Sequencing of Euscaphis konishii Endocarp Transcriptome Points to Molecular Mechanisms of Endocarp Coloration

 ,
,
Abstract
:
1. Introduction
2. Results
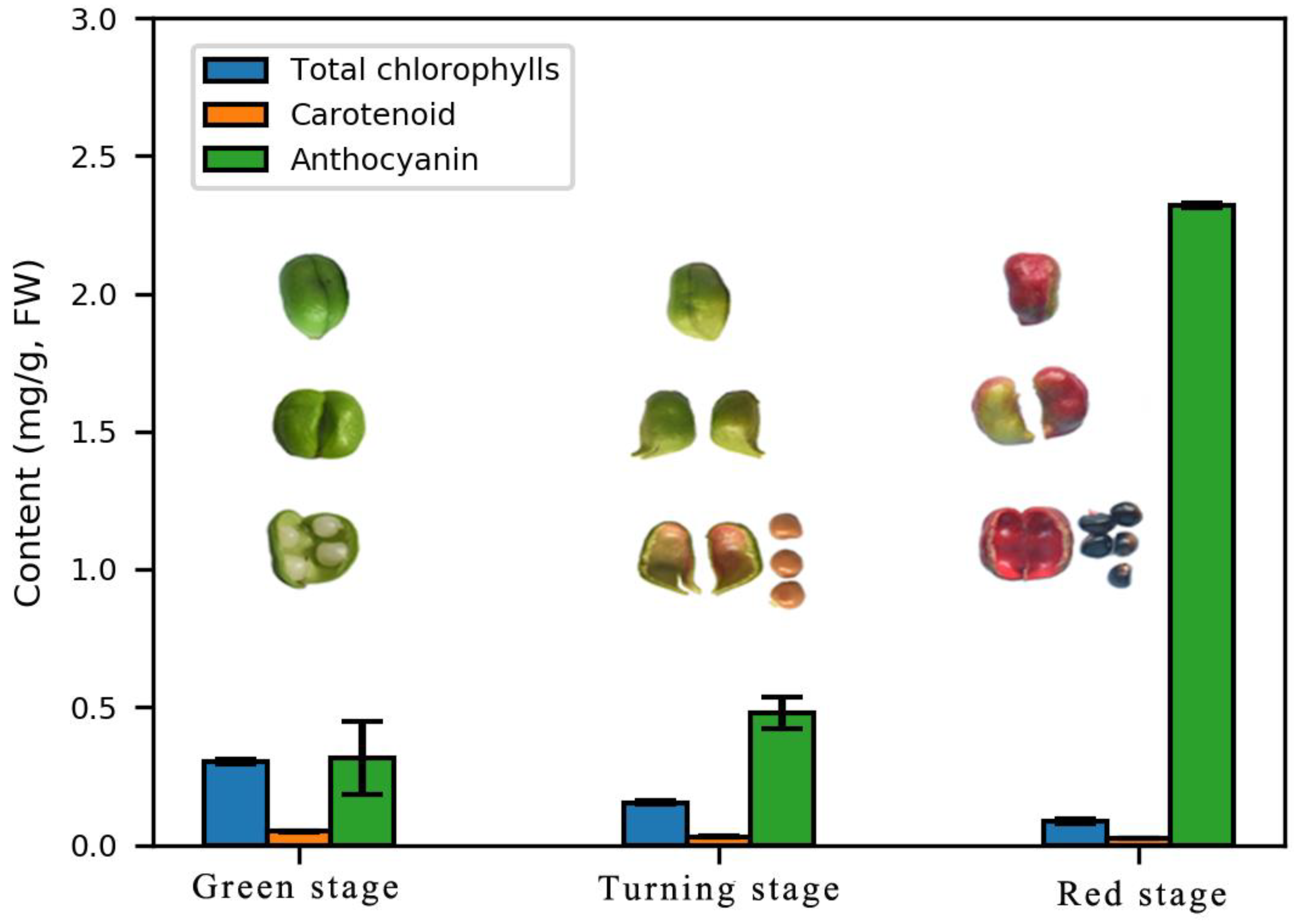
2.1. Changes in Pigment Content
2.2. Illumina Sequencing and De Novo Assembly
2.3. Functional Annotation of the Unigenes
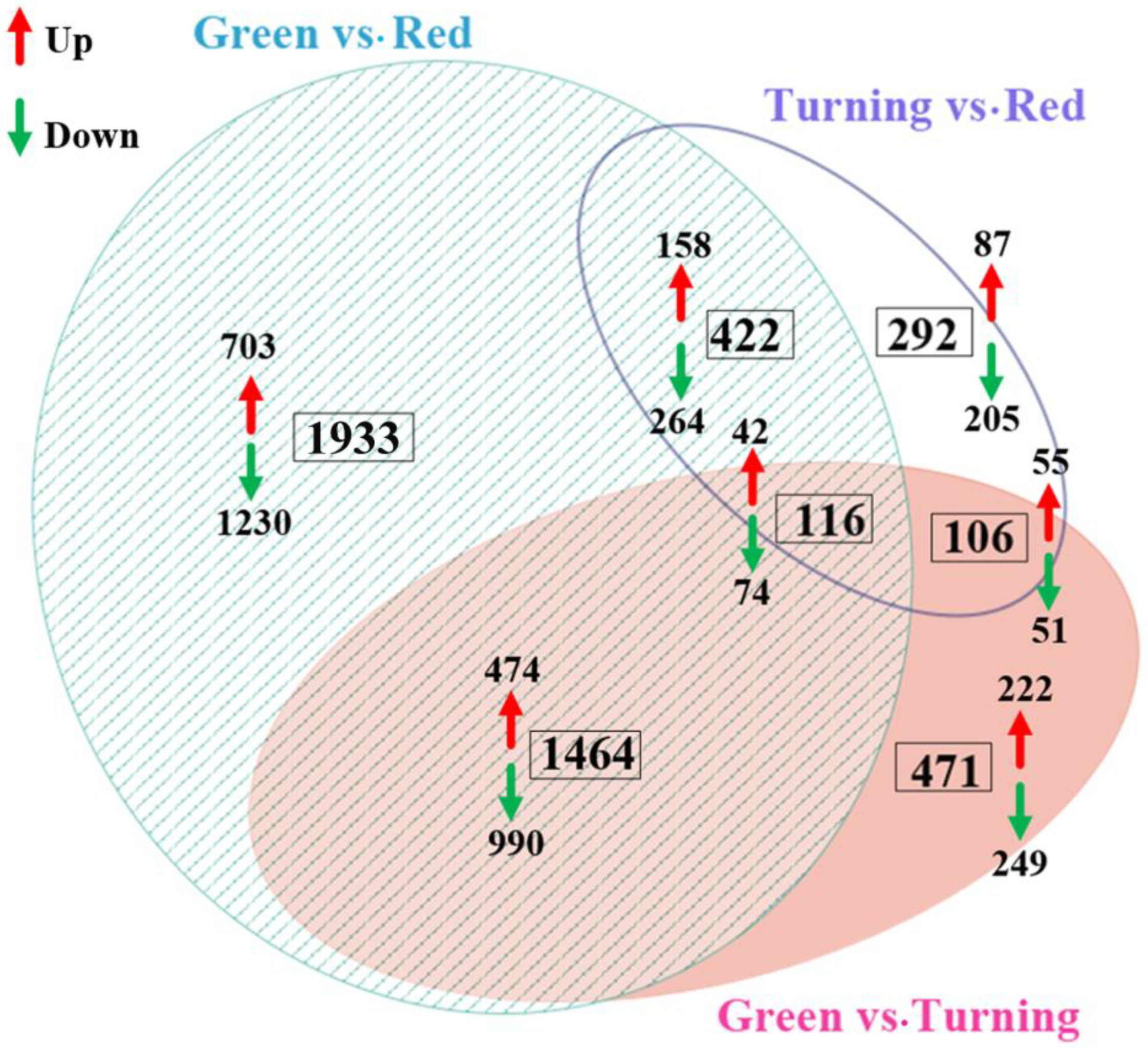
2.4. Differentially Expressed Genes between Fruit with Varied Color
2.5. GO Annotation of DEGs
2.6. Co-Expression Analysis and KEGG Enrichment of DEGs
2.7. Identification of Transcription Factors
2.8. DEGs Related to Anthocyanin Biosynthesis
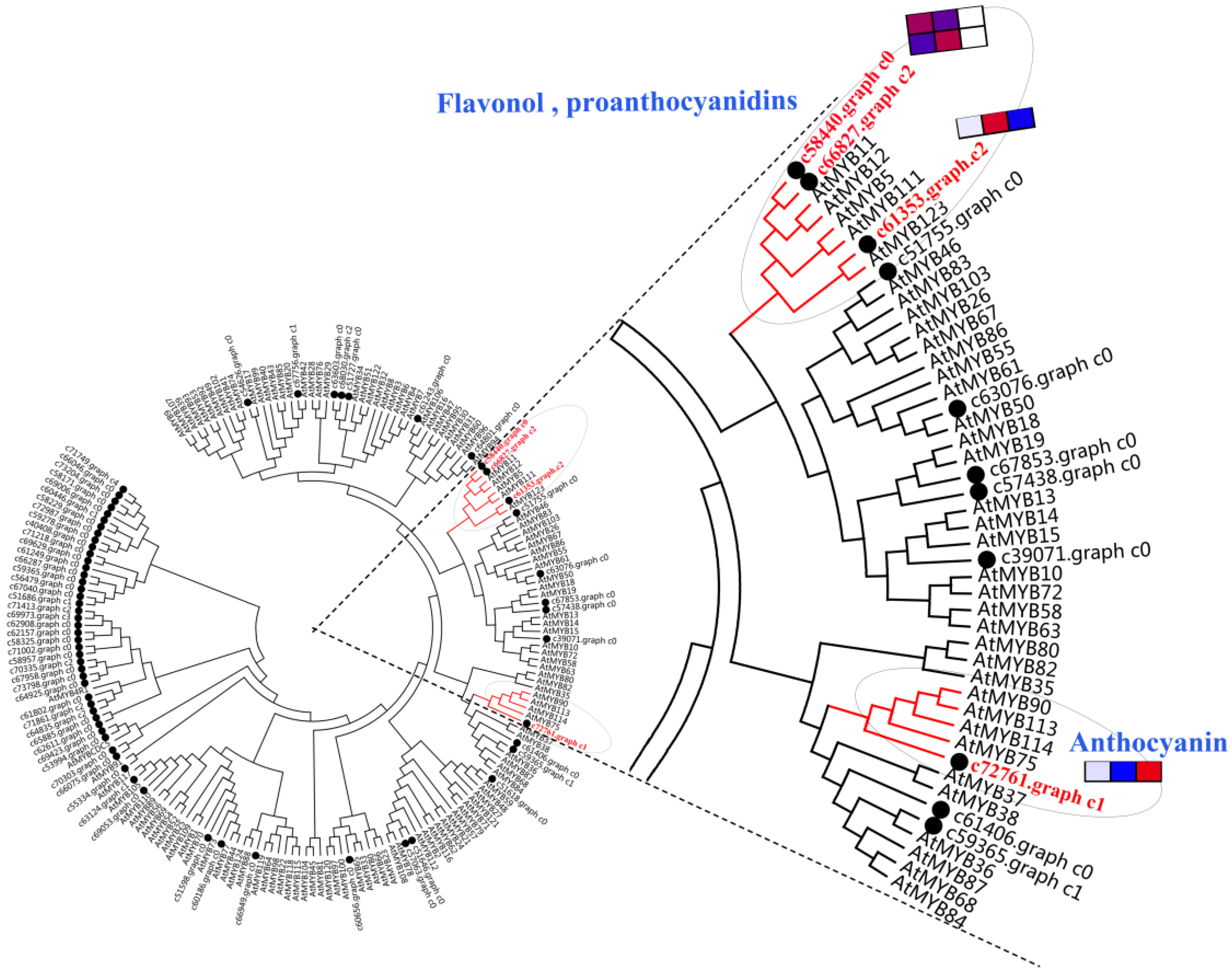
2.9. Analysis of R2R3-MYB Gene Family
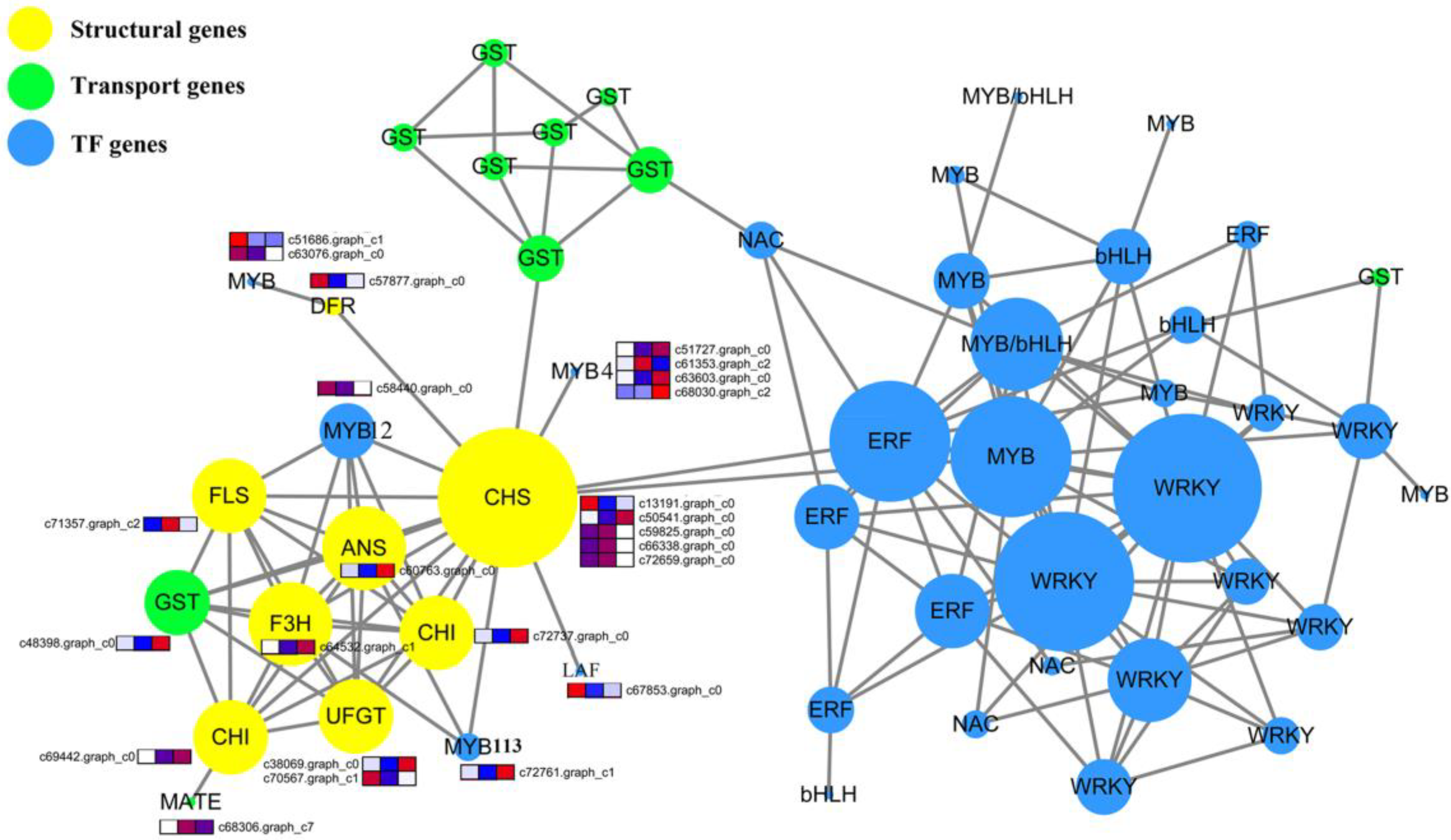
2.10. Protein–Protein Interaction Network Construction and Candidate Gene Selection
2.11. Genes Involved in Carotenoid Biosynthesis and Chlorophyll Degradation
2.12. Quantitative Real-Time PCR Validation of Differentially Expressed Transcripts from RNA-Seq
3. Discussion
3.1. Candidate Genes Involved in Anthocyanin Biosynthesis
3.2. Candidate Genes Involved in Carotenoid Biosynthesis and Chlorophyll Degradation
4. Conclusions
5. Material and Methods
5.1. Plant Material
5.2. Total Anthocyanin, Carotenoid, and Chlorophyll Determination
5.3. RNA Extraction and cDNA Synthesis
5.4. Library Construction and Transcriptome Sequencing
5.5. RNA-Sequencing Data Analysis
5.6. RT-qPCR Validation
5.7. Phylogenetic Analysis
5.8. PPI Network Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Grotewold, E. The genetics and biochemistry of floral pigments. Annu. Rev. Plant Biol. 2006, 57, 761–780. [Google Scholar] [CrossRef] [PubMed]
- And, P.G.K.; Baker, H.G. Insects as flower visitors and pollinators. Ann. Rev. Entomol. 2003, 28, 407–453. [Google Scholar]
- Tornielli, G.; Koes, R.; Quattrocchio, F. Petunia. Evolutionary, Developmental and Physiological Genetics. In The Genetics of Flower Color; Gerats, T., Strommer, J., Eds.; Springer: New York, NY, USA, 2009; pp. 269–299. [Google Scholar]
- Clegg, M.T.; Durbin, M.L.; Ayala, F.J.; Fitch, W.M.; Clegg, M.T. Flower color variation: A model for the experimental study of evolution. Proc. Natl. Acad. Sci. USA 2000, 97, 7016–7023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, Y.; Hoshino, A. Recent advances in flower color variation and patterning of Japanese morning glory and petunia. Breed. Sci. 2018, 68, 128–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brugliera, F.; Tao, G.Q.; Tems, U.; Kalc, G.; Mouradova, E.; Price, K.; Stevenson, K.; Nakamura, N.; Stacey, I.; Katsumoto, Y.; et al. Violet/blue chrysanthemums––metabolic engineering of the anthocyanin biosynthetic pathway results in novel petal colors. Plant Cell Physiol. 2013, 54, 1696–1710. [Google Scholar] [CrossRef] [PubMed]
- Noda, N. Recent advances in the research and development of blue flowers. Breed. Sci. 2018, 68, 79–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, J.; Kanno, Y.; Itoh, Y.; Tsugawa, H.; Suzuki, M. Plant biochemistry: Anthocyanin biosynthesis in roses. Nature 2005, 435, 757–758. [Google Scholar] [CrossRef] [PubMed]
- Honda, C.; Bessho, H.; Murai, M.; Iwanami, H.; Moriya, S.; Abe, K.; Wada, M.; Tanaka, Y.M.; Hayama, H.; Tatsuki, M. Effect of temperature on anthocyanin synthesis and ethylene production in the fruit of early– and medium–maturing apple cultivars during ripening stages. Hortscience 2014, 49, 1510–1517. [Google Scholar]
- El-Kereamy, A.; Chervin, C.; Roustan, J.P.; Cheynier, V.; Souquet, J.M.; Moutounet, M.; Raynal, J.; Ford, C.; Latche, A.; Pech, J.C.; et al. Exogenous ethylene stimulates the long-term expression of genes related to anthocyanin biosynthesis in grape berries. Physiol. Plant. 2010, 119, 175–182. [Google Scholar] [CrossRef]
- Holzwarth, M.; Korhummel, S.; Kammerer, D.R.; Carle, R. Thermal inactivation of strawberry polyphenoloxidase and its impact on anthocyanin and color retention in strawberry (fragaria x ananassa duch.) purees. Eur. Food Res. Technol. 2012, 235, 1171–1180. [Google Scholar] [CrossRef]
- Enstone, D.E.; Peterson, C.A.; Gijzen, M. Soybean Hydrophobic Protein is Present in a Matrix Secreted by the Endocarp Epidermis during Seed Development. Sci. Rep. 2015, 5, 15074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dardick, C.; Callahan, A.M. Evolution of the fruit endocarp: Molecular mechanisms underlying adaptations in seed protection and dispersal strategies. Front. Plant Sci. 2014, 5, 284. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.H.; Zhang, Q.M.; Wang, J.; Jian, S.G.; Ren, H. The ecological and biological characteristics of Euscaphis konishii. Ecol. Environ. 2009, 18, 306–309. [Google Scholar]
- Park, S.Y.; Yu, J.W.; Park, J.S.; Li, J.; Yoo, S.C.; Lee, N.Y.; Lee, S.K.; Jeong, S.W.; Seo, H.S.; Koh, H.J.; et al. The Senescence–Induced Staygreen Protein Regulates Chlorophyll Degradation. Plant Cell 2007, 19, 1649–1664. [Google Scholar] [CrossRef] [PubMed]
- Espley, R.V.; Hellens, R.P.; Jo, P.; Stevenson, D.E.; Sumathi, K.A.; Allan, A.C. Red colouration in apple fruit is due to the activity of the MYB transcription factor, MdMYB10. Plant J. 2007, 49, 414–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Du, H.; Zhai, R.; Song, L.; Ma, F.; Xu, L. Transcriptome analysis reveals candidate genes related to color fading of ‘red bartlett’ (Pyrus communis L.). Front. Plant Sci. 2017, 8, 445. [Google Scholar] [CrossRef] [PubMed]
- Rahim, M.A.; Busatto, N.; Trainotti, L. Regulation of anthocyanin biosynthesis in peach fruits. Planta 2014, 240, 913–929. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.Y.; Zou, X.X.; Huang, W.; Zhang, X.H.; Chen, Z.M.; Sun, W.H.; Li, Y.L.; Chen, L.Y.; Zou, S.Q. Study on the Changes of Contents of Pigments of Euscaphis konishii Hayata Fruit during Fruit Development. Nonwood For. Res. 2018, 3, 100–106. [Google Scholar]
- Tanaka, Y.; Sasaki, N.; Ohmiya, A. Biosynthesis of plant pigments: Anthocyanins, betalains and carotenoids. Plant J. 2010, 54, 733–749. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Dai, C.; Li, Y.; Feng, J.; Liu, Z.; Kang, C. Reduced anthocyanins in petiolescodes for a GST anthocyanin transporter that is essential for the foliage and fruit coloration in strawberry. J. Exp. Bot. 2018, 69, 2595–2608. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Gao, L.; Wang, H.; Chen, X.; Wang, Y.; Yang, H.; Wei, C.; Wan, X.; Xia, T. The R2R3–MYB, bHLH, WD40, and related transcription factors in flavonoid biosynthesis. Funct. Integr. Genom. 2013, 13, 75–98. [Google Scholar] [CrossRef] [PubMed]
- Grotewold, E. The science of flavonoids. In The Regulation of Flavonoid Biosynthesis; Springer: New York, NY, USA, 2006; pp. 97–141. [Google Scholar]
- Hörtensteiner, S.; Kräutler, B. Chlorophyll breakdown in higher plants and algae. Biochim. Biophys. Acta 2011, 56, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Matile, P.; Hörtensteiner, S.; Thomas, H. Chlorophyll degradation. Annu. Rev. Plant Biol. 1999, 50, 67–95. [Google Scholar] [CrossRef] [PubMed]
- Takamiya, K.I.; Tsuchiya, T.; Ohta, H. Degradation pathway(s) of chlorophyll: What has gene cloning revealed? Trends Plant Sci. 2000, 5, 426–431. [Google Scholar] [CrossRef]
- Balazadeh, S. Stay–green not always stays green. Mol. Plant 2014, 7, 1264–1266. [Google Scholar] [CrossRef] [PubMed]
- Lai, B.; Hu, B.; Qin, Y.H.; Zhao, J.T.; Wang, H.C.; Hu, G.B. Transcriptomic analysis of litchi chinensis pericarp during maturation with a focus on chlorophyll degradation and flavonoid biosynthesis. BMC Genom. 2015, 16, 225. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Huang, Y.; Zhang, Y.; Wang, X.; Yang, H.; Yu, O.; Dai, W.; Fang, C. Transcriptome profiling of fruit development and maturation in Chinese white pear (Pyrus bretschneideri, Rehd). BMC Genom. 2013, 14, 823. [Google Scholar] [CrossRef] [PubMed]
- Springob, K.; Nakajima, J.; Yamazaki, M.; Saito, K. Recent advances in the biosynthesis and accumulation of anthocyanins. Nat. Prod. Rep. 2003, 20, 288–303. [Google Scholar] [CrossRef] [PubMed]
- Stracke, R.; Ishihara, H.; Huep, G.; Barsch, A.; Mehrtens, F.; Niehaus, K.; Weisshaar, B. Differential regulation of closely related R2R3-MYB transcription factors controls flavonol accumulation in different parts of the Arabidopsis thaliana seedling. Plant J. 2007, 50, 660–677. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Zhao, M.; Leavitt, J.M.; Lloyd, A.M. Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLH/MUB transcriptional complex in Arabidopsis seedlings. Plant J. 2008, 53, 814–827. [Google Scholar] [CrossRef] [PubMed]
- Lepiniec, L.; Debeaujon, I.; Routaboul, J.M.; Baudry, A.; Pourcel, L.; Nesi, N.; Caboche, M. Genetics and biochemistry of seed flavonoids. Annu. Rev. Plant Biol. 2006, 57, 405–430. [Google Scholar] [CrossRef] [PubMed]
- Petroni, K.; Tonelli, C. Recent advances on the regulation of anthocyanin synthesis in reproductive organs. Plant Sci. 2011, 181, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.L.; Bolitho, K.; Grafton, K.; Kortstee, A.; Karunairetnam, S.; McGhie, T.K.; Espley, R.V.; Hellens, R.P.; Allan, A.C. An R2R3 MYB transcription factor associated with regulation of the anthocyanin biosynthetic pathway in Rosaceae. BMC Plant Biol. 2010, 10, 50. [Google Scholar]
- Gomez, C.; Conejero, G.; Torregrosa, L.; Cheynier, V.; Terrier, N.; Ageorges, A. In vivo grapevine anthocyanin transport involves vesicle-mediated trafficking and the contribution of anthoMATE transporters and GST. Plant J. 2011, 67, 960–970. [Google Scholar] [CrossRef] [PubMed]
- Alfenito, M.R.; Souer, E.; Goodman, C.D.; Buell, R.; Mol, J.; Koes, R.; Walbot, V. Functional complementation of anthocyanin sequestration in the vacuole by widely divergent glutathiones–transferases. Plant Cell 1998, 10, 1135–1149. [Google Scholar] [CrossRef] [PubMed]
- Francisco, R.M.; Regalado, A.; Ageorges, A.; Burla, B.J.; Bassin, B.; Eisenach, C.; Zarrouk, O.; Vialet, S.; Marlin, T.; Chaves, M.M.; et al. Abcc1, an ATP binding cassette protein from grape berry, transports anthocyanidin 3–O–Glucosides. Plant Cell 2013, 25, 1840–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, C.; Terrier, N.; Torregrosa, L.; Vialet, S.; Fournier-Level, A.; Verriès, C.; Souquet, J.M.; Mazauric, J.P.; Klein, M.; Cheynier, V.; et al. Grapevine MATE–type proteins act as vacuolar H+–dependent acylated anthocyanin transporters. Plant Physiol. 2009, 150, 402–415. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Li, H.; Huang, J.R. Arabidopsis TT19 functions as a carrier to transport anthocyanin from the cytosol to tonoplasts. Mol. Plant 2012, 5, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Zhao, J.; Lai, B.; Qin, Y.; Wang, H.; Hu, G. LcGTS4 is an anthocyanin–related glutathione s–transferase gene in litchi chinensis sonn. Plant Cell Rep. 2016, 35, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Marinova, K.; Pourcel, L.; Weder, B.; Schwarz, M.; Barron, D.; Routaboul, J.M.; Debeaujon, I.; Klein, M. The Arabidopsis MATE transporter TT12 acts as a vacuolar flavonoid/H+–antiporter active in Proanthocyanidin–accumulating cells of the seed coat. Plant Cell 2007, 19, 2023–2038. [Google Scholar] [CrossRef] [PubMed]
- Cutanda-Perez, M.C.; Ageorges, A.; Gomez, C.; Vialet, S.; Terrier, N.; Romieu, C.; Torregrosa, L. Ectopic expression of VlmybA1, in grapevine activates a narrow set of genes involved in anthocyanin synthesis and transport. Plant Mol. Biol. 2009, 69, 633–648. [Google Scholar] [CrossRef] [PubMed]
- Eckhardt, U.; Grimm, B.; Hörtensteiner, S. Recent advances in chlorophyll biosynthesis and breakdown in higher plants. Plant Mol. Biol. 2004, 56, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hörtensteiner, S. Chlorophyll degradation during senescence. Annu. Rev. Plant Biol. 2006, 50, 55–77. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Sun, Y.; Zhao, G.; Liao, X.; Hu, X.; Wu, J.; Wang, Z. Optimization of ultrasound–assisted extraction of anthocyanins in red raspberries and identification of anthocyanins in extract using high–performance liquid chromatography–mass spectrometry. Ultrason. Sonochem. 2007, 14, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.F. Experimental Guidance for Plant Physiology; Higher Education Press: Beijing, China, 2006; pp. 74–223. [Google Scholar]
- Kenneth, J.; Livak, T.D. Analysis of relative gene expression data using Real–time quantitative PCR and the 2 –∆∆ct method. Method 2001, 25, 402–408. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. Mega7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| #ID | Function Annotation | FPKM | Log2 (FC) | ||||
|---|---|---|---|---|---|---|---|
| Green | Turning | Red | G vs. T | G vs. R | T vs. R | ||
| Carotenoid biosynthesis | |||||||
| c64566.graph_c0 | Geranylgeranyl pyrophosphate synthase (GGPPS) | 20.07 | 6.83 | 7.35 | −1.46 | −1.28 | 0.19 |
| c49043.graph_c0 | beta-carotene hydroxylase (BOH) | 28.14 | 9.83 | 4.58 | −1.45 | −2.5 | −1.04 |
| c68245.graph_c1 | zeaxanthin epoxidase (ZEP) | 60.19 | 3.2 | 4.5 | −4.18 | −3.62 | 0.57 |
| c69135.graph_c0 | zeaxanthin epoxidase (ZEP) | 17.99 | 27.12 | 46.58 | 0.64 | 1.49 | 0.86 |
| c64983.graph_c0 | 9-cis-epoxycarotenoid dioxygenase (NCEDs) | 10.53 | 2.88 | 1.45 | −1.78 | −3.22 | −1.44 |
| c65617.graph_c0 | 9-cis-epoxycarotenoid dioxygenase (NCEDs) | 49.88 | 16.86 | 8.44 | −1.5 | −2.44 | −0.94 |
| Chlorophyll degradation | |||||||
| c48268.graph_c0 | Chlorophyllase (CLH) | 43.88 | 14.36 | 51.63 | −1.46 | 0.45 | 1.92 |
| c56088.graph_c0 | Chlorophyllase (CLH) | 16.83 | 73.89 | 114.68 | 2.18 | 2.87 | 0.71 |
| c66184.graph_c4 | Chlorophyllase (CLH) | 2.24 | 0 | 0 | / | / | / |
| c69667.graph_c2 | Chlorophyllase (CLH) | 26.13 | 33.15 | 70.04 | 0.34 | 1.38 | 1.06 |
| c70181.graph_c0 | Mg-chelatase (MCS) | 6.77 | 17.3 | 15.64 | 1.44 | 1.34 | −0.08 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, X.; Sun, W.; Zou, X.; Liu, B.; Huang, W.; Chen, Z.; Li, Y.; Qiu, M.-Y.; Liu, Z.-J.; Mao, Y.; et al. Sequencing of Euscaphis konishii Endocarp Transcriptome Points to Molecular Mechanisms of Endocarp Coloration. Int. J. Mol. Sci. 2018, 19, 3209. https://doi.org/10.3390/ijms19103209
Yuan X, Sun W, Zou X, Liu B, Huang W, Chen Z, Li Y, Qiu M-Y, Liu Z-J, Mao Y, et al. Sequencing of Euscaphis konishii Endocarp Transcriptome Points to Molecular Mechanisms of Endocarp Coloration. International Journal of Molecular Sciences. 2018; 19(10):3209. https://doi.org/10.3390/ijms19103209
Chicago/Turabian StyleYuan, Xueyan, Weihong Sun, Xiaoxing Zou, Bobin Liu, Wei Huang, Zeming Chen, Yanlei Li, Meng-Yuan Qiu, Zhong-Jian Liu, Yanling Mao, and et al. 2018. "Sequencing of Euscaphis konishii Endocarp Transcriptome Points to Molecular Mechanisms of Endocarp Coloration" International Journal of Molecular Sciences 19, no. 10: 3209. https://doi.org/10.3390/ijms19103209