Heat Shock Proteins as a Potential Therapeutic Target in the Treatment of Gestational Diabetes Mellitus: What We Know so Far

 , , ,
, , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. A Crosslink between Gestational Diabetes Mellitus, Obesity, and Inflammation—A Brief Summary
3. The Role of Unfolded Protein Response in Insulin Signaling and Pancreatic β-Cell Function
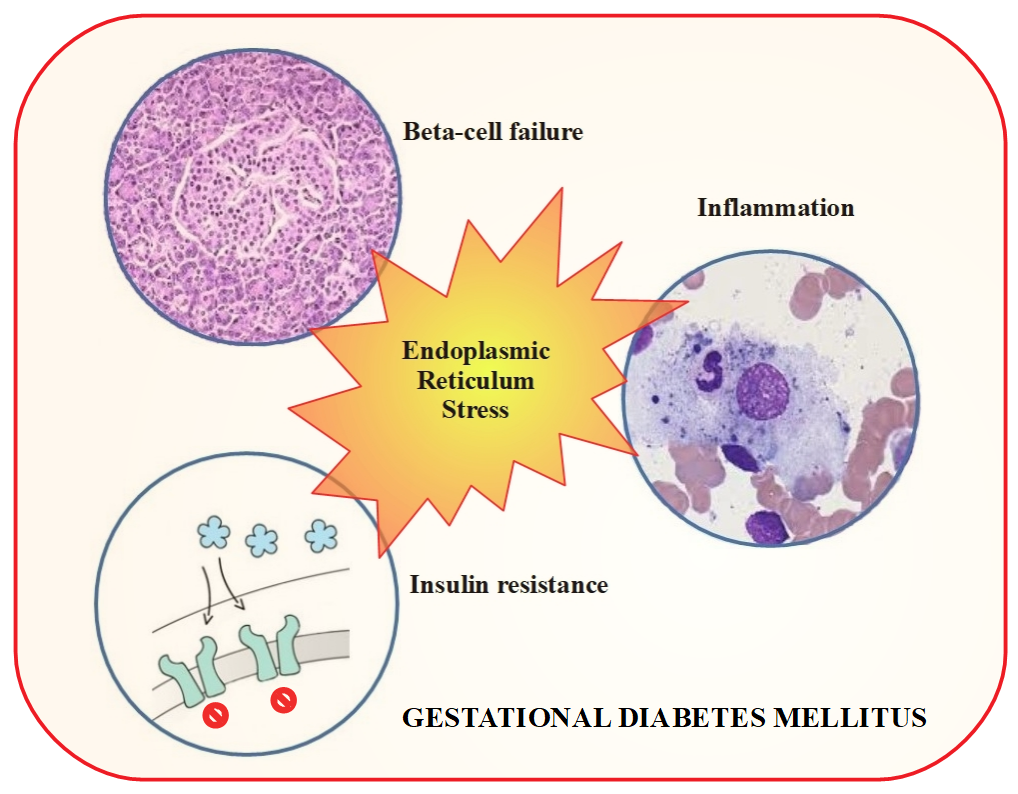
4. Gestational Diabetes Mellitus and Chaperones
5. The 70-kDa-Family (HSP70) of Chaperones
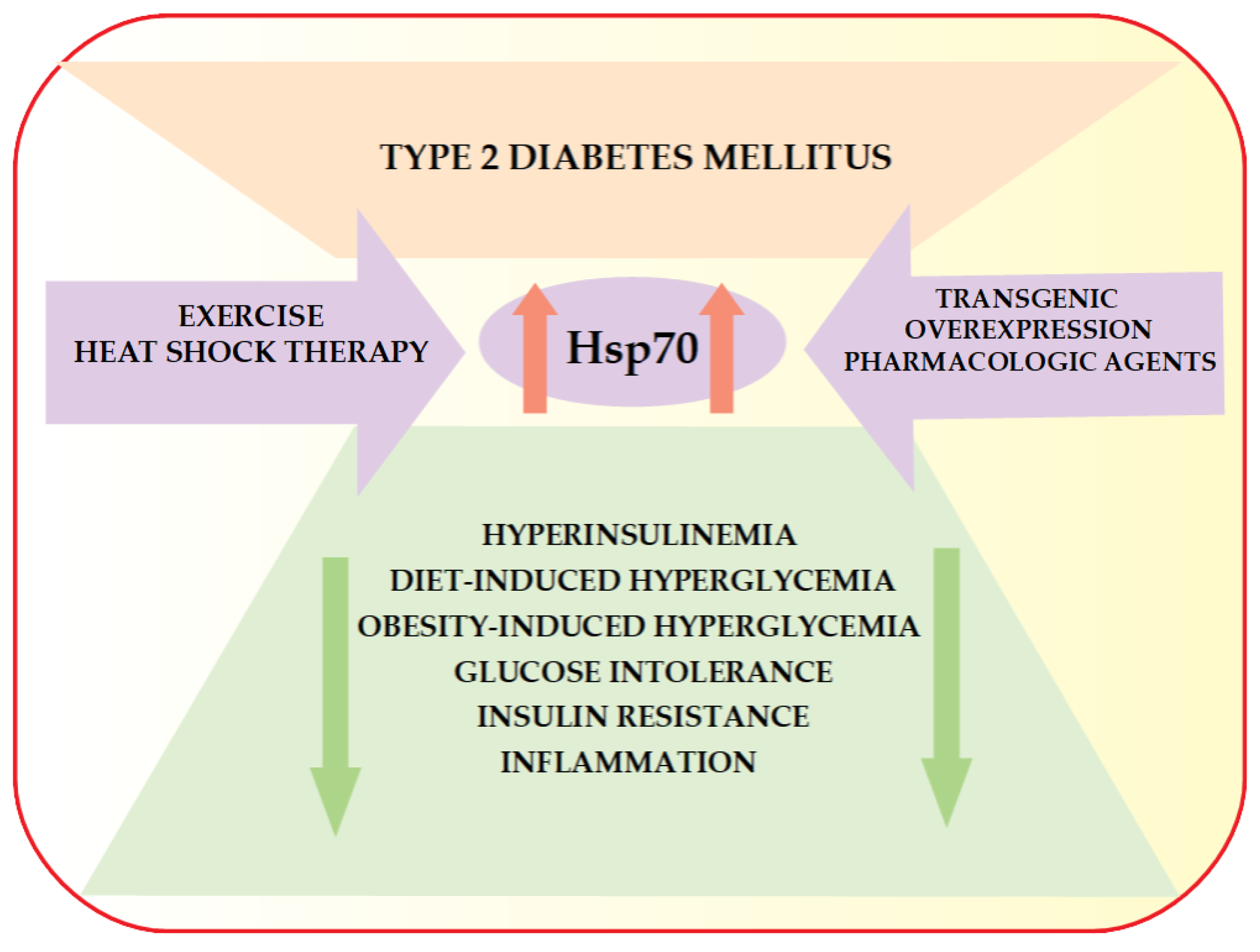
6. HSP70 and Type 2 Diabetes Mellitus
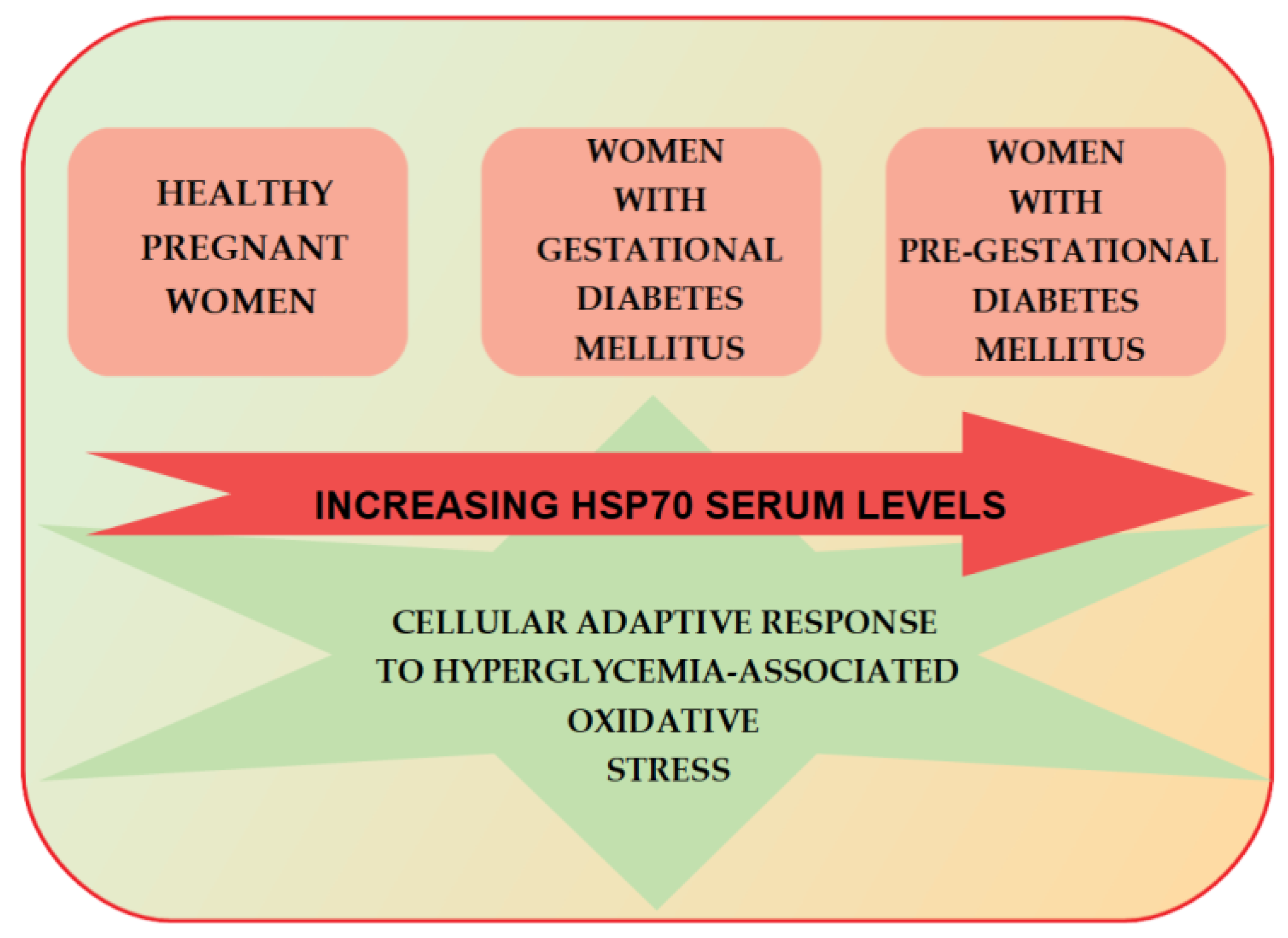
7. HSP70 and Gestational Diabetes Mellitus
8. Enhanced Endoplasmic Reticulum Stress and Gestational Diabetes Mellitus
9. Placental Endoplasmic Reticulum Stress in Gestational Diabetes Mellitus
10. Concluding Remarks
Funding
Conflicts of Interest
References
- American Diabetes Association. Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 2009, 32 (Suppl. 1), S62–S67. [Google Scholar] [CrossRef] [PubMed]
- Colagiuri, S.; Falavigna, M.; Agarwal, M.M.; Boulvain, M.; Coetzee, E.; Hod, M.; Meltzer, S.J.; Metzger, B.; Omori, Y.; Rasa, I.; et al. Strategies for implementing the WHO diagnostic criteria and classification of hyperglycaemia first detected in pregnancy. Diabetes Res. Clin. Pract. 2014, 103, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Zhen, X.M.; Li, X.; Chen, C. Longer-term outcomes in offspring of GDM mothers treated with metformin versus insulin. Diabetes Res. Clin. Pract. 2018, 144, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Hur, K.Y.; Lee, M.S. New mechanisms of metformin action: Focusing on mitochondria and the gut. J. Diabetes Investig. 2015, 6, 600–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaked, M.; Ketzinel-Gilad, M.; Cerasi, E.; Kaiser, N.; Leibowitz, G. AMP-activated protein kinase (AMPK) mediates nutrient regulation of thioredoxin-interacting protein (TXNIP) in pancreatic beta-cells. PLoS ONE 2011, 6, e28804. [Google Scholar] [CrossRef] [PubMed]
- Chai, T.F.; Hong, S.Y.; He, H.; Zheng, L.; Hagen, T.; Luo, Y.; Yu, F.X. A potential mechanism of metformin-mediated regulation of glucose homeostasis: Inhibition of Thioredoxin-interacting protein (Txnip) gene expression. Cell. Signal. 2012, 24, 1700–1705. [Google Scholar] [CrossRef] [PubMed]
- Sitia, R.; Braakman, I. Quality control in the endoplasmic reticulum protein factory. Nature 2003, 426, 891–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.Y.; Sappenfield, W.; Sharma, A.J.; Wilson, H.G.; Bish, C.L.; Salihu, H.M.; England, L.J. Racial/ethnic differences in the prevalence of gestational diabetes mellitus and maternal overweight and obesity, by nativity, Florida, 2004–2007. Obesity 2013, 21, E33–E40. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, T.A.; Xiang, A.H. Gestational diabetes mellitus. J. Clin. Investig. 2005, 115, 485–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalupahana, N.S.; Moustaid-Moussa, N.; Claycombe, K.J. Immunity as a link between obesity and insulin resistance. Mol. Asp. Med. 2012, 33, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.E.; Kirwan, J.P.; Jing, M.; Presley, L.; Catalano, P.M. Increased skeletal muscle tumor necrosis factor-alpha and impaired insulin signaling persist in obese women with gestational diabetes mellitus 1 year postpartum. Diabetes 2008, 57, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Aye, I.; Lager, S.; Ramirez, V.I.; Gaccioli, F.; Dudley, D.J.; Jansson, T.; Powell, T.L. Increasing maternal body mass index is associated with systemic inflammation in the mother and the activation of distinct placental inflammatory pathways. Biol. Reprod. 2014, 90, 129. [Google Scholar] [CrossRef] [PubMed]
- Ozyer, Ş.; Engin-Ustun, Y.; Uzunlar, O.; Katar, C.; Danışman, N. Inflammation and glycemic tolerance status in pregnancy: The role of maternal adiposity. Gynecol. Obstet. Investig. 2014, 78, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, E.; Ashouri, J.; Wolter, S.; Doerrie, A.; Dittrich-Breiholz, O.; Schneider, H.; Wagner, E.F.; Troppmair, J.; Mackman, N.; Kracht, M. Transcriptional regulation of EGR-1 by the interleukin-1-JNK-MKK7-c-Jun pathway. J. Biol. Chem. 2008, 283, 12120–12128. [Google Scholar] [CrossRef] [PubMed]
- Kleiblova, P.; Dostalova, I.; Bartlova, M.; Lacinova, Z.; Ticha, I.; Krejci, V.; Springer, D.; Kleibl, Z.; Haluzik, M. Expression of adipokines and estrogen receptors in adipose tissue and placenta of patients with gestational diabetes mellitus. Mol. Cell. Endocrinol. 2010, 314, 150–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stirm, L.; Kovárová, M.; Perschbacher, S.; Michlmaier, R.; Fritsche, L.; Siegel-Axel, D.; Schleicher, E.; Peter, A.; Pauluschke-Fröhlich, J.; Brucker, S.; et al. BMI-independent effects of gestational diabetes on human placenta. J. Clin. Endocrinol. Metab. 2018. [Google Scholar] [CrossRef] [PubMed]
- Aires, M.B.; Dos Santos, A.C. Effects of maternal diabetes on trophoblast cells. World J. Diabetes 2015, 6, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Desoye, G. The Human Placenta in Diabetes and Obesity: Friend or Foe? The 2017 Norbert Freinkel Award Lecture. Diabetes Care 2018, 41, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Zhang, K.; Kaufman, R.J. The unfolded protein response—A stress signaling pathway of the endoplasmic reticulum. J. Chem. Neuroanat. 2004, 28, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 2005, 35, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, Y.; Kaneto, H.; Kawamori, D.; Yoshiuchi, K.; Hatazaki, M.; Matsuoka, T.A.; Ozawa, K.; Ogawa, S.; Hori, M.; Yamasaki, Y.; et al. Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J. Biol. Chem. 2005, 280, 847–851. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Wang, C.H. ER stress in adipocytes and insulin resistance: Mechanisms and significance (Review). Mol. Med. Rep. 2014, 10, 2234–2240. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.W.; Chanda, D.; Yang, J.; Oh, H.; Kim, S.S.; Yoon, Y.S.; Hong, S.; Park, K.G.; Lee, I.K.; Choi, C.S.; et al. Regulation of hepatic gluconeogenesis by an ER-bound transcription factor, CREBH. Cell Metab. 2010, 11, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Görgün, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Görgün, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Kars, M.; Yang, L.; Gregor, M.F.; Mohammed, B.S.; Pietka, T.A.; Finck, B.N.; Patterson, B.W.; Horton, J.D.; Mittendorfer, B.; Hotamisligil, G.S.; et al. Tauroursodeoxycholic Acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes 2010, 59, 1899–1905. [Google Scholar] [CrossRef] [PubMed]
- Brozzi, F.; Eizirik, D.L. ER stress and the decline and fall of pancreatic beta cells in type 1 diabetes. Upsal. J. Med. Sci. 2016, 121, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Back, S.H.; Kaufman, R.J. Endoplasmic reticulum stress and type 2 diabetes. Annu. Rev. Biochem. 2012, 81, 767–793. [Google Scholar] [CrossRef] [PubMed]
- Costes, S. Targeting protein misfolding to protect pancreatic beta-cells in type 2 diabetes. Curr. Opin. Pharmacol. 2018, 43, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Cadavez, L.; Montane, J.; Alcarraz-Vizán, G.; Visa, M.; Vidal-Fŕbrega, L.; Servitja, J.M.; Novials, A. Chaperones ameliorate beta cell dysfunction associated with human islet amyloid polypeptide overexpression. PLoS ONE 2014, 9, e101797. [Google Scholar] [CrossRef] [PubMed]
- Irvine, A.G.; Wallis, A.K.; Sanghera, N.; Rowe, M.L.; Ruddock, L.W.; Howard, M.J.; Williamson, R.A.; Blindauer, C.A.; Freedman, R.B. Protein disulfide-isomerase interacts with a substrate protein at all stages along its folding pathway. PLoS ONE 2014, 9, e82511. [Google Scholar] [CrossRef] [PubMed]
- Khadir, A.; Kavalakatt, S.; Abubaker, J.; Cherian, P.; Madhu, D.; Al-Khairi, I.; Abu-Farha, M.; Warsame, S.; Elkum, N.; Dehbi, M.; et al. Physical exercise alleviates ER stress in obese humans through reduction in the expression and release of GRP78 chaperone. Metabolism 2016, 65, 1409–1420. [Google Scholar] [CrossRef] [PubMed]
- Sage, A.T.; Holtby-Ottenhof, S.; Shi, Y.; Damjanovic, S.; Sharma, A.M.; Werstuck, G.H. Metabolic syndrome and acute hyperglycemia are associated with endoplasmic reticulum stress in human mononuclear cells. Obesity 2012, 20, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, A009191. [Google Scholar] [CrossRef] [PubMed]
- Garamvölgyi, Z.; Prohászka, Z.; Rigó, J., Jr.; Kecskeméti, A.; Molvarec, A. Increased circulating heat shock protein 70 (HSPA1A) levels in gestational diabetes mellitus: A pilot study. Cell Stress Chaperones 2015, 20, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Liong, S.; Lappas, M. Endoplasmic reticulum stress is increased in adipose tissue of women with gestational diabetes. PLoS ONE 2015, 10, e0122633. [Google Scholar] [CrossRef] [PubMed]
- Liong, S.; Lappas, M. Endoplasmic reticulum stress regulates inflammation and insulin resistance in skeletal muscle from pregnant women. Mol. Cell. Endocrinol. 2016, 425, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Lappas, M. Activation of inflammasomes in adipose tissue of women with gestational diabetes. Mol. Cell. Endocrinol. 2014, 382, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Yung, H.W.; Alnćs-Katjavivi, P.; Jones, C.J.; El-Bacha, T.; Golic, M.; Staff, A.C.; Burton, G.J. Placental endoplasmic reticulum stress in gestational diabetes: The potential for therapeutic intervention with chemical chaperones and antioxidants. Diabetologia 2016, 59, 2240–2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radons, J. The human HSP70 family of chaperones: Where do we stand? Cell Stress Chaperones 2016, 21, 379–404. [Google Scholar] [CrossRef] [PubMed]
- Daugaard, M.; Rohde, M.; Jäättelä, M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinctfunctions. FEBS Lett. 2007, 581, 3702–3710. [Google Scholar] [CrossRef] [PubMed]
- Amorim, F.T.; Fonseca, I.T.; Machado-Moreira, C.A.; Magalhăes Fde, C. Insights into the role of heat shock protein 72 to whole-body heat acclimation in humans. Temperature 2015, 2, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Bellini, S.; Barutta, F.; Mastrocola, R.; Imperatore, L.; Bruno, G.; Gruden, G. Heat Shock Proteins in Vascular Diabetic Complications: Review and Future Perspective. Int. J. Mol. Sci. 2017, 18, 2709. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Nguyen, A.K.; Henstridge, D.C.; Holmes, A.G.; Chan, M.H.; Mesa, J.L.; Lancaster, G.I.; Southgate, R.J.; Bruce, C.R.; Duffy, S.J.; et al. HSP72 protects against obesity-induced insulin resistance. Proc. Natl. Acad. Sci. USA 2008, 105, 1739–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuzuki, T.; Kobayashi, H.; Yoshihara, T.; Kakigi, R.; Ichinoseki-Sekine, N.; Naito, H. Attenuation of exercise-induced heat shock protein 72 expression blunts improvements in whole-body insulin resistance in rats with type 2 diabetes. Cell Stress Chaperones 2017, 22, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupte, A.A.; Bomhoff, G.L.; Swerdlow, R.H.; Geiger, P.C. Heat treatment improves glucose tolerance and prevents skeletal muscle insulin resistance in rats fed a high-fat diet. Diabetes 2009, 58, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Henstridge, D.C.; Bruce, C.R.; Drew, B.G.; Tory, K.; Kolonics, A.; Estevez, E.; Chung, J.; Watson, N.; Gardner, T.; Lee-Young, R.S.; et al. Activating HSP72 in rodent skeletal muscle increases mitochondrial number and oxidative capacity and decreases insulin resistance. Diabetes 2014, 63, 1881–1894. [Google Scholar] [CrossRef] [PubMed]
- Xing, B.; Wang, L.; Li, Q.; Cao, Y.; Dong, X.; Liang, J.; Wu, X. Hsp70 plays an important role in high-fat diet induced gestational hyperglycemia in mice. J. Physiol. Biochem. 2015, 71, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Bruce, C.R.; Carey, A.L.; Hawley, J.A.; Febbraio, M.A. Intramuscular heat shock protein 72 and heme oxygenase-1 mRNA are reduced in patients with type 2 diabetes: Evidence that insulin resistance is associated with a disturbed antioxidant defense mechanism. Diabetes 2003, 52, 2338–2345. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Lee, J.S.; Huh, S.H.; Seo, J.S.; Choi, E.J. Hsp72 functions as a natural inhibitory protein of c-Jun N-terminal kinase. EMBO J. 2001, 20, 446–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meldrum, K.K.; Burnett, A.L.; Meng, X.; Misseri, R.; Shaw, M.B.; Gearhart, J.P.; Meldrum, D.R. Liposomal delivery of heat shock protein 72 into renal tubular cells blocks nuclear factor-kappaBactivation, tumor necrosis factor-alpha production, and subsequent ischemia-induced apoptosis. Circ. Res. 2003, 92, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Borges, T.J.; Lopes, R.L.; Pinho, N.G.; Machado, F.D.; Souza, A.P.; Bonorino, C. Extracellular Hsp70 inhibits pro-inflammatory cytokine production by IL-10 driven down-regulation of C/EBPβ and C/EBPδ. Int. J. Hyperth. 2013, 29, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Ono, K.; Kitano, S.; Matsuyama, R.; Goto, R.; Suico, M.A.; Kawasaki, S.; Igata, M.; Kawashima, J.; Motoshima, H.; et al. Mild Electrical Stimulation with Heat Shock Reduces Visceral Adiposity and Improves Metabolic Abnormalities in Subjects with Metabolic Syndrome or Type 2 Diabetes: Randomized Crossover Trials. EBioMedicine 2014, 1, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, F.F.; Haines, D.; Dashti, A.A.; El-Shazly, S.; Al-Najjar, F. Correlation between heat shock proteins, adiponectin, and T lymphocyte cytokine expression in type 2 diabetics. Cell Stress Chaperones 2018. [Google Scholar] [CrossRef] [PubMed]
- Nakhjavani, M.; Morteza, A.; Khajeali, L.; Esteghamati, A.; Khalilzadeh, O.; Asgarani, F.; Outeiro, T.F. Increased serum HSP70 levels are associated with the duration of diabetes. Cell Stress Chaperones 2010, 15, 959–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, L.; Martinus, R.D. Hyperglycaemia and oxidative stress upregulate HSP60 & HSP70 expression in HeLa cells. Springerplus 2013, 2, 431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jafarnejad, A.; Bathaie, S.Z.; Nakhjavani, M.; Hassan, M.Z.; Banasadegh, S. The improvement effect of L-Lys as a chemical chaperone on STZ-induced diabetic rats, protein structure and function. Diabetes Metab. Res. Rev. 2008, 24, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Mirmiranpour, H.; Khaghani, S.; Bathaie, S.Z.; Nakhjavani, M.; Kebriaeezadeh, A.; Ebadi, M.; Gerayesh-Nejad, S.; Zangooei, M. The Preventive Effect of L-Lysine on Lysozyme Glycation in Type 2 Diabetes. Acta Med. Iran. 2016, 54, 24–31. [Google Scholar] [PubMed]
- Rosas, P.C.; Nagaraja, G.M.; Kaur, P.; Panossian, A.; Wickman, G.; Garcia, L.R.; Al-Khamis, F.A.; Asea, A.A. Hsp72 (HSPA1A) Prevents Human Islet Amyloid Polypeptide Aggregation and Toxicity: A New Approach for Type 2 Diabetes Treatment. PLoS ONE 2016, 11, e0149409. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, S.; Doulaveris, G.; Orfanelli, T.; Arantes, M.; Damasceno, D.; Calderon, I.; Rudge, M.V.; Witkin, S.S. Induction of the 72 kDa heat shock protein by glucose ingestion in black pregnant women. Cell Stress Chaperones 2013, 18, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Simar, D.; Jacques, A.; Caillaud, C. Heat shock proteins induction reduces stress kinases activation, potentially improving insulin signalling in monocytes from obese subjects. Cell Stress Chaperones 2012, 17, 615–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, X.Y.; Wang, H.; Bai, F.; Zhou, X.; Li, S.P.; Ren, L.P.; Sun, R.Q.; Xue, C.C.; Jiang, H.L.; Hu, L.H.; et al. Identification of matrine as a promising novel drug for hepatic steatosis and glucose intolerance with HSP72 as an upstream target. Br. J. Pharmacol. 2015, 172, 4303–4318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colomiere, M.; Permezel, M.; Lappas, M. Diabetes and obesity during pregnancy alter insulin signalling and glucose transporter expression in maternal skeletal muscle and subcutaneous adipose tissue. J. Mol. Endocrinol. 2010, 44, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Meng, S.; Cao, J.; He, Q.; Xiong, L.; Chang, E.; Radovick, S.; Wondisford, F.E.; He, L. Metformin activates AMP-activated protein kinase by promoting formation of the αβγ heterotrimeric complex. J. Biol. Chem. 2015, 290, 3793–3802. [Google Scholar] [CrossRef] [PubMed]
- Liong, S.; Lappas, M. Activation of AMPK improves inflammation and insulin resistance in adipose tissue and skeletal muscle from pregnant women. J. Physiol. Biochem. 2015, 71, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Morisaki, H.; Toyama, K.; Sugimoto, N.; Shintani, T.; Tandelilin, A.; Hirase, T.; Holmes, E.W.; Morisaki, T. AMPD1: A novel therapeutic target for reversing insulin resistance. BMC Endocr. Disord. 2014, 14, 96. [Google Scholar] [CrossRef] [PubMed]
- Tandelilin, A.A.; Hirase, T.; Hudoyo, A.W.; Cheng, J.; Toyama, K.; Morisaki, H.; Morisaki, T. AMPD1 regulates mTORC1-p70 S6 kinase axis in the control of insulin sensitivity in skeletal muscle. BMC Endocr. Disord. 2015, 15, 11. [Google Scholar] [CrossRef]
- Hudoyo, A.W.; Hirase, T.; Tandelillin, A.; Honda, M.; Shirai, M.; Cheng, J.; Morisaki, H.; Morisaki, T. Role of AMPD2 in impaired glucose tolerance induced by high fructose diet. Mol. Genet. Metab. Rep. 2017, 13, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, J.; Parakhia, R.A.; Ochs, R.S. Metformin activates AMP kinase through inhibition of AMP deaminase. J. Biol. Chem. 2011, 286, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Beuers, U. Drug insight: Mechanisms and sites of action of ursodeoxycholic acid in cholestasis. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Challier, J.C.; Basu, S.; Bintein, T.; Minium, J.; Hotmire, K.; Catalano, P.M.; Hauguel-de Mouzon, S. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta 2008, 29, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Hauguel-de Mouzon, S.; Guerre-Millo, M. The placenta cytokine network and inflammatory signals. Placenta 2006, 27, 794–798. [Google Scholar] [CrossRef] [PubMed]
- Schmatz, M.; Madan, J.; Marino, T.; Davis, J. Maternal obesity: The interplay between inflammation, mother and fetus. J. Perinatol. 2010, 30, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Jansson, N.; Rosario, F.J.; Gaccioli, F.; Lager, S.; Jones, H.N.; Roos, S.; Jansson, T.; Powell, T.L. Activation of placental mTOR signaling and amino acid transporters in obese women giving birth to large babies. J. Clin. Endocrinol. Metab. 2013, 98, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Yung, H.W.; Korolchuk, S.; Tolkovsky, A.M.; Charnock-Jones, D.S.; Burton, G.J. Endoplasmic reticulum stress exacerbates ischemia-reperfusion-induced apoptosis through attenuation of Akt protein synthesis in human choriocarcinoma cells. FASEB J. 2007, 21, 872–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biri, A.; Onan, A.; Devrim, E.; Babacan, F.; Kavutcu, M.; Durak, I. Oxidant status in maternal and cord plasma and placental tissue in gestational diabetes. Placenta 2006, 27, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Araújo, J.R.; Ramalho, C.; Correia-Branco, A.; Faria, A.; Ferraz, T.; Keating, E.; Martel, F. A parallel increase in placental oxidative stress and antioxidant defenses occurs in pre-gestational type 1 but not gestational diabetes. Placenta 2013, 34, 1095–1098. [Google Scholar] [CrossRef] [PubMed]
- Gauster, M.; Majali-Martinez, A.; Maninger, S.; Gutschi, E.; Greimel, P.H.; Ivanisevic, M.; Djelmis, J.; Desoye, G.; Hiden, U. Maternal Type 1 diabetes activates stress response in early placenta. Placenta 2017, 50, 110–116. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skórzyńska-Dziduszko, K.E.; Kimber-Trojnar, Ż.; Patro-Małysza, J.; Stenzel-Bembenek, A.; Oleszczuk, J.; Leszczyńska-Gorzelak, B. Heat Shock Proteins as a Potential Therapeutic Target in the Treatment of Gestational Diabetes Mellitus: What We Know so Far. Int. J. Mol. Sci. 2018, 19, 3205. https://doi.org/10.3390/ijms19103205
Skórzyńska-Dziduszko KE, Kimber-Trojnar Ż, Patro-Małysza J, Stenzel-Bembenek A, Oleszczuk J, Leszczyńska-Gorzelak B. Heat Shock Proteins as a Potential Therapeutic Target in the Treatment of Gestational Diabetes Mellitus: What We Know so Far. International Journal of Molecular Sciences. 2018; 19(10):3205. https://doi.org/10.3390/ijms19103205
Chicago/Turabian StyleSkórzyńska-Dziduszko, Katarzyna E., Żaneta Kimber-Trojnar, Jolanta Patro-Małysza, Agnieszka Stenzel-Bembenek, Jan Oleszczuk, and Bożena Leszczyńska-Gorzelak. 2018. "Heat Shock Proteins as a Potential Therapeutic Target in the Treatment of Gestational Diabetes Mellitus: What We Know so Far" International Journal of Molecular Sciences 19, no. 10: 3205. https://doi.org/10.3390/ijms19103205