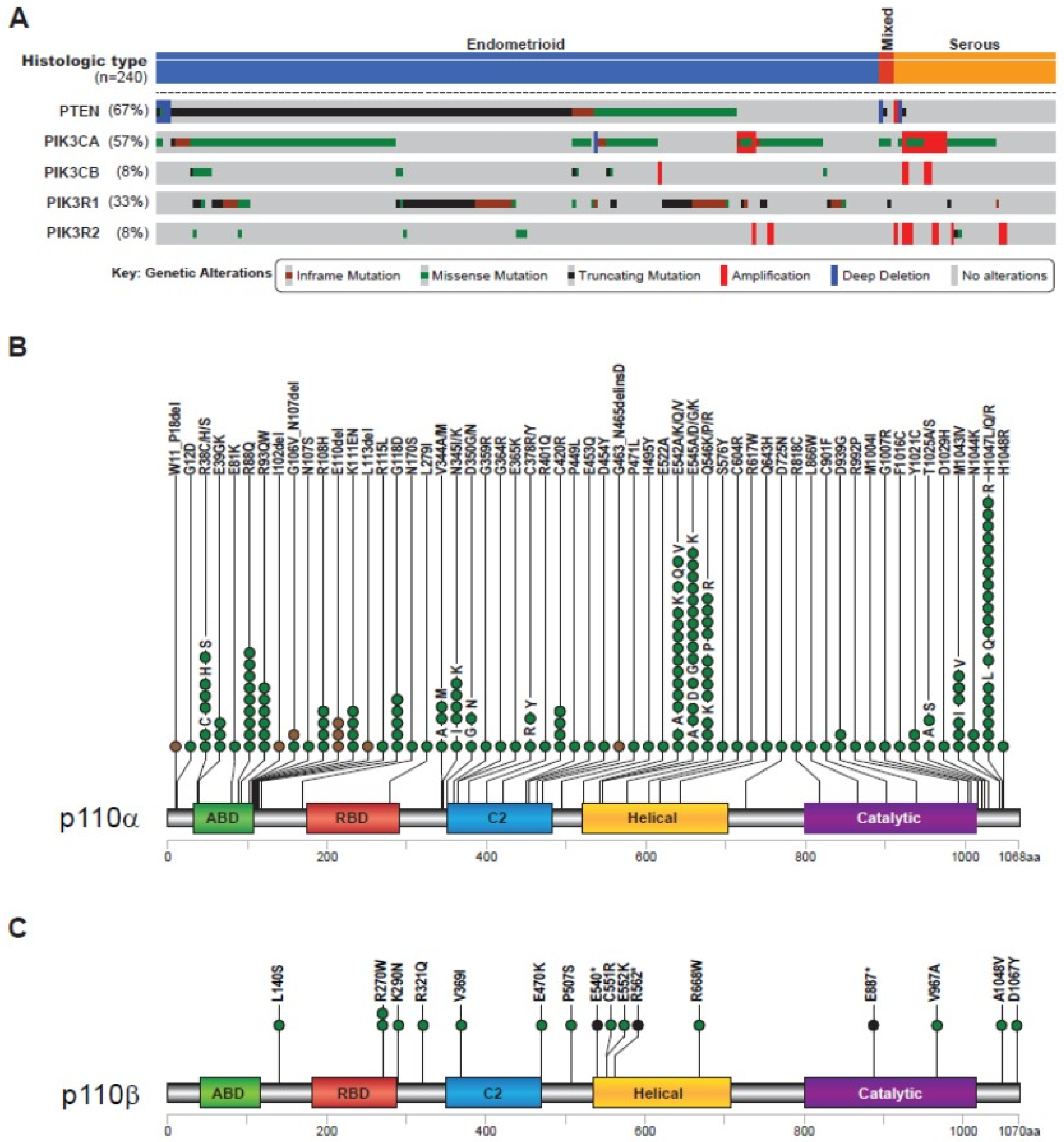
2.2. Alteration of PI3K PIK3CA and PIK3CB in Endometrial Cancer
3. Targeting p110α and p110β in Endometrial Cancer
3.1. PI3K Inhibitor Studies in Endometrial Cancer Cell Lines
3.2. Clinical Trials in Endometrial Cancer
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| GPCR | G protein coupled receptor |
| PI3K | Phosphoinositide 3-kinase |
| PtdIns | Phosphatidylinositol |
| PtdIns(4,5)P2 | Phosphatidylinositol (4,5) bisphosphate |
| PtdIns(3,4,5)P3 | Phosphatidylinositol (3,4,5) triphosphate |
| PTEN | Phosphatase and tensin homologue |
| RTK | Receptor tyrosine kinase |
| SH2 | Src homology 2 |
Appendix A


References
- Vanhaesebroeck, B.; Stephens, L.; Hawkins, P. PI3K signalling: The path to discovery and understanding. Nat. Rev. Mol. Cell Boil. 2012, 13, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Chalhoub, N.; Baker, S.J. PTEN and the PI3-kinase pathway in cancer. Annu. Rev. Pathol. 2009, 4, 127–150. [Google Scholar] [CrossRef]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Jean, S.; Kiger, A.A. Classes of phosphoinositide 3-kinases at a glance. J. Cell Sci. 2014, 127, 923–928. [Google Scholar] [CrossRef] [Green Version]
- Vanhaesebroeck, B.; Waterfield, M.D. Signaling by distinct classes of phosphoinositide 3-kinases. Exp. Cell Res. 1999, 253, 239–254. [Google Scholar] [CrossRef]
- Stephens, L.R.; Hughes, K.T.; Irvine, R.F. Pathway of phosphatidylinositol(3,4,5)-trisphosphate synthesis in activated neutrophils. Nature 1991, 351, 33–39. [Google Scholar] [CrossRef]
- Sasaki, T.; Takasuga, S.; Sasaki, J.; Kofuji, S.; Eguchi, S.; Yamazaki, M.; Suzuki, A. Mammalian phosphoinositide kinases and phosphatases. Prog. Lipid Res. 2009, 48, 307–343. [Google Scholar] [CrossRef] [PubMed]
- Auger, K.R.; Serunian, L.A.; Soltoff, S.P.; Libby, P.; Cantley, L.C. PDGF-dependent tyrosine phosphorylation stimulates production of novel polyphosphoinositides in intact cells. Cell 1989, 57, 167–175. [Google Scholar] [CrossRef]
- Whitman, M.; Downes, C.P.; Keeler, M.; Keller, T.; Cantley, L. Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature 1988, 332, 644–646. [Google Scholar] [CrossRef] [PubMed]
- Backer, J.M. The intricate regulation and complex functions of the Class III phosphoinositide 3-kinase Vps34. Biochem. J. 2016, 473, 2251–2271. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.T.; Stephens, L.R. Emerging evidence of signalling roles for PI(3,4)P2 in Class I and II PI3K-regulated pathways. Biochem. Soc. Trans. 2016, 44, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Sud, M.; Fahy, E.; Cotter, D.; Brown, A.; Dennis, E.A.; Glass, C.K.; Merrill, A.H., Jr.; Murphy, R.C.; Raetz, C.R.; Russell, D.W.; et al. LMSD: LIPID MAPS structure database. Nucleic Acids Res. 2007, 35, D527–532. [Google Scholar] [CrossRef] [PubMed]
- Fahy, E.; Subramaniam, S.; Murphy, R.C.; Nishijima, M.; Raetz, C.R.; Shimizu, T.; Spener, F.; van Meer, G.; Wakelam, M.J.; Dennis, E.A. Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 2009, 50, S9–S14. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.E.; Williams, R.L. Synergy in activating class I PI3Ks. Trends Biochem. Sci. 2015, 40, 88–100. [Google Scholar] [CrossRef]
- Dbouk, H.A.; Vadas, O.; Shymanets, A.; Burke, J.E.; Salamon, R.S.; Khalil, B.D.; Barrett, M.O.; Waldo, G.L.; Surve, C.; Hsueh, C.; et al. G protein-coupled receptor-mediated activation of p110beta by Gbetagamma is required for cellular transformation and invasiveness. Sci. Signal. 2012, 5, ra89. [Google Scholar] [CrossRef]
- Vadas, O.; Dbouk, H.A.; Shymanets, A.; Perisic, O.; Burke, J.E.; Abi Saab, W.F.; Khalil, B.D.; Harteneck, C.; Bresnick, A.R.; Nurnberg, B.; et al. Molecular determinants of PI3Kgamma-mediated activation downstream of G-protein-coupled receptors (GPCRs). Proc. Natl. Acad. Sci. USA 2013, 110, 18862–18867. [Google Scholar] [CrossRef]
- Jimenez, C.; Hernandez, C.; Pimentel, B.; Carrera, A.C. The p85 regulatory subunit controls sequential activation of phosphoinositide 3-kinase by Tyr kinases and Ras. J. Biol. Chem. 2002, 277, 41556–41562. [Google Scholar] [CrossRef] [PubMed]
- Lietzke, S.E.; Bose, S.; Cronin, T.; Klarlund, J.; Chawla, A.; Czech, M.P.; Lambright, D.G. Structural basis of 3-phosphoinositide recognition by pleckstrin homology domains. Mol. Cell 2000, 6, 385–394. [Google Scholar] [CrossRef]
- Park, W.S.; Heo, W.D.; Whalen, J.H.; O’Rourke, N.A.; Bryan, H.M.; Meyer, T.; Teruel, M.N. Comprehensive identification of PIP3-regulated PH domains from C. elegans to H. sapiens by model prediction and live imaging. Mol. Cell 2008, 30, 381–392. [Google Scholar] [CrossRef] [PubMed]
- James, S.R.; Downes, C.P.; Gigg, R.; Grove, S.J.; Holmes, A.B.; Alessi, D.R. Specific binding of the Akt-1 protein kinase to phosphatidylinositol 3,4,5-trisphosphate without subsequent activation. Biochem. J. 1996, 315, 709–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgering, B.M.; Coffer, P.J. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature 1995, 376, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Gan, W.; Chin, Y.R.; Ogura, K.; Guo, J.; Zhang, J.; Wang, B.; Blenis, J.; Cantley, L.C.; Toker, A.; et al. PtdIns(3,4,5)P3-Dependent Activation of the mTORC2 Kinase Complex. Cancer Discov. 2015, 5, 1194–1209. [Google Scholar] [CrossRef] [Green Version]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Maehama, T.; Dixon, J.E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef]
- Damen, J.E.; Liu, L.; Rosten, P.; Humphries, R.K.; Jefferson, A.B.; Majerus, P.W.; Krystal, G. The 145-kDa protein induced to associate with Shc by multiple cytokines is an inositol tetraphosphate and phosphatidylinositol 3,4,5-triphosphate 5-phosphatase. Proc. Natl. Acad. Sci. USA 1996, 93, 1689–1693. [Google Scholar] [CrossRef]
- Pesesse, X.; Deleu, S.; De Smedt, F.; Drayer, L.; Erneux, C. Identification of a second SH2-domain-containing protein closely related to the phosphatidylinositol polyphosphate 5-phosphatase SHIP. Biochem. Biophys. Res. Commun. 1997, 239, 697–700. [Google Scholar] [CrossRef] [PubMed]
- Maffucci, T. An introduction to phosphoinositides. Curr. Top. Microbiol. Immunol. 2012, 362, 1–42. [Google Scholar] [PubMed]
- Kok, K.; Geering, B.; Vanhaesebroeck, B. Regulation of phosphoinositide 3-kinase expression in health and disease. Trends Biochem. Sci. 2009, 34, 115–127. [Google Scholar] [CrossRef]
- Ilic, N.; Roberts, T.M. Comparing the roles of the p110alpha and p110beta isoforms of PI3K in signaling and cancer. Curr. Top. Microbiol. Immunol. 2010, 347, 55–77. [Google Scholar]
- Singh, P.; Dar, M.S.; Dar, M.J. p110alpha and p110beta isoforms of PI3K signaling: Are they two sides of the same coin? FEBS Lett. 2016, 590, 3071–3082. [Google Scholar] [CrossRef] [PubMed]
- Bi, L.; Okabe, I.; Bernard, D.J.; Nussbaum, R.L. Early embryonic lethality in mice deficient in the p110beta catalytic subunit of PI 3-kinase. Mamm. Genome 2002, 13, 169–172. [Google Scholar] [PubMed]
- Bi, L.; Okabe, I.; Bernard, D.J.; Wynshaw-Boris, A.; Nussbaum, R.L. Proliferative defect and embryonic lethality in mice homozygous for a deletion in the p110alpha subunit of phosphoinositide 3-kinase. J. Biol. Chem. 1999, 274, 10963–10968. [Google Scholar] [CrossRef] [PubMed]
- Foukas, L.C.; Claret, M.; Pearce, W.; Okkenhaug, K.; Meek, S.; Peskett, E.; Sancho, S.; Smith, A.J.; Withers, D.J.; Vanhaesebroeck, B. Critical role for the p110alpha phosphoinositide-3-OH kinase in growth and metabolic regulation. Nature 2006, 441, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Graupera, M.; Guillermet-Guibert, J.; Foukas, L.C.; Phng, L.K.; Cain, R.J.; Salpekar, A.; Pearce, W.; Meek, S.; Millan, J.; Cutillas, P.R.; et al. Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature 2008, 453, 662–666. [Google Scholar] [CrossRef]
- Guillermet-Guibert, J.; Smith, L.B.; Halet, G.; Whitehead, M.A.; Pearce, W.; Rebourcet, D.; Leon, K.; Crepieux, P.; Nock, G.; Stromstedt, M.; et al. Novel Role for p110beta PI 3-Kinase in Male Fertility through Regulation of Androgen Receptor Activity in Sertoli Cells. PLoS Genet. 2015, 11, e1005304. [Google Scholar] [CrossRef]
- Benistant, C.; Chapuis, H.; Roche, S. A specific function for phosphatidylinositol 3-kinase alpha (p85alpha-p110alpha) in cell survival and for phosphatidylinositol 3-kinase beta (p85alpha-p110beta) in de novo DNA synthesis of human colon carcinoma cells. Oncogene 2000, 19, 5083–5090. [Google Scholar] [CrossRef] [PubMed]
- Marques, M.; Kumar, A.; Cortes, I.; Gonzalez-Garcia, A.; Hernandez, C.; Moreno-Ortiz, M.C.; Carrera, A.C. Phosphoinositide 3-kinases p110alpha and p110beta regulate cell cycle entry, exhibiting distinct activation kinetics in G1 phase. Mol. Cell Biol. 2008, 28, 2803–2814. [Google Scholar] [CrossRef]
- Marques, M.; Kumar, A.; Poveda, A.M.; Zuluaga, S.; Hernandez, C.; Jackson, S.; Pasero, P.; Carrera, A.C. Specific function of phosphoinositide 3-kinase beta in the control of DNA replication. Proc. Natl. Acad. Sci. USA 2009, 106, 7525–7530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, T.; Krakstad, C.; Tangen, I.L.; Hoivik, E.A.; Pollock, P.M.; Salvesen, H.B.; Lewis, A.E. Endometrial cancer cells exhibit high expression of p110β and its selective inhibition induces variable responses on PI3K signaling, cell survival and proliferation. Oncotarget 2017, 8, 3881–3894. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Redondo-Muñoz, J.; Perez-García, V.; Cortes, I.; Chagoyen, M.; Carrera, A.C. Nuclear but Not Cytosolic Phosphoinositide 3-Kinase Beta Has an Essential Function in Cell Survival. Mol. Cell. Boil. 2011, 31, 2122–2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, T.; Altankhuyag, A.; Dobrovolska, O.; Turcu, D.C.; Lewis, A.E. A polybasic motif in ErbB3-binding protein 1 (EBP1) has key functions in nucleolar localization and polyphosphoinositide interaction. Biochem. J. 2016, 473, 2033–2047. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Fernandez-Capetillo, O.; Carrera, A.C. Nuclear phosphoinositide 3-kinase beta controls double-strand break DNA repair. Proc. Natl. Acad. Sci. USA 2010, 107, 7491–7496. [Google Scholar] [CrossRef]
- Burke, J.E.; Williams, R.L. Dynamic steps in receptor tyrosine kinase mediated activation of class IA phosphoinositide 3-kinases (PI3K) captured by H/D exchange (HDX-MS). Adv. Biol. Regul. 2013, 53, 97–110. [Google Scholar] [CrossRef]
- Zhang, X.; Vadas, O.; Perisic, O.; Anderson, K.E.; Clark, J.; Hawkins, P.T.; Stephens, L.R.; Williams, R.L. Structure of lipid kinase p110beta/p85beta elucidates an unusual SH2-domain-mediated inhibitory mechanism. Mol. Cell 2011, 41, 567–578. [Google Scholar] [CrossRef]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef]
- Huang, C.H.; Mandelker, D.; Schmidt-Kittler, O.; Samuels, Y.; Velculescu, V.E.; Kinzler, K.W.; Vogelstein, B.; Gabelli, S.B.; Amzel, L.M. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science 2007, 318, 1744–1748. [Google Scholar] [CrossRef] [PubMed]
- Mandelker, D.; Gabelli, S.B.; Schmidt-Kittler, O.; Zhu, J.; Cheong, I.; Huang, C.H.; Kinzler, K.W.; Vogelstein, B.; Amzel, L.M. A frequent kinase domain mutation that changes the interaction between PI3Kalpha and the membrane. Proc. Natl. Acad. Sci. USA 2009, 106, 16996–17001. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.E.; Perisic, O.; Masson, G.R.; Vadas, O.; Williams, R.L. Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110alpha (PIK3CA). Proc. Natl. Acad. Sci. USA 2012, 109, 15259–15264. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, B.S.; Janakiraman, V.; Kljavin, N.M.; Chaudhuri, S.; Stern, H.M.; Wang, W.; Kan, Z.; Dbouk, H.A.; Peters, B.A.; Waring, P.; et al. Somatic mutations in p85alpha promote tumorigenesis through class IA PI3K activation. Cancer Cell 2009, 16, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Hillmann, P.; Hofmann, B.T.; Hart, J.R.; Vogt, P.K. Cancer-derived mutations in the regulatory subunit p85alpha of phosphoinositide 3-kinase function through the catalytic subunit p110alpha. Proc. Natl. Acad. Sci. USA 2010, 107, 15547–15552. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, L.M.; Spangle, J.M.; Ohlson, C.E.; Cheng, H.; Roberts, T.M.; Cantley, L.C.; Zhao, J.J. PI3K-p110alpha mediates the oncogenic activity induced by loss of the novel tumor suppressor PI3K-p85alpha. Proc. Natl. Acad. Sci. USA 2017, 114, 7095–7100. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Denley, A.; Vanhaesebroeck, B.; Vogt, P.K. Oncogenic transformation induced by the p110β, -γ, and -δ isoforms of class I phosphoinositide 3-kinase. Proc. Natl. Acad. Sci. USA 2006, 103, 1289–1294. [Google Scholar] [CrossRef]
- Dbouk, H.A.; Pang, H.; Fiser, A.; Backer, J.M. A biochemical mechanism for the oncogenic potential of the p110beta catalytic subunit of phosphoinositide 3-kinase. Proc. Natl. Acad. Sci. USA 2010, 107, 19897–19902. [Google Scholar] [CrossRef]
- Ciraolo, E.; Iezzi, M.; Marone, R.; Marengo, S.; Curcio, C.; Costa, C.; Azzolino, O.; Gonella, C.; Rubinetto, C.; Wu, H.; et al. Phosphoinositide 3-kinase p110beta activity: Key role in metabolism and mammary gland cancer but not development. Sci. Signal. 2008, 1, ra3. [Google Scholar] [CrossRef]
- Guillermet-Guibert, J.; Bjorklof, K.; Salpekar, A.; Gonella, C.; Ramadani, F.; Bilancio, A.; Meek, S.; Smith, A.J.; Okkenhaug, K.; Vanhaesebroeck, B. The p110beta isoform of phosphoinositide 3-kinase signals downstream of G protein-coupled receptors and is functionally redundant with p110gamma. Proc. Natl. Acad. Sci. USA 2008, 105, 8292–8297. [Google Scholar] [CrossRef]
- Jia, S.; Liu, Z.; Zhang, S.; Liu, P.; Zhang, L.; Lee, S.H.; Zhang, J.; Signoretti, S.; Loda, M.; Roberts, T.M.; et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature 2008, 454, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Khalil, B.D.; Hsueh, C.; Cao, Y.; Abi Saab, W.F.; Wang, Y.; Condeelis, J.S.; Bresnick, A.R.; Backer, J.M. GPCR Signaling Mediates Tumor Metastasis via PI3Kbeta. Cancer Res. 2016, 76, 2944–2953. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, R.; de Krijger, I.; Fritsch, K.; George, R.; Reason, B.; Kumar, M.S.; Diefenbacher, M.; Stamp, G.; Downward, J. RAS and RHO families of GTPases directly regulate distinct phosphoinositide 3-kinase isoforms. Cell 2013, 153, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Dbouk, H.A.; Khalil, B.D.; Wu, H.; Shymanets, A.; Nurnberg, B.; Backer, J.M. Characterization of a tumor-associated activating mutation of the p110beta PI 3-kinase. PLoS ONE 2013, 8, e63833. [Google Scholar] [CrossRef] [PubMed]
- Pazarentzos, E.; Giannikopoulos, P.; Hrustanovic, G.; St John, J.; Olivas, V.R.; Gubens, M.A.; Balassanian, R.; Weissman, J.; Polkinghorn, W.; Bivona, T.G. Oncogenic activation of the PI3-kinase p110beta isoform via the tumor-derived PIK3Cbeta(D1067V) kinase domain mutation. Oncogene 2016, 35, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Whale, A.D.; Colman, L.; Lensun, L.; Rogers, H.L.; Shuttleworth, S.J. Functional characterization of a novel somatic oncogenic mutation of PIK3CB. Signal. Transduct. Target. Ther. 2017, 2, 17063. [Google Scholar] [CrossRef] [Green Version]
- Mateo, J.; Ganji, G.; Lemech, C.; Burris, H.A.; Han, S.-W.; Swales, K.E.; Decordova, S.; DeYoung, M.P.; Smith, D.A.; Kalyana-Sundaram, S.; et al. A first-time-in-human study of GSK2636771, a phosphoinositide 3 kinase beta-selective inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2017, 23, 5981–5992. [Google Scholar] [CrossRef]
- Kim, E.; Ilic, N.; Shrestha, Y.; Zou, L.; Kamburov, A.; Zhu, C.; Yang, X.; Lubonja, R.; Tran, N.; Nguyen, C.; et al. Systematic Functional Interrogation of Rare Cancer Variants Identifies Oncogenic Alleles. Cancer Discov. 2016, 6, 714–726. [Google Scholar] [CrossRef] [Green Version]
- Juric, D.; Castel, P.; Griffith, M.; Griffith, O.L.; Won, H.H.; Ellis, H.; Ebbesen, S.H.; Ainscough, B.J.; Ramu, A.; Iyer, G.; et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kalpha inhibitor. Nature 2014. [Google Scholar] [CrossRef]
- Costa, C.; Ebi, H.; Martini, M.; Beausoleil, S.A.; Faber, A.C.; Jakubik, C.T.; Huang, A.; Wang, Y.; Nishtala, M.; Hall, B.; et al. Measurement of PIP3 levels reveals an unexpected role for p110beta in early adaptive responses to p110alpha-specific inhibitors in luminal breast cancer. Cancer Cell 2015, 27, 97–108. [Google Scholar] [CrossRef]
- Nakanishi, Y.; Walter, K.; Spoerke, J.M.; O’Brien, C.; Huw, L.Y.; Hampton, G.M.; Lackner, M.R. Activating Mutations in PIK3CB Confer Resistance to PI3K Inhibition and Define a Novel Oncogenic Role for p110beta. Cancer Res. 2016, 76, 1193–1203. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, S.; Milanezi, F.; Costa, J.L.; Amendoeira, I.; Schmitt, F. PIKing the right isoform: The emergent role of the p110beta subunit in breast cancer. Virchows Arch. 2010, 456, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Wee, S.; Wiederschain, D.; Maira, S.M.; Loo, A.; Miller, C.; de Beaumont, R.; Stegmeier, F.; Yao, Y.M.; Lengauer, C. PTEN-deficient cancers depend on PIK3CB. Proc. Natl. Acad. Sci. USA 2008, 105, 13057–13062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, J.; Liu, Q.; Xie, S.; Carlson, C.; Von, T.; Vogel, K.; Riddle, S.; Benes, C.; Eck, M.; Roberts, T.; et al. Functional characterization of an isoform-selective inhibitor of PI3K-p110beta as a potential anticancer agent. Cancer Discov. 2012, 2, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Yuzugullu, H.; Baitsch, L.; Von, T.; Steiner, A.; Tong, H.; Ni, J.; Clayton, L.K.; Bronson, R.; Roberts, T.M.; Gritsman, K.; et al. A PI3K p110beta-Rac signalling loop mediates Pten-loss-induced perturbation of haematopoiesis and leukaemogenesis. Nat. Commun. 2015, 6, 8501. [Google Scholar] [CrossRef] [PubMed]
- Berenjeno, I.M.; Guillermet-Guibert, J.; Pearce, W.; Gray, A.; Fleming, S.; Vanhaesebroeck, B. Both p110alpha and p110beta isoforms of PI3K can modulate the impact of loss-of-function of the PTEN tumour suppressor. Biochem. J. 2012, 442, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, P.; Spangle, J.M.; Von, T.; Roberts, T.M.; Lin, N.U.; Krop, I.E.; Winer, E.P.; Zhao, J.J. PI3K-p110alpha mediates resistance to HER2-targeted therapy in HER2+, PTEN-deficient breast cancers. Oncogene 2016, 35, 3607–3612. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gao, X.L.; Schmit, F.; Adelmant, G.; Eck, M.J.; Marto, J.A.; Zhao, J.J.; Roberts, T.M. CRKL Mediates p110 beta-Dependent PI3K Signaling in PTEN-Deficient Cancer Cells. Cell Rep. 2017, 20, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Geering, B.; Cutillas, P.R.; Nock, G.; Gharbi, S.I.; Vanhaesebroeck, B. Class IA phosphoinositide 3-kinases are obligate p85-p110 heterodimers. Proc. Natl. Acad. Sci. USA 2007, 104, 7809–7814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backer, J.M. The regulation of class IA PI 3-kinases by inter-subunit interactions. Curr. Top. Microbiol. Immunol. 2010, 346, 87–114. [Google Scholar] [PubMed]
- Fruman, D.A. Regulatory subunits of class IA PI3K. Curr. Top. Microbiol. Immunol. 2010, 346, 225–244. [Google Scholar] [PubMed]
- Le Gallo, M.; Bell, D.W. The emerging genomic landscape of endometrial cancer. Clin. Chem. 2014, 60, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Morice, P.; Leary, A.; Creutzberg, C.; Abu-Rustum, N.; Darai, E. Endometrial cancer. Lancet 2016, 387, 1094–1108. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Bokhman, J.V. Two pathogenetic types of endometrial carcinoma. Gynecol. Oncol. 1983, 15, 10–17. [Google Scholar] [CrossRef]
- Murali, R.; Soslow, R.A.; Weigelt, B. Classification of endometrial carcinoma: More than two types. Lancet Oncol. 2014, 15, e268–e278. [Google Scholar] [CrossRef]
- Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; Benz, C.C.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [Green Version]
- Myers, A.P. New strategies in endometrial cancer: Targeting the PI3K/mTOR pathway--the devil is in the details. Clin. Cancer Res. 2013, 19, 5264–5274. [Google Scholar] [CrossRef]
- Levine, R.L.; Cargile, C.B.; Blazes, M.S.; van Rees, B.; Kurman, R.J.; Ellenson, L.H. PTEN mutations and microsatellite instability in complex atypical hyperplasia, a precursor lesion to uterine endometrioid carcinoma. Cancer Res. 1998, 58, 3254–3258. [Google Scholar]
- Miyake, T.; Yoshino, K.; Enomoto, T.; Takata, T.; Ugaki, H.; Kim, A.; Fujiwara, K.; Miyatake, T.; Fujita, M.; Kimura, T. PIK3CA gene mutations and amplifications in uterine cancers, identified by methods that avoid confounding by PIK3CA pseudogene sequences. Cancer Lett. 2008, 261, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Cheung, L.W.; Hennessy, B.T.; Li, J.; Yu, S.; Myers, A.P.; Djordjevic, B.; Lu, Y.; Stemke-Hale, K.; Dyer, M.D.; Zhang, F.; et al. High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov. 2011, 1, 170–185. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.L.; Xiu, J.; Chatterjee-Paer, S.; Buckley de Meritens, A.; Burke, W.M.; Tergas, A.I.; Wright, J.D.; Hou, J.Y. Distinct molecular landscapes between endometrioid and nonendometrioid uterine carcinomas. Int. J. Cancer 2017, 140, 1396–1404. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutter, G.L.; Lin, M.C.; Fitzgerald, J.T.; Kum, J.B.; Baak, J.P.; Lees, J.A.; Weng, L.P.; Eng, C. Altered PTEN expression as a diagnostic marker for the earliest endometrial precancers. J. Natl. Cancer Inst. 2000, 92, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Enomoto, T.; Fujita, M.; Wada, H.; Yoshino, K.; Ozaki, K.; Nakamura, T.; Murata, Y. Mutational analysis of the PTEN gene in endometrial carcinoma and hyperplasia. Am. J. Clin. Pathol. 2001, 115, 32–38. [Google Scholar] [CrossRef]
- Hayes, M.P.; Wang, H.; Espinal-Witter, R.; Douglas, W.; Solomon, G.J.; Baker, S.J.; Ellenson, L.H. PIK3CA and PTEN mutations in uterine endometrioid carcinoma and complex atypical hyperplasia. Clin. Cancer Res. 2006, 12, 5932–5935. [Google Scholar] [CrossRef]
- Berg, A.; Hoivik, E.A.; Mjos, S.; Holst, F.; Werner, H.M.; Tangen, I.L.; Taylor-Weiner, A.; Gibson, W.J.; Kusonmano, K.; Wik, E.; et al. Molecular profiling of endometrial carcinoma precursor, primary and metastatic lesions suggests different targets for treatment in obese compared to non-obese patients. Oncotarget 2015, 6, 1327–1339. [Google Scholar] [CrossRef]
- Gibson, W.J.; Hoivik, E.A.; Halle, M.K.; Taylor-Weiner, A.; Cherniack, A.D.; Berg, A.; Holst, F.; Zack, T.I.; Werner, H.M.; Staby, K.M.; et al. The genomic landscape and evolution of endometrial carcinoma progression and abdominopelvic metastasis. Nat. Genet. 2016, 48, 848–855. [Google Scholar] [CrossRef] [Green Version]
- Forbes, S.A.; Bindal, N.; Bamford, S.; Cole, C.; Kok, C.Y.; Beare, D.; Jia, M.; Shepherd, R.; Leung, K.; Menzies, A.; et al. COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011, 39, D945–D950. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Stokoe, D.; Taketani, Y.; McCormick, F. High frequency of coexistent mutations of PIK3CA and PTEN genes in endometrial carcinoma. Cancer Res. 2005, 65, 10669–10673. [Google Scholar] [CrossRef] [PubMed]
- Rudd, M.L.; Price, J.C.; Fogoros, S.; Godwin, A.K.; Sgroi, D.C.; Merino, M.J.; Bell, D.W. A unique spectrum of somatic PIK3CA (p110alpha) mutations within primary endometrial carcinomas. Clin. Cancer Res. 2011, 17, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, E.; Wu, R.C.; Guan, B.; Wu, G.; Zhang, J.; Wang, Y.; Song, L.; Yuan, X.; Wei, L.; Roden, R.B.; et al. Identification of molecular pathway aberrations in uterine serous carcinoma by genome-wide analyses. J. Natl. Cancer Inst. 2012, 104, 1503–1513. [Google Scholar] [CrossRef] [PubMed]
- Le Gallo, M.; O’Hara, A.J.; Rudd, M.L.; Urick, M.E.; Hansen, N.F.; O’Neil, N.J.; Price, J.C.; Zhang, S.; England, B.M.; Godwin, A.K.; et al. Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat. Genet. 2012, 44, 1310. [Google Scholar] [CrossRef] [PubMed]
- Mjos, S.; Werner, H.M.J.; Birkeland, E.; Holst, F.; Berg, A.; Halle, M.K.; Tangen, I.L.; Kusonmano, K.; Mauland, K.K.; Oyan, A.M.; et al. PIK3CA exon9 mutations associate with reduced survival, and are highly concordant between matching primary tumors and metastases in endometrial cancer. Sci. Rep. 2017, 7, 10240. [Google Scholar] [CrossRef] [PubMed]
- Salvesen, H.B.; Carter, S.L.; Mannelqvist, M.; Dutt, A.; Getz, G.; Stefansson, I.M.; Raeder, M.B.; Sos, M.L.; Engelsen, I.B.; Trovik, J.; et al. Integrated genomic profiling of endometrial carcinoma associates aggressive tumors with indicators of PI3 kinase activation. Proc. Natl. Acad. Sci. USA 2009, 106, 4834–4839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konopka, B.; Janiec-Jankowska, A.; Kwiatkowska, E.; Najmola, U.; Bidzinski, M.; Olszewski, W.; Goluda, C. PIK3CA mutations and amplification in endometrioid endometrial carcinomas: Relation to other genetic defects and clinicopathologic status of the tumors. Hum. Pathol. 2011, 42, 1710–1719. [Google Scholar] [CrossRef] [PubMed]
- An, H.J.; Cho, N.H.; Yang, H.S.; Kwak, K.B.; Kim, N.K.; Oh, D.Y.; Lee, S.W.; Kim, H.O.; Koh, J.J. Targeted RNA interference of phosphatidylinositol 3-kinase p110-beta induces apoptosis and proliferation arrest in endometrial carcinoma cells. J. Pathol. 2007, 212, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Salvesen, H.B.; Haldorsen, I.S.; Trovik, J. Markers for individualised therapy in endometrial carcinoma. Lancet Oncol. 2012, 13, e353–e361. [Google Scholar] [CrossRef]
- Rodriguez-Freixinos, V.; Karakasis, K.; Oza, A.M. New Targeted Agents in Endometrial Cancer: Are We Really Making Progress? Curr. Oncol. Rep. 2016, 18, 23. [Google Scholar] [CrossRef] [PubMed]
- Aslan, O.; Cremona, M.; Morgan, C.; Cheung, L.W.; Mills, G.B.; Hennessy, B.T. Preclinical evaluation and reverse phase protein Array-based profiling of PI3K and MEK inhibitors in endometrial carcinoma in vitro. BMC Cancer 2018, 18, 168. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Warne, P.; Lambros, M.; Reis-Filho, J.; Downward, J. PI3K pathway dependencies in endometrioid endometrial cancer cell lines. Clin. Cancer Res. 2013. [Google Scholar] [CrossRef] [PubMed]
- Heudel, P.E.; Fabbro, M.; Roemer-Becuwe, C.; Kaminsky, M.C.; Arnaud, A.; Joly, F.; Roche-Forestier, S.; Meunier, J.; Foa, C.; You, B.; et al. Phase II study of the PI3K inhibitor BKM120 in patients with advanced or recurrent endometrial carcinoma: A stratified type I-type II study from the GINECO group. Br. J. Cancer 2017, 116, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Matulonis, U.; Vergote, I.; Backes, F.; Martin, L.P.; McMeekin, S.; Birrer, M.; Campana, F.; Xu, Y.; Egile, C.; Ghamande, S. Phase II study of the PI3K inhibitor pilaralisib (SAR245408; XL147) in patients with advanced or recurrent endometrial carcinoma. Gynecol. Oncol. 2015, 136, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Juric, D.; Rodon, J.; Tabernero, J.; Janku, F.; Burris, H.A.; Schellens, J.H.M.; Middleton, M.R.; Berlin, J.; Schuler, M.; Gil-Martin, M.; et al. Phosphatidylinositol 3-Kinase alpha-Selective Inhibition With Alpelisib (BYL719) in PIK3CA-Altered Solid Tumors: Results From the First-in-Human Study. J. Clin. Oncol. 2018, 36, 1291–1299. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property Description | p110α | p110β |
|---|---|---|
| Gene name | PIK3CA | PIK3CB |
| Regulatory subunit 1 | p85α, p55α, p50α | p85α, p55α, p50α |
| p85β | p85β | |
| p55γ | p55γ | |
| Cellular localisation | cytoplasm | cytoplasm, nucleoplasm, nucleolus |
| Receptor activation | RTKs | GPCRs (dominant) and RTKs |
| Mutations in carcinomas | frequent | rare |
| Drug Name | Molecular Condition for Trial | Types of Cancer | Phase | ID Number |
|---|---|---|---|---|
| GSK2636771 | PTEN deficiency | advanced solid tumours | I | NCT01458067 (completed and results published [67]) |
| GSK2636771 | PTEN loss, mutation or deletion | Advanced-stage refractory solid cancers | II | NCT02465060 (recruiting patients) |
| AZD8186 | PTEN loss, mutation or deletion | Advanced-stage refractory solid cancers | II | NCT02465060 (recruiting patients) |
| AZD8186 (with docetaxel) | PTEN loss or mutation, or PIK3CB Mutation | Advanced-stage solid cancers metastatic or unresectable | I | NCT03218826 (recruiting patients) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazloumi Gavgani, F.; Smith Arnesen, V.; Jacobsen, R.G.; Krakstad, C.; Hoivik, E.A.; Lewis, A.E. Class I Phosphoinositide 3-Kinase PIK3CA/p110α and PIK3CB/p110β Isoforms in Endometrial Cancer. Int. J. Mol. Sci. 2018, 19, 3931. https://doi.org/10.3390/ijms19123931
Mazloumi Gavgani F, Smith Arnesen V, Jacobsen RG, Krakstad C, Hoivik EA, Lewis AE. Class I Phosphoinositide 3-Kinase PIK3CA/p110α and PIK3CB/p110β Isoforms in Endometrial Cancer. International Journal of Molecular Sciences. 2018; 19(12):3931. https://doi.org/10.3390/ijms19123931
Chicago/Turabian StyleMazloumi Gavgani, Fatemeh, Victoria Smith Arnesen, Rhîan G. Jacobsen, Camilla Krakstad, Erling A. Hoivik, and Aurélia E. Lewis. 2018. "Class I Phosphoinositide 3-Kinase PIK3CA/p110α and PIK3CB/p110β Isoforms in Endometrial Cancer" International Journal of Molecular Sciences 19, no. 12: 3931. https://doi.org/10.3390/ijms19123931