cGMP Imaging in Brain Slices Reveals Brain Region-Specific Activity of NO-Sensitive Guanylyl Cyclases (NO-GCs) and NO-GC Stimulators

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
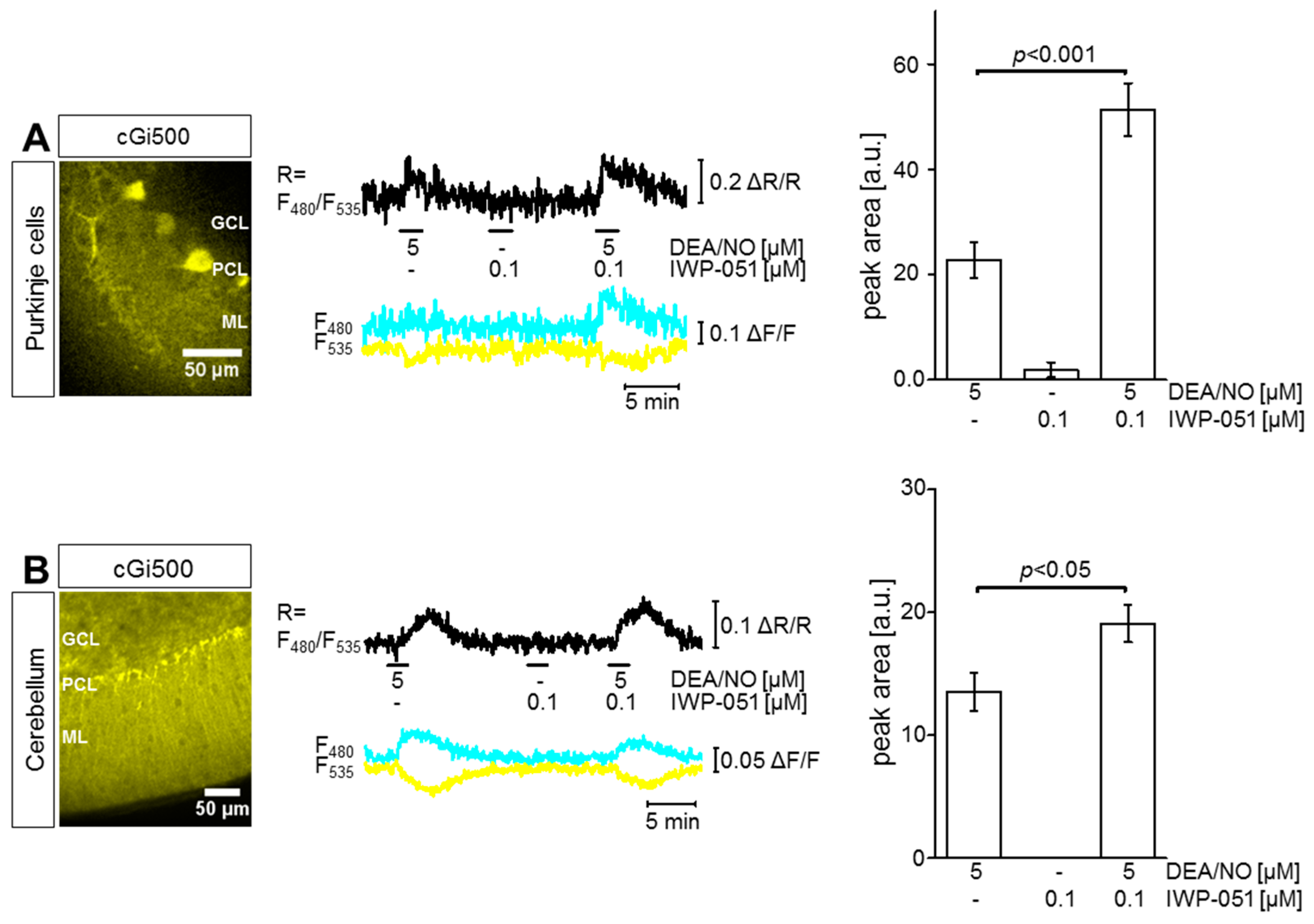
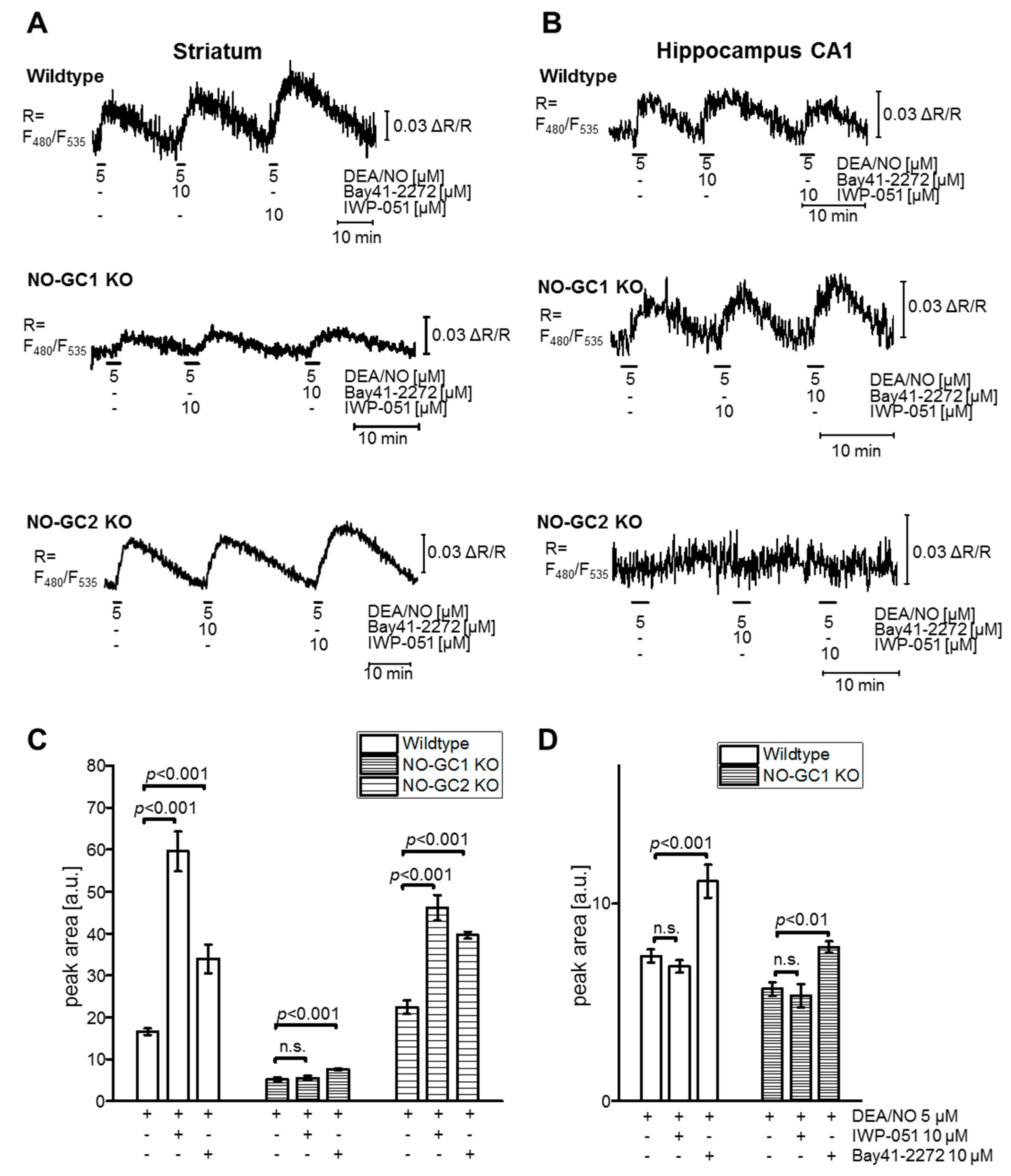
2.1. IWP-051 Potentiates NO-Induced cGMP Signals in Purkinje Cells and Granule Neurons of the Cerebellum as Well as in the Striatum
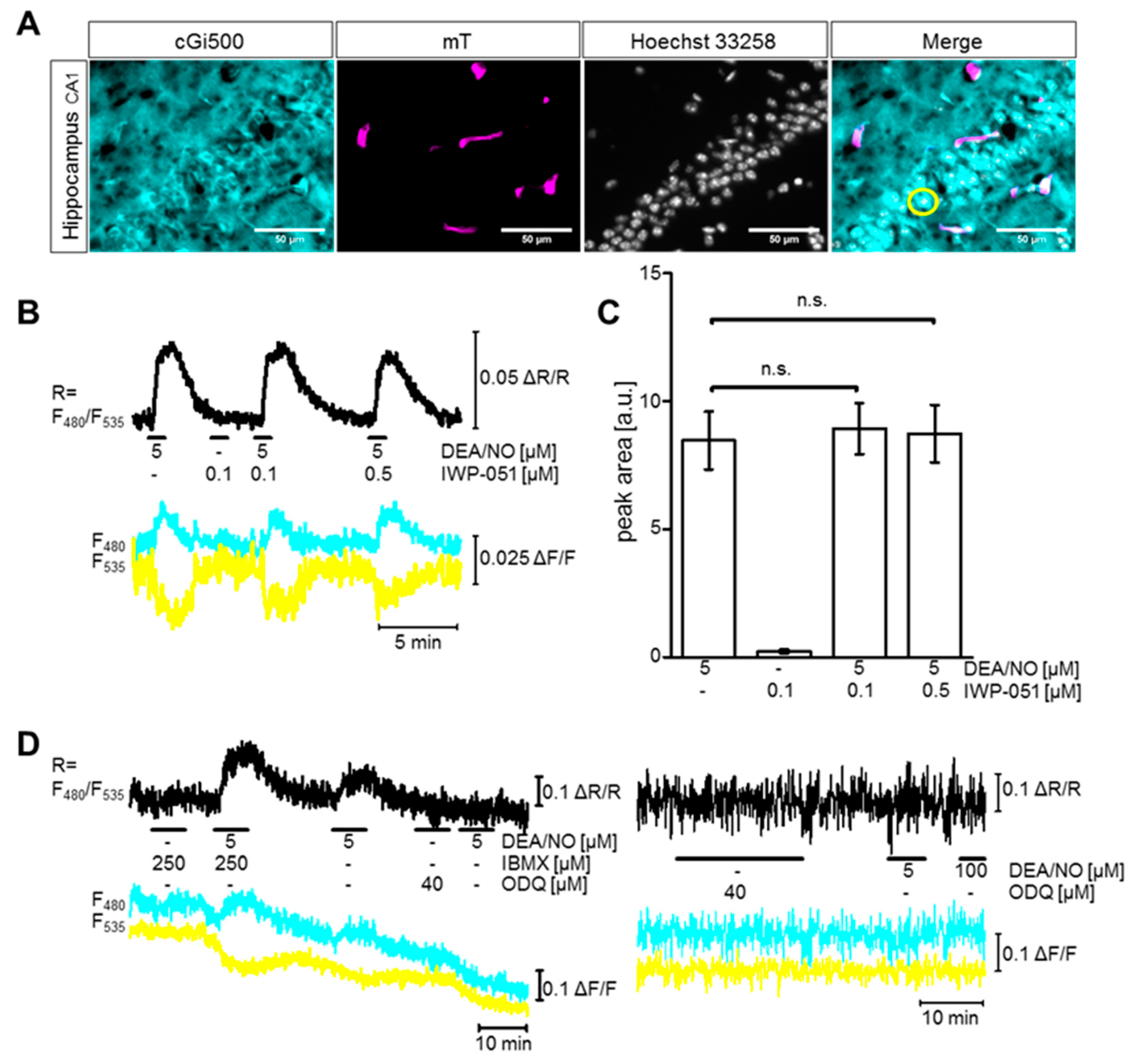
2.2. IWP-051 Has No Detectable Effect on NO-Induced cGMP Signals in the Hippocampal CA1 Area of Acute Brain Slices
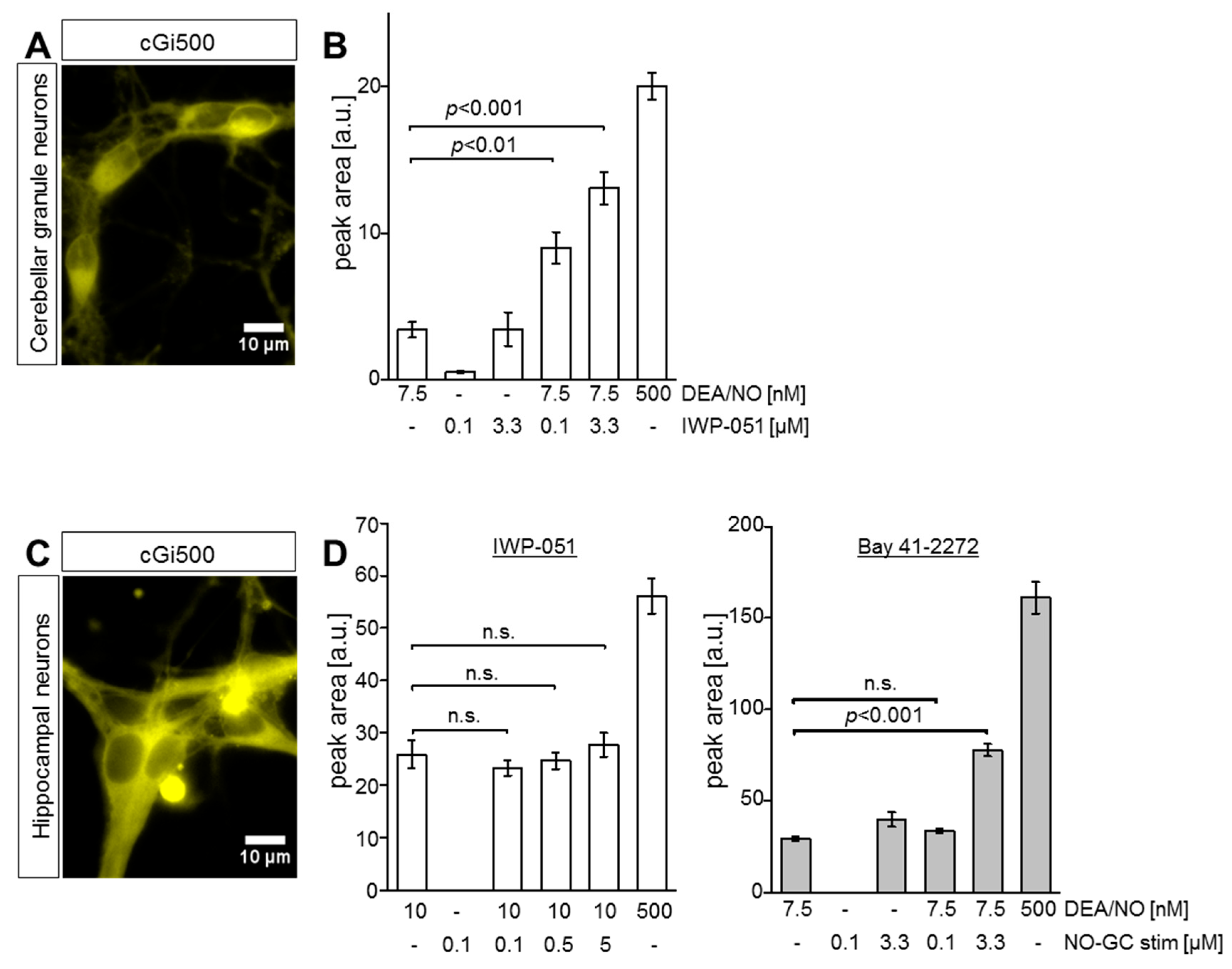
2.3. IWP-051 Potentiates NO-Induced cGMP Signals in Primary Cerebellar Granule Neurons, But Not in Primary Hippocampal Neurons
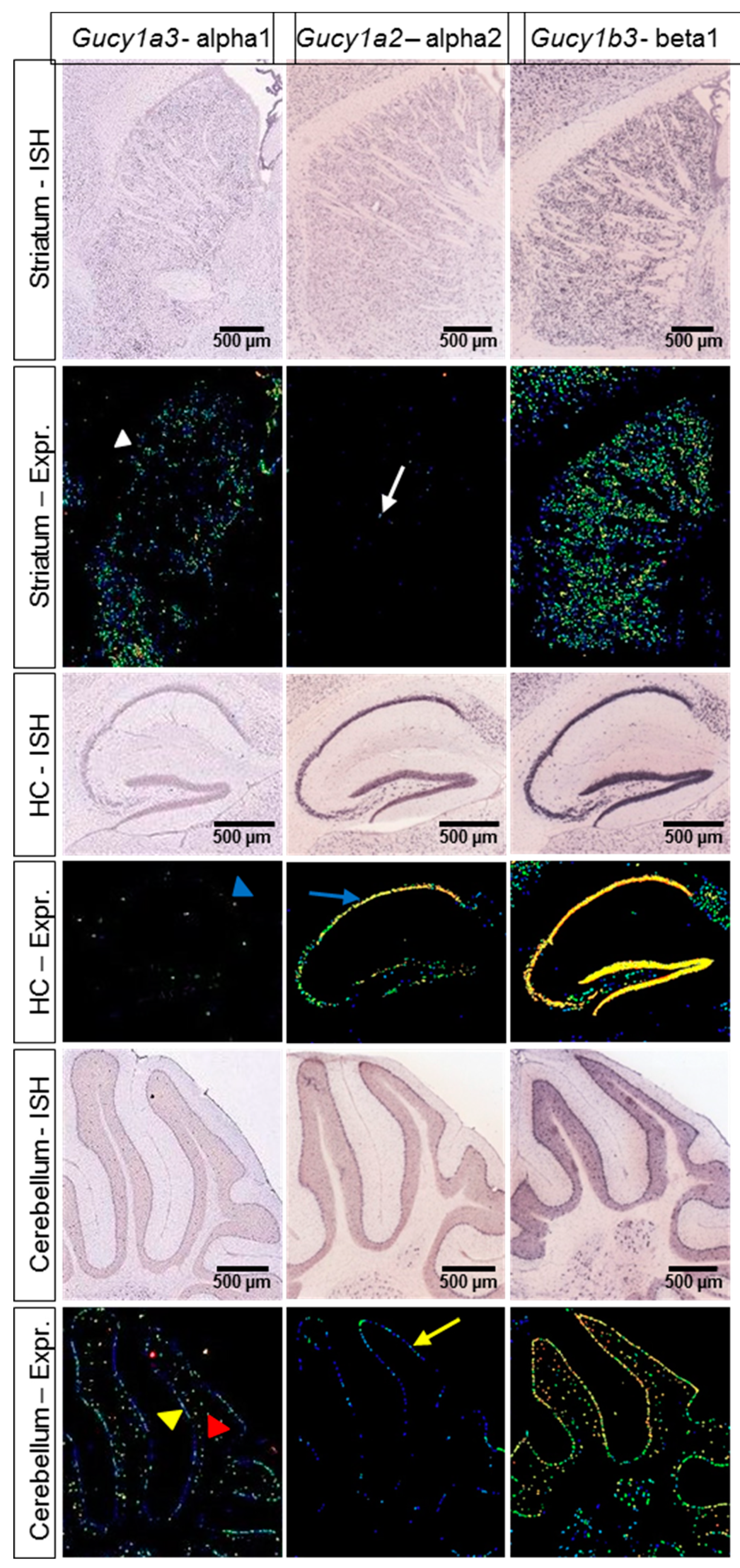
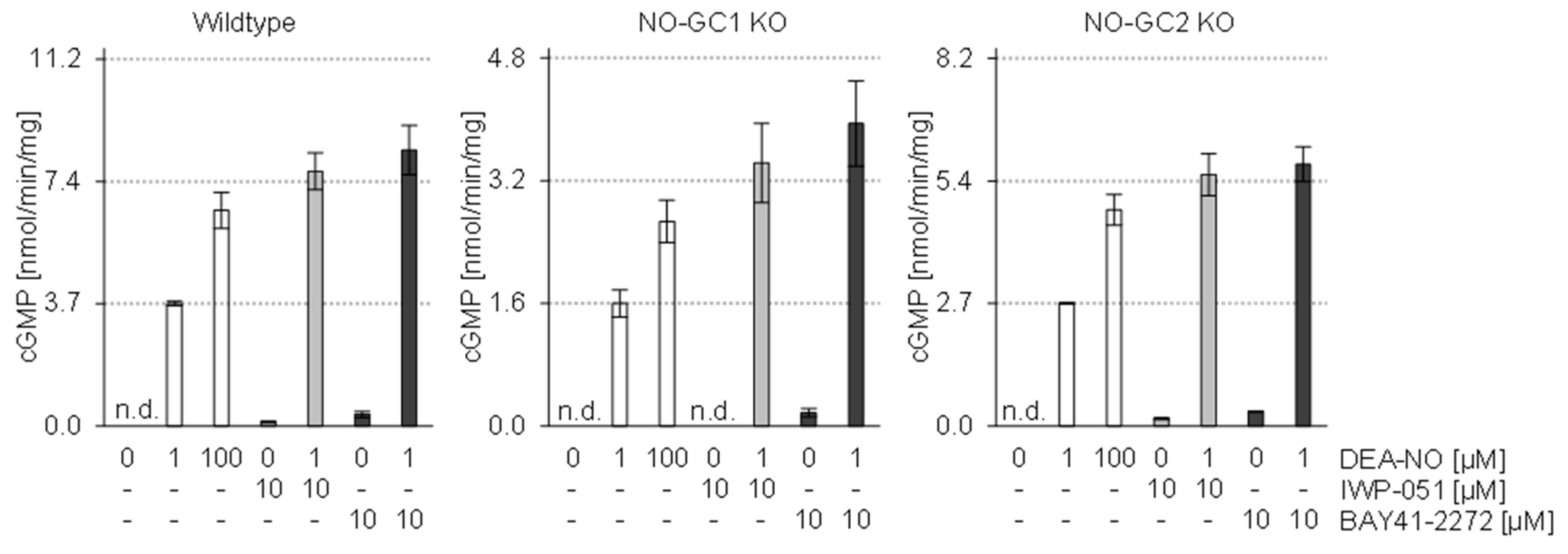
2.4. Expression Data and cGMP Imaging in Knockout Models Indicate a NO-GC1-Specific Activity of IWP-051
2.5. In Vitro Analysis of Brain Homogenates Does Not Show Isoform-Specific Activity of IWP-051
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Drugs
4.3. Cell Culture
4.4. Preparation of Acute Brain Slices and Fixed Brain Sections
4.5. Imaging
4.6. Data Base Research
4.7. Determination of cGMP-Forming Activities
4.8. Analysis and Statistics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ANP | Atrial natriuretic peptide |
| BSA | Bovine serum albumin |
| CAG | CMV immediate early enhancer, chicken β-actin and rabbit β-globin |
| CFP | Cyan fluorescent protein |
| cGi | cGMP indicator |
| cGMP | Cyclic 3’ 5’ guanosine monophosphate |
| CGN | Cerebellar granule neuron |
| CNP | C-type natriuretic peptide |
| DEA/NO | 2-(N,N-diethylamino)-diazenolate-2-oxide diethylammonium salt |
| DMEM | Dulbecco’s modified Eagle medium |
| FRET | Förster/Fluorescence resonance energy transfer |
| GCL | granule cell layer |
| HN | Hippocampal neuron |
| IBMX | 3-isobutyl-1-methylxanthin |
| NO | Nitric oxide |
| NO-GC | NO-sensitive guanylyl cyclase |
| ODQ | 1H-[1,2,4]oxadiazolo[4 ,3-a]quinoxalin-1-one |
| PBS | Phosphate buffered saline |
| PDE | Phosphodiesterase |
| ROI | Region of interest |
| YFP | Yellow fluorescent protein |
References
- Feil, R.; Kemp-Harper, B. cGMP signalling: From bench to bedside. EMBO Rep. 2006, 7, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Kemp-Harper, B.; Feil, R. Meeting report: cGMP matters. Sci. Signal. 2008, 1, pe12. [Google Scholar] [CrossRef] [PubMed]
- Friebe, A.; Koesling, D. The function of NO-sensitive guanylyl cyclase: What we can learn from genetic mouse models. Nitric Oxide 2009, 21, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Pyriochou, A.; Papapetropoulos, A. Soluble guanylyl cyclase: More secrets revealed. Cell. Signal. 2005, 17, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M. Molecular physiology of membrane guanylyl cyclase receptors. Physiol. Rev. 2016, 96, 751–804. [Google Scholar] [CrossRef] [PubMed]
- Russwurm, M.; Behrends, S.; Harteneck, C.; Koesling, D. Functional properties of a naturally occurring isoform of soluble guanylyl cyclase. Biochem. J. 1998, 335, 125–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wedel, B.; Humbert, P.; Harteneck, C.; Foerster, J.; Malkewitz, J.; Böhme, E.; Schultz, G.; Koesling, D. Mutation of His-105 in the beta 1 subunit yields a nitric oxide-insensitive form of soluble guanylyl cyclase. Proc. Natl. Acad. Sci. USA 1994, 91, 2592–2596. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.M.; Schramm, M.; Schröder, H.; Wunder, F.; Stasch, J.-P. Identification of residues crucially involved in the binding of the heme moiety of soluble guanylate cyclase. J. Biol. Chem. 2004, 279, 3025–3032. [Google Scholar] [CrossRef] [PubMed]
- Wolin, M.S.; Wood, K.S.; Ignarro, L.J. Guanylate cyclase from bovine lung. A kinetic analysis of the regulation of the purified soluble enzyme by protoporphyrin IX, heme, and nitrosyl-heme. J. Biol. Chem. 1982, 257, 13312–13320. [Google Scholar] [PubMed]
- Stone, J.R.; Marletta, M.A. Soluble guanylate cyclase from bovine lung: Activation with nitric oxide and carbon monoxide and spectral characterization of the ferrous and ferric states. Biochemistry 1994, 33, 5636–5640. [Google Scholar] [CrossRef] [PubMed]
- Russwurm, M.; Koesling, D. NO activation of guanylyl cyclase. EMBO J. 2004, 23, 4443–4450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cary, S.P.L.; Winger, J.A.; Marletta, M.A. Tonic and acute nitric oxide signaling through soluble guanylate cyclase is mediated by nonheme nitric oxide, ATP, and GTP. Proc. Natl. Acad. Sci. USA 2005, 102, 13064–13069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlossmann, J.; Schinner, E. cGMP becomes a drug target. Naunyn-Schmiedebergs Arch. Pharmacol. 2012, 385, 243–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chirkov, Y.Y.; Horowitz, J.D. Impaired tissue responsiveness to organic nitrates and nitric oxide: A new therapeutic frontier? Pharmacol. Ther. 2007, 116, 287–305. [Google Scholar] [CrossRef] [PubMed]
- Friebe, A.; Schultz, G.; Koesling, D. Sensitizing soluble guanylyl cyclase to become a highly CO-sensitive enzyme. EMBO J. 1996, 15, 6863–6868. [Google Scholar] [PubMed]
- Russwurm, M.; Mergia, E.; Mullershausen, F.; Koesling, D. Inhibition of deactivation of NO-sensitive guanylyl cyclase accounts for the sensitizing effect of YC-1. J. Biol. Chem. 2002, 277, 24883–24888. [Google Scholar] [CrossRef] [PubMed]
- Stasch, J.-P.; Pacher, P.; Evgenov, O.V. Soluble guanylate cyclase as an emerging therapeutic target in cardiopulmonary disease. Circulation 2011, 123, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Evgenov, O.V.; Pacher, P.; Schmidt, P.M.; Haskó, G.; Schmidt, H.H.H.W.; Stasch, J.-P. NO-independent stimulators and activators of soluble guanylate cyclase: Discovery and therapeutic potential. Nat. Rev. Drug Discov. 2006, 5, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Stasch, J.-P.; Becker, E.M.; Alonso-Alija, C.; Apeler, H.; Dembowsky, K.; Feurer, A.; Gerzer, R.; Minuth, T.; Perzborn, E.; Pleiß, U.; et al. NO-independent regulatory site on soluble guanylate cyclase. Nature 2001, 410, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Mullershausen, F.; Russwurm, M.; Friebe, A.; Koesling, D. Inhibition of phosphodiesterase type 5 by the activator of nitric oxide-sensitive guanylyl cyclase BAY 41-2272. Circulation 2004, 109, 1711–1713. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, E.; Stasch, J.-P. Effects of the sGC stimulator BAY 41-2272 are not mediated by phosphodiesterase 5 inhibition * Response. Circulation 2004, 110, e320–e321. [Google Scholar] [CrossRef] [PubMed]
- Schlaich, M.P.; Parnell, M.M.; Ahlers, B.A.; Finch, S.; Marshall, T.; Zhang, W.-Z.; Kaye, D.M. Impaired L-arginine transport and endothelial function in hypertensive and genetically predisposed normotensive subjects. Circulation 2004, 110, 3680–3686. [Google Scholar] [CrossRef] [PubMed]
- Taddei, S.; Virdis, A.; Mattei, P.; Ghiadoni, L.; Sudano, I.; Salvetti, A. Defective L-arginine-nitric oxide pathway in offspring of essential hypertensive patients. Circulation 1996, 94, 1298–1303. [Google Scholar] [CrossRef] [PubMed]
- Evgenov, O.V.; Kohane, D.S.; Bloch, K.D.; Stasch, J.-P.; Volpato, G.P.; Bellas, E.; Evgenov, N.V.; Buys, E.S.; Gnoth, M.J.; Graveline, A.R.; et al. Inhaled agonists of soluble guanylate cyclase induce selective pulmonary vasodilation. Am. J. Respir. Crit. Care Med. 2007, 176, 1138–1145. [Google Scholar] [CrossRef] [PubMed]
- Mittendorf, J.; Weigand, S.; Alonso-Alija, C.; Bischoff, E.; Feurer, A.; Gerisch, M.; Kern, A.; Knorr, A.; Lang, D.; Muenter, K.; et al. Discovery of riociguat (BAY 63-2521): A potent, oral stimulator of soluble guanylate cyclase for the treatment of pulmonary hypertension. ChemMedChem 2009, 4, 853–865. [Google Scholar] [CrossRef] [PubMed]
- Binder, C.; Zotter-Tufaro, C.; Bonderman, D. Riociguat for the treatment of pulmonary hypertension: A safety evaluation. Expert Opin. Drug Saf. 2016, 15, 1671–1677. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, E.; Jena, A.; George, M. Riociguat: Something new in pulmonary hypertension therapeutics? J. Pharmacol. Pharmacother. 2015, 6, 3–6. [Google Scholar] [PubMed]
- Nakai, T.; Perl, N.R.; Barden, T.C.; Carvalho, A.; Fretzen, A.; Germano, P.; Im, G.-Y.J.; Jin, H.; Kim, C.; Lee, T.W.-H.; et al. Discovery of IWP-051, a novel orally bioavailable sGC stimulator with once-daily dosing potential in humans. ACS Med. Chem. Lett. 2016, 7, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Cossenza, M.; Socodato, R.; Portugal, C.C.; Domith, I.C.L.; Gladulich, L.F.H.; Encarnação, T.G.; Calaza, K.C.; Mendonça, H.R.; Campello-Costa, P.; Paes-de-Carvalho, R. Nitric oxide in the nervous system: Biochemical, developmental, and neurobiological aspects. Vitam. Horm. 2014, 96, 79–125. [Google Scholar] [PubMed]
- Kleppisch, T.; Feil, R. cGMP signalling in the mammalian brain: Role in synaptic plasticity and behaviour. Handb. Exp. Pharmacol. 2009, 191, 549–579. [Google Scholar]
- Thunemann, M.; Wen, L.; Hillenbrand, M.; Vachaviolos, A.; Feil, S.; Ott, T.; Han, X.; Fukumura, D.; Jain, R.K.; Russwurm, M.; et al. Transgenic mice for cGMP imaging. Circ. Res. 2013, 113, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Russwurm, M.; Mullershausen, F.; Friebe, A.; Jäger, R.; Russwurm, C.; Koesling, D. Design of fluorescence resonance energy transfer (FRET)-based cGMP indicators: A systematic approach. Biochem. J. 2007, 407, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Thunemann, M.; Fomin, N.; Krawutschke, C.; Russwurm, M.; Feil, R. Visualization of cGMP with cGi biosensors. Methods Mol. Biol. 2013, 1020, 89–120. [Google Scholar] [PubMed]
- Thunemann, M.; Schmidt, K.; de Wit, C.; Han, X.; Jain, R.K.; Fukumura, D.; Feil, R. Correlative intravital imaging of cGMP signals and vasodilation in mice. Front. Physiol. 2014, 5, 394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Threlfell, S.; West, A.R. Modulation of striatal neuron activity by cyclic nucleotide signalling and phosphodiesterase inhibition. Basal Ganglia 2013, 3, 137–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lein, E.S.; Hawrylycz, M.J.; Ao, N.; Ayres, M.; Bensinger, A.; Bernard, A.; Boe, A.F.; Boguski, M.S.; Brockway, K.S.; Byrnes, E.J.; et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 2007, 445, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Gibb, B.J.; Garthwaite, J. Subunits of the nitric oxide receptor, soluble guanylyl cyclase, expressed in rat brain. Eur. J. Neurosci. 2001, 13, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Mergia, E.; Russwurm, M.; Zoidl, G.; Koesling, D. Major occurrence of the new alpha2beta1 isoform of NO-sensitive guanylyl cyclase in brain. Cell Signal. 2003, 15, 189–195. [Google Scholar] [CrossRef]
- Keravis, T.; Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) isozymes as targets of the intracellular signalling network: Benefits of PDE inhibitors in various diseases and perspectives for future therapeutic developments. Br. J. Pharmacol. 2012, 165, 1288–1305. [Google Scholar] [CrossRef] [PubMed]
- Shafiee-Nick, R.; Afshari, A.R.; Mousavi, S.H.; Rafighdoust, A.; Askari, V.R.; Mollazadeh, H.; Fanoudi, S.; Mohtashami, E.; Rahimi, V.B.; Mohebbi, M.; et al. A comprehensive review on the potential therapeutic benefits of phosphodiesterase inhibitors on cardiovascular diseases. Biomed. Pharmacother. 2017, 94, 541–556. [Google Scholar] [CrossRef] [PubMed]
- Sandner, P. From molecules to patients: Exploring the therapeutic role of soluble guanylate cyclase stimulators. Biol. Chem. 2018, 399, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Sandner, P.; Stasch, J.P. Anti-fibrotic effects of soluble guanylate cyclase stimulators and activators: A review of the preclinical evidence. Respir. Med. 2017, 122, S1–S9. [Google Scholar] [CrossRef] [PubMed]
- Feil, R.; Hartmann, J.; Luo, C.; Wolfsgruber, W.; Schilling, K.; Feil, S.; Barski, J.J.; Meyer, M.; Konnerth, A.; De Zeeuw, C.I.; et al. Impairment of LTD and cerebellar learning by Purkinje cell-specific ablation of cGMP-dependent protein kinase I. J. Cell Biol. 2003, 163, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.L.; Emmett, M.R.; Wood, J.A. Involvement of granule, basket and stellate neurons but not Purkinje or Golgi cells in cerebellar cGMP increases in vivo. Life Sci. 1994, 54, 615–620. [Google Scholar] [CrossRef]
- Southam, E.; Morris, R.; Garthwaite, J. Sources and targets of nitric oxide in rat cerebellum. Neurosci. Lett. 1992, 137, 241–244. [Google Scholar] [CrossRef]
- Kleppisch, T.; Pfeifer, A.; Klatt, P.; Ruth, P.; Montkowski, A.; Fässler, R.; Hofmann, F. Long-term potentiation in the hippocampal CA1 region of mice lacking cGMP-dependent kinases is normal and susceptible to inhibition of nitric oxide synthase. J. Neurosci. 1999, 19, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Kleppisch, T.; Wolfsgruber, W.; Feil, S.; Allmann, R.; Wotjak, C.T.; Goebbels, S.; Nave, K.-A.; Hofmann, F.; Feil, R. Hippocampal cGMP-dependent protein kinase I supports an age- and protein synthesis-dependent component of long-term potentiation but is not essential for spatial reference and contextual memory. J. Neurosci. 2003, 23, 6005–6012. [Google Scholar] [CrossRef] [PubMed]
- Mergia, E.; Koesling, D.; Friebe, A. Genetic mouse models of the NO receptor ‘soluble’ guanylyl cyclases. Handb. Exp. Pharmacol. 2009, 191, 33–46. [Google Scholar]
- Russwurm, M.; Wittau, N.; Koesling, D. Guanylyl cyclase/PSD-95 interaction. J. Biol. Chem. 2001, 276, 44647–44652. [Google Scholar] [CrossRef] [PubMed]
- Barski, J.J.; Dethleffsen, K.; Meyer, M. Cre recombinase expression in cerebellar Purkinje cells. Genesis 2000, 28, 93–98. [Google Scholar] [CrossRef]
- Tronche, F.; Kellendonk, C.; Kretz, O.; Gass, P.; Anlag, K.; Orban, P.C.; Bock, R.; Klein, R.; Schütz, G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet. 1999, 23, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Mergia, E.; Friebe, A.; Dangel, O.; Russwurm, M.; Koesling, D. Spare guanylyl cyclase NO receptors ensure high NO sensitivity in the vascular system. J. Clin. Investig. 2006, 116, 1731–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, H.; Peters, S.; Frank, K.; Wen, L.; Feil, R.; Rathjen, F.G. Dorsal root ganglion axon bifurcation tolerates increased cyclic GMP levels: The role of phosphodiesterase 2A and scavenger receptor Npr3. Eur. J. Neurosci. 2016, 44, 2991–3000. [Google Scholar] [CrossRef] [PubMed]
- Russwurm, M.; Koesling, D. Purification and characterization of NO-sensitive guanylyl cyclase. Methods Enzymol. 2005, 396, 492–501. [Google Scholar] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peters, S.; Paolillo, M.; Mergia, E.; Koesling, D.; Kennel, L.; Schmidtko, A.; Russwurm, M.; Feil, R. cGMP Imaging in Brain Slices Reveals Brain Region-Specific Activity of NO-Sensitive Guanylyl Cyclases (NO-GCs) and NO-GC Stimulators. Int. J. Mol. Sci. 2018, 19, 2313. https://doi.org/10.3390/ijms19082313
Peters S, Paolillo M, Mergia E, Koesling D, Kennel L, Schmidtko A, Russwurm M, Feil R. cGMP Imaging in Brain Slices Reveals Brain Region-Specific Activity of NO-Sensitive Guanylyl Cyclases (NO-GCs) and NO-GC Stimulators. International Journal of Molecular Sciences. 2018; 19(8):2313. https://doi.org/10.3390/ijms19082313
Chicago/Turabian StylePeters, Stefanie, Michael Paolillo, Evanthia Mergia, Doris Koesling, Lea Kennel, Achim Schmidtko, Michael Russwurm, and Robert Feil. 2018. "cGMP Imaging in Brain Slices Reveals Brain Region-Specific Activity of NO-Sensitive Guanylyl Cyclases (NO-GCs) and NO-GC Stimulators" International Journal of Molecular Sciences 19, no. 8: 2313. https://doi.org/10.3390/ijms19082313