Quantum Mechanics/Molecular Mechanics Studies on the Relative Reactivities of Compound I and II in Cytochrome P450 Enzymes

 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
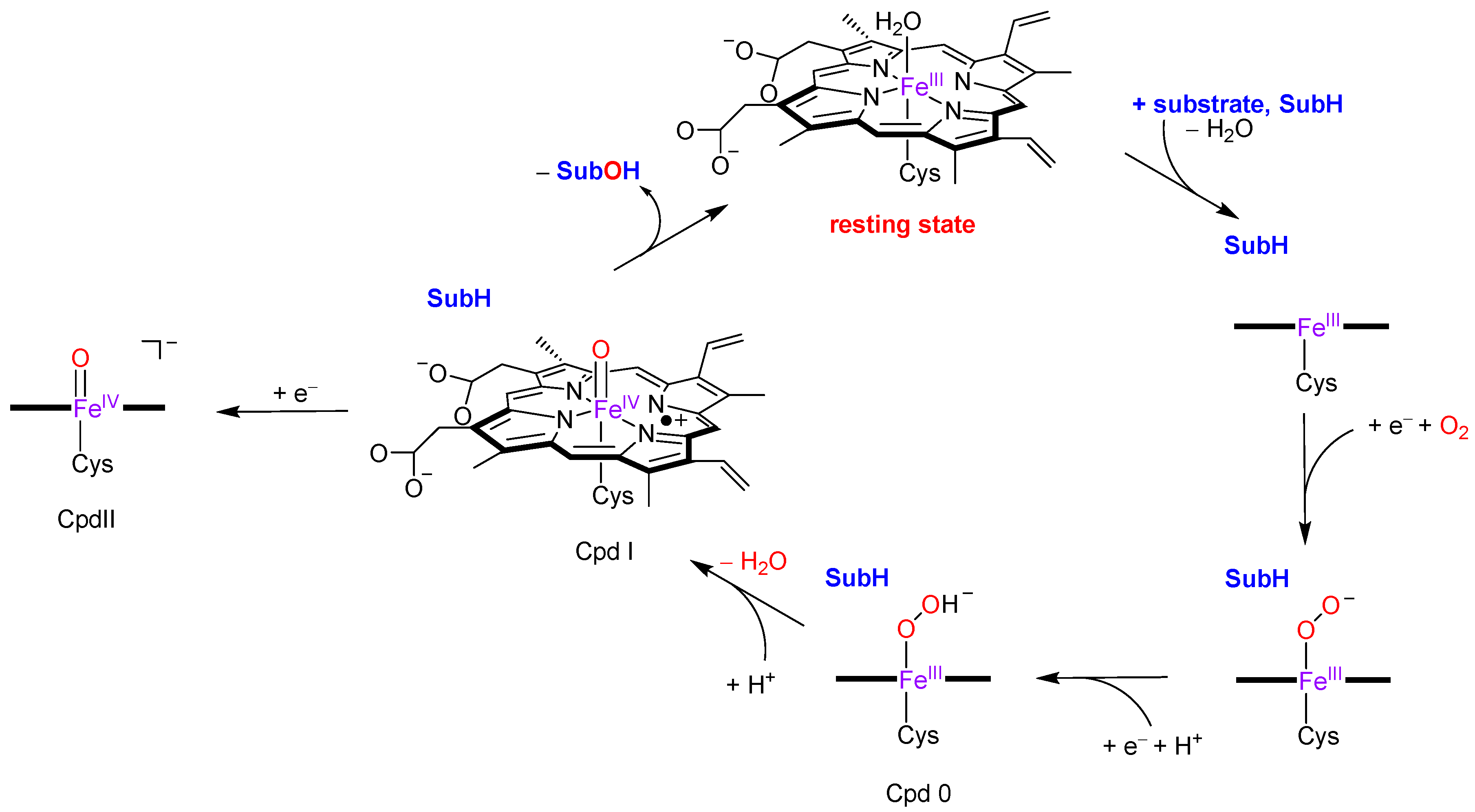
1. Introduction
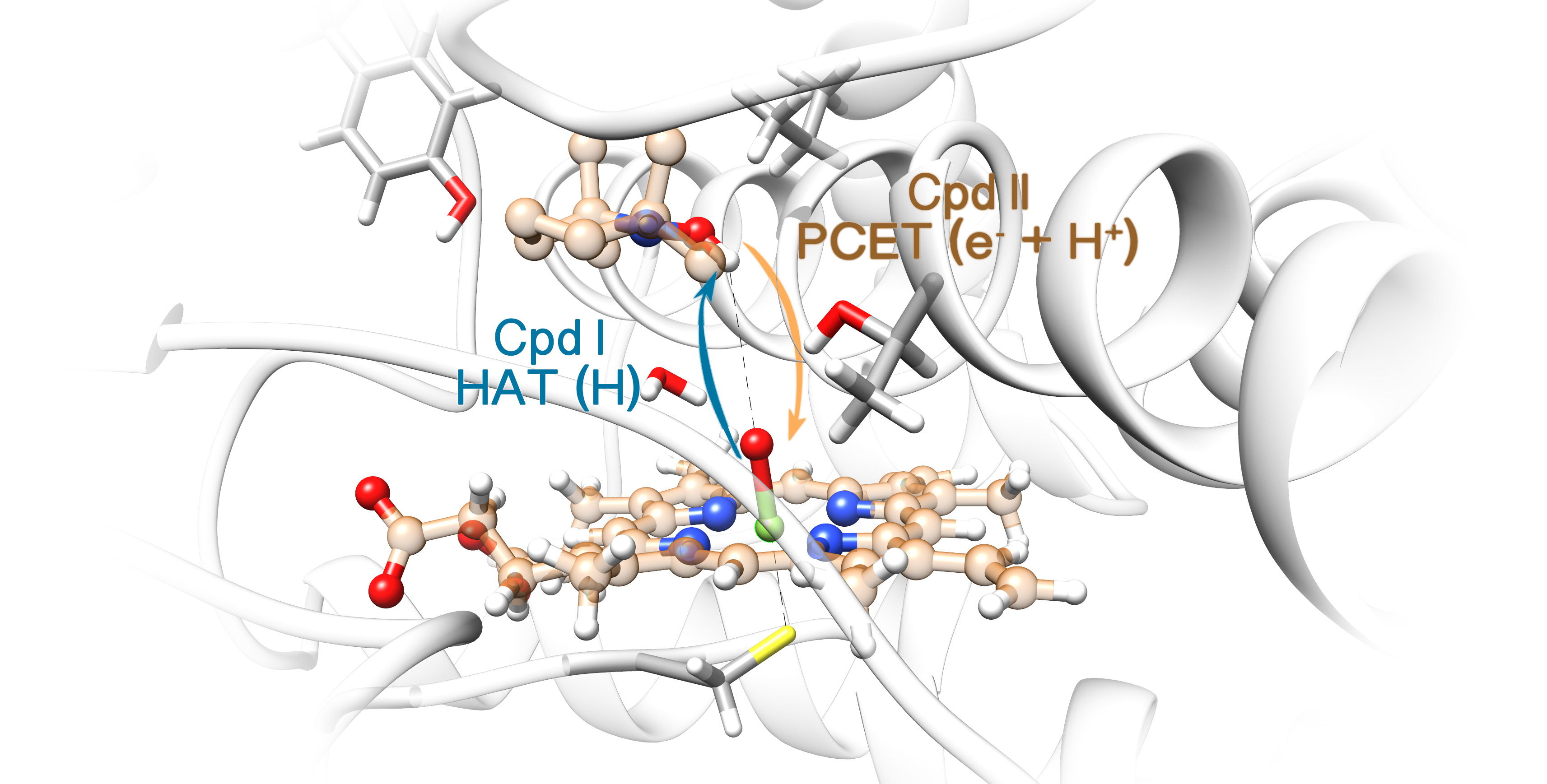
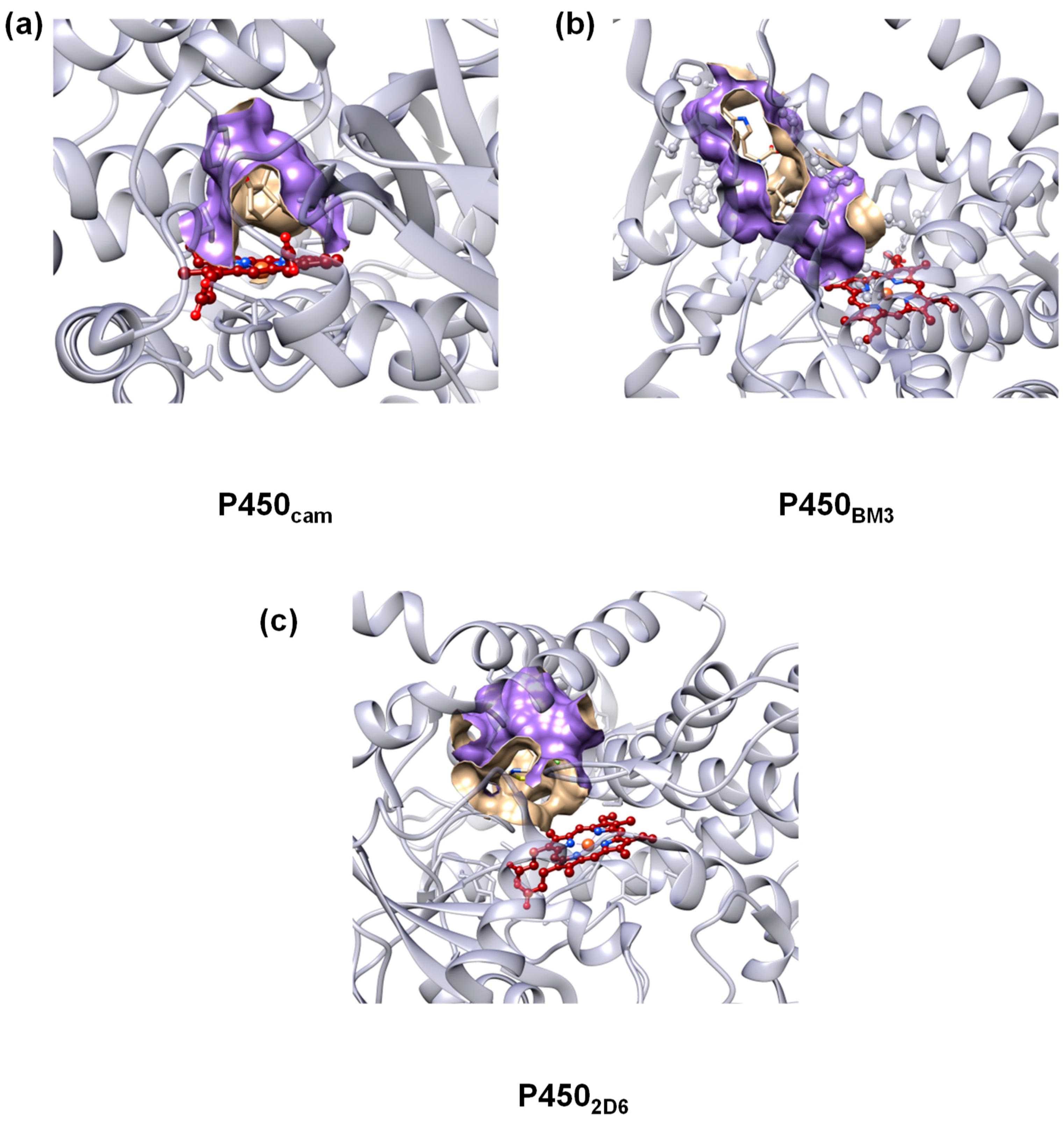
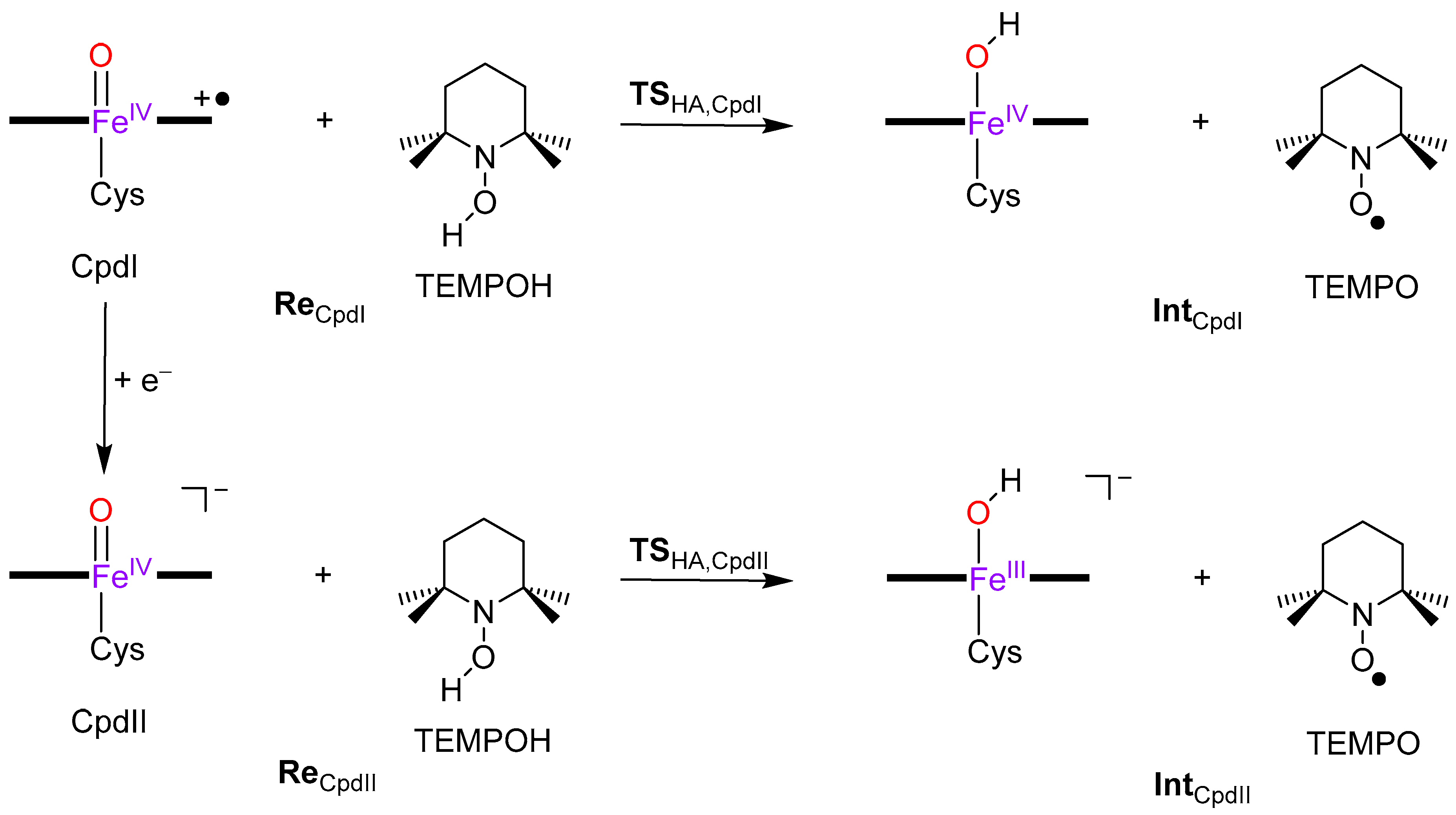
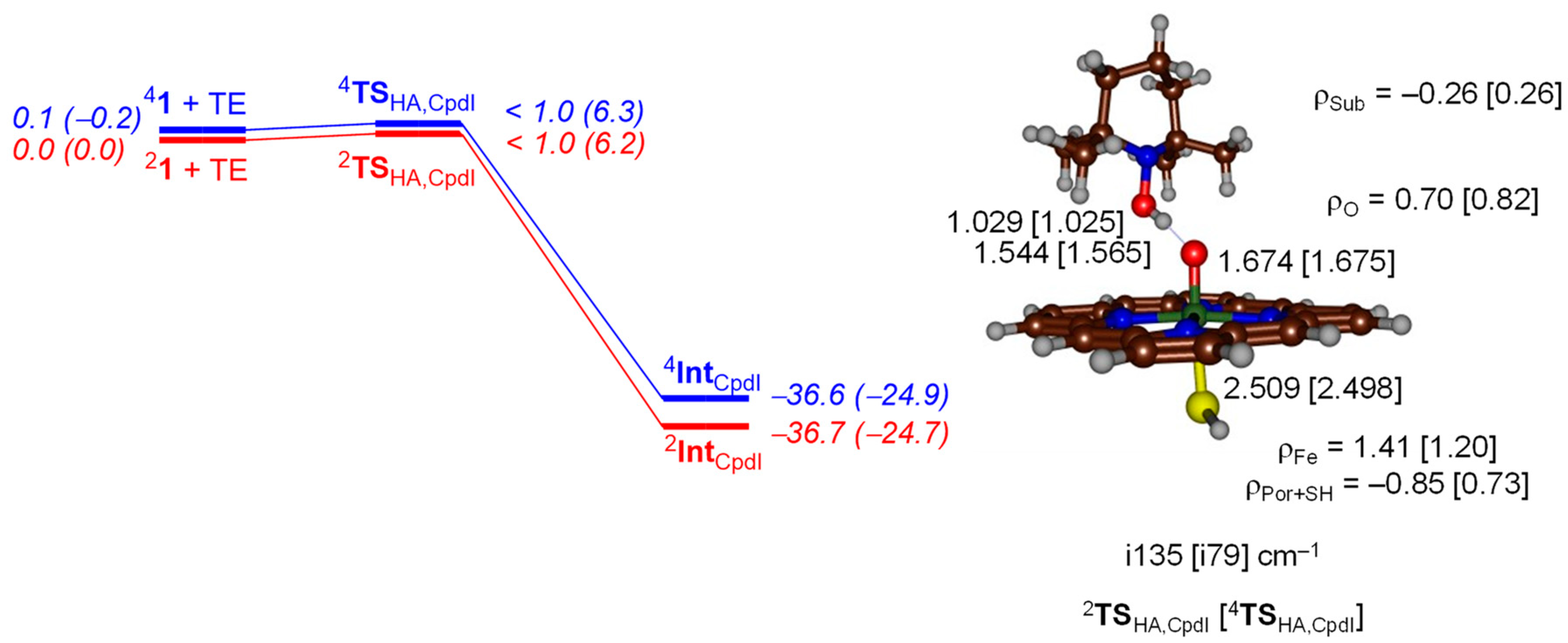
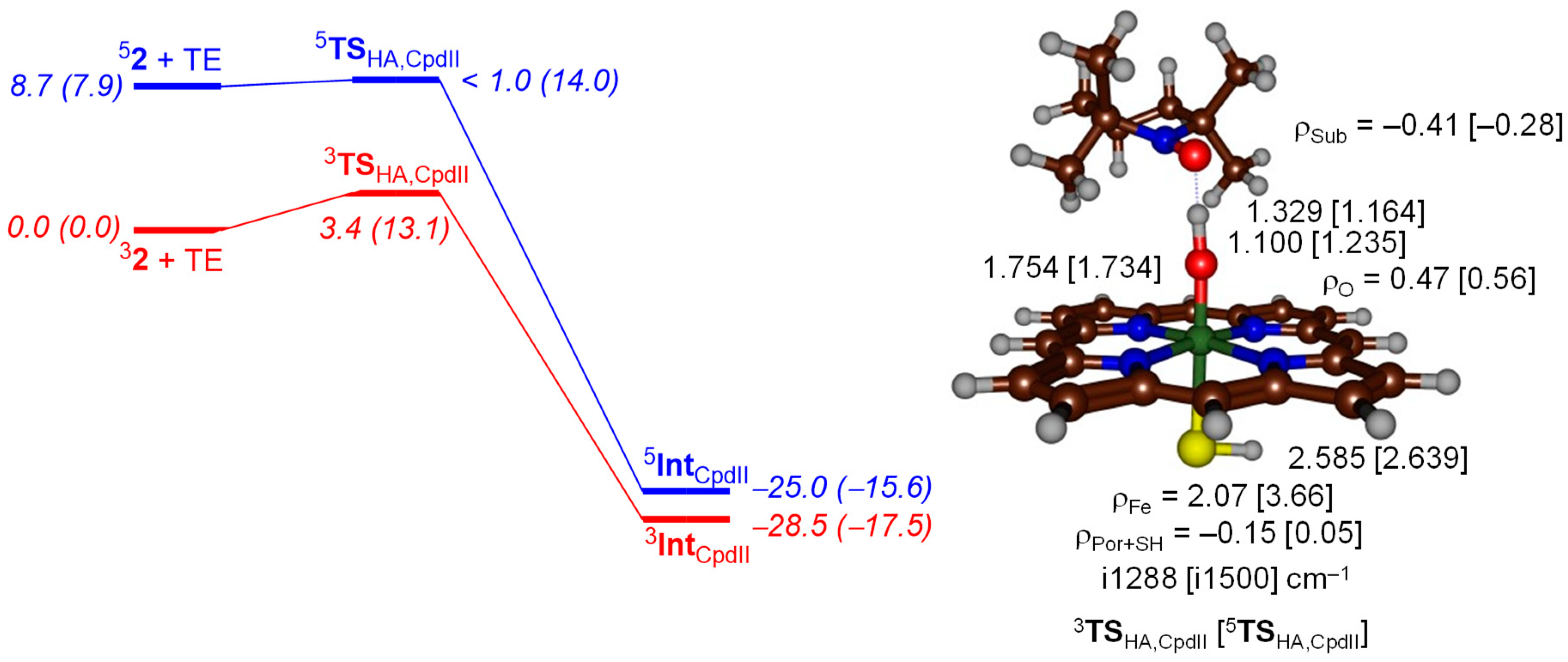
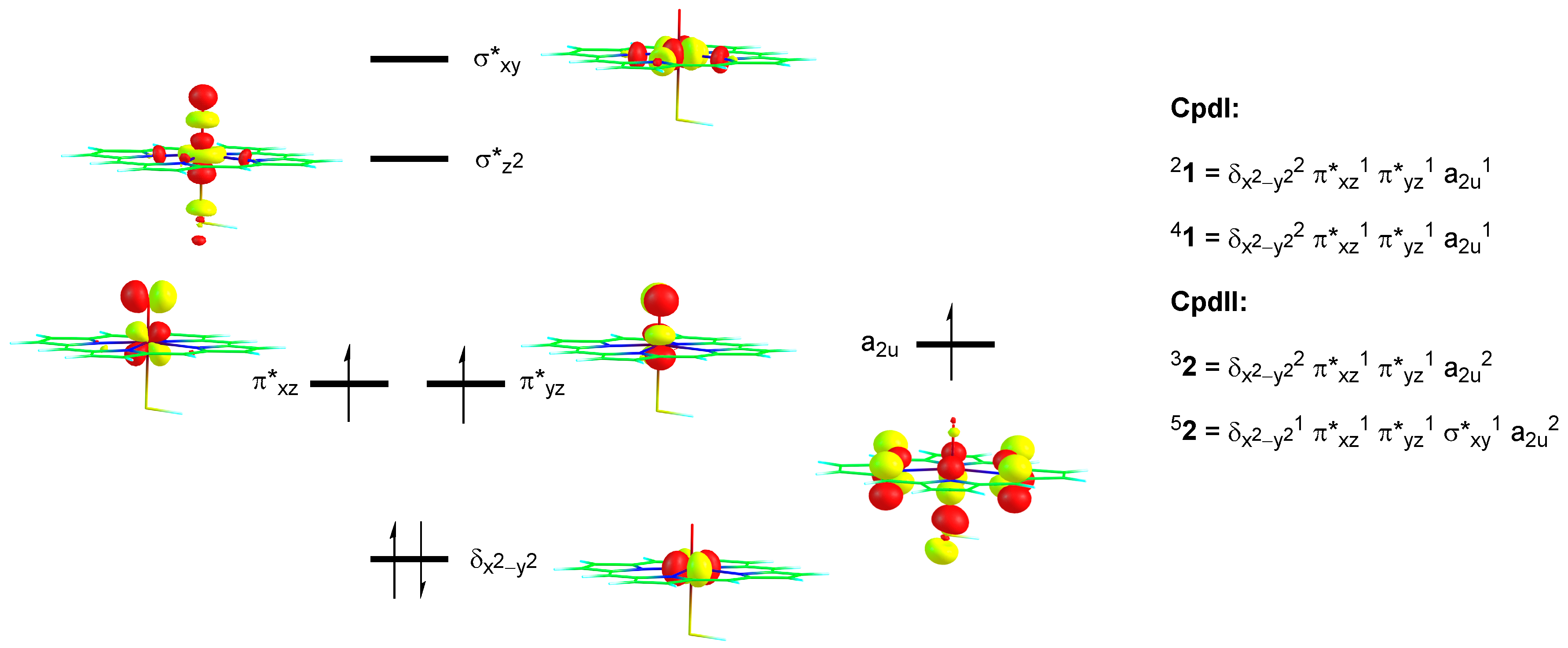
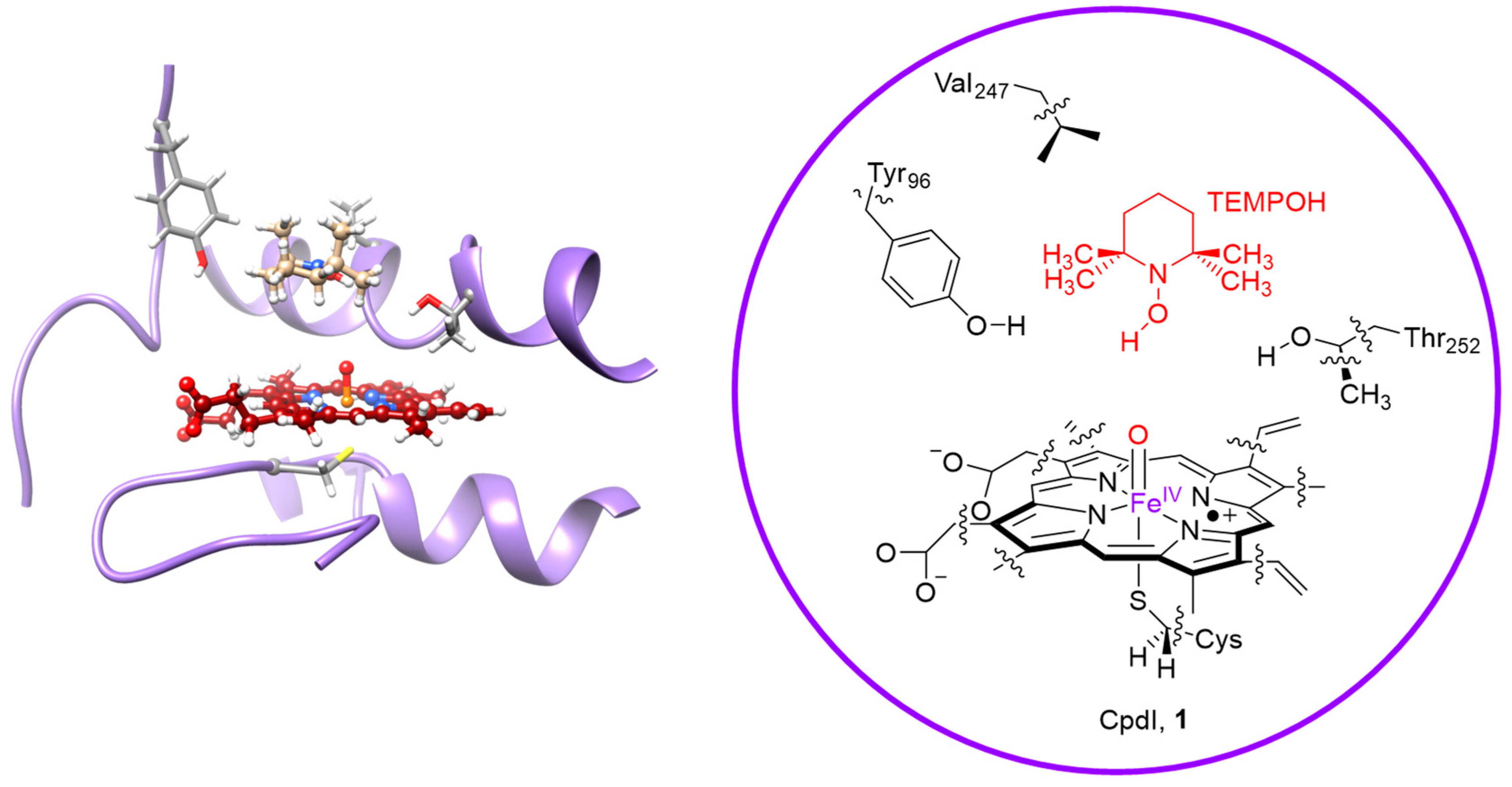
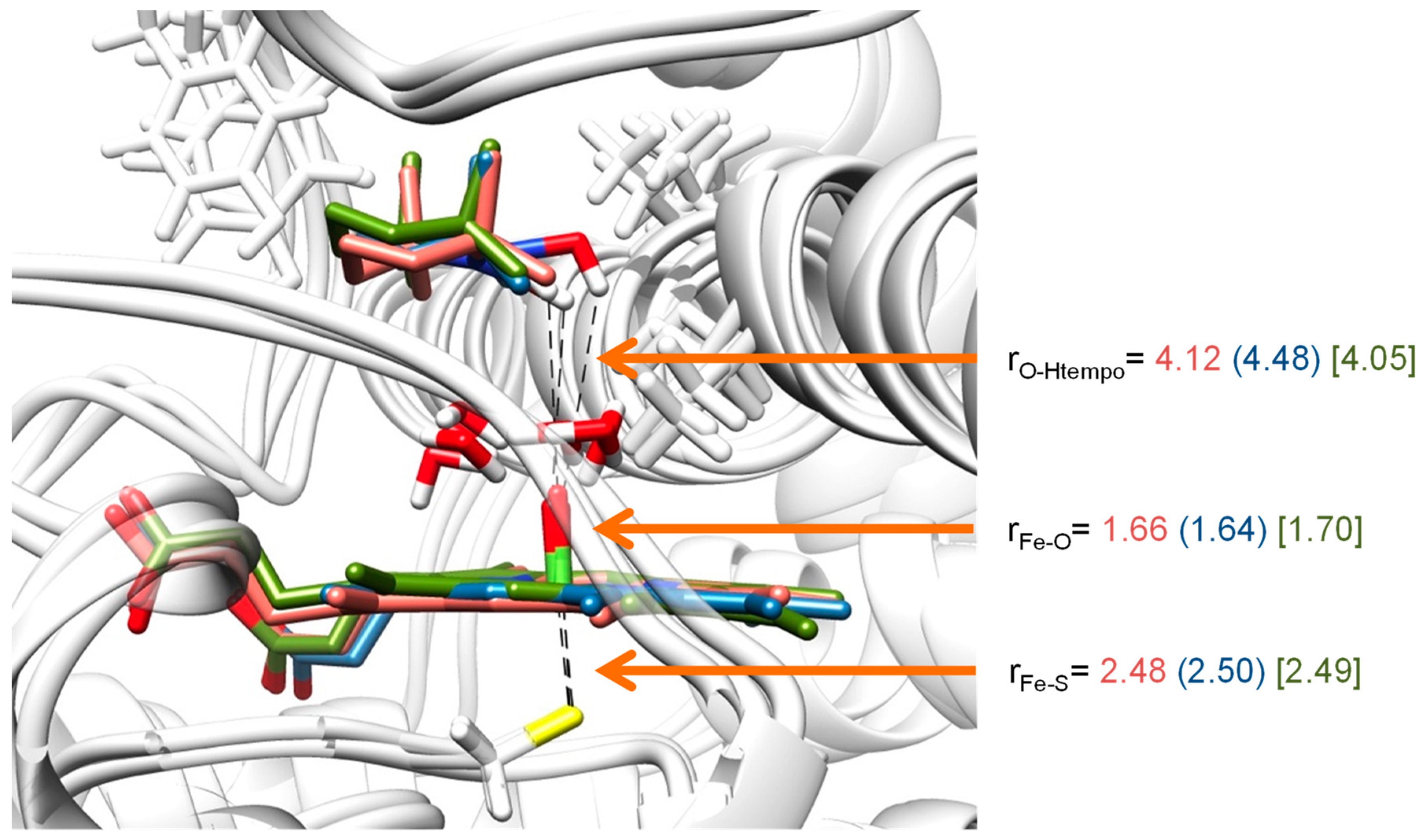
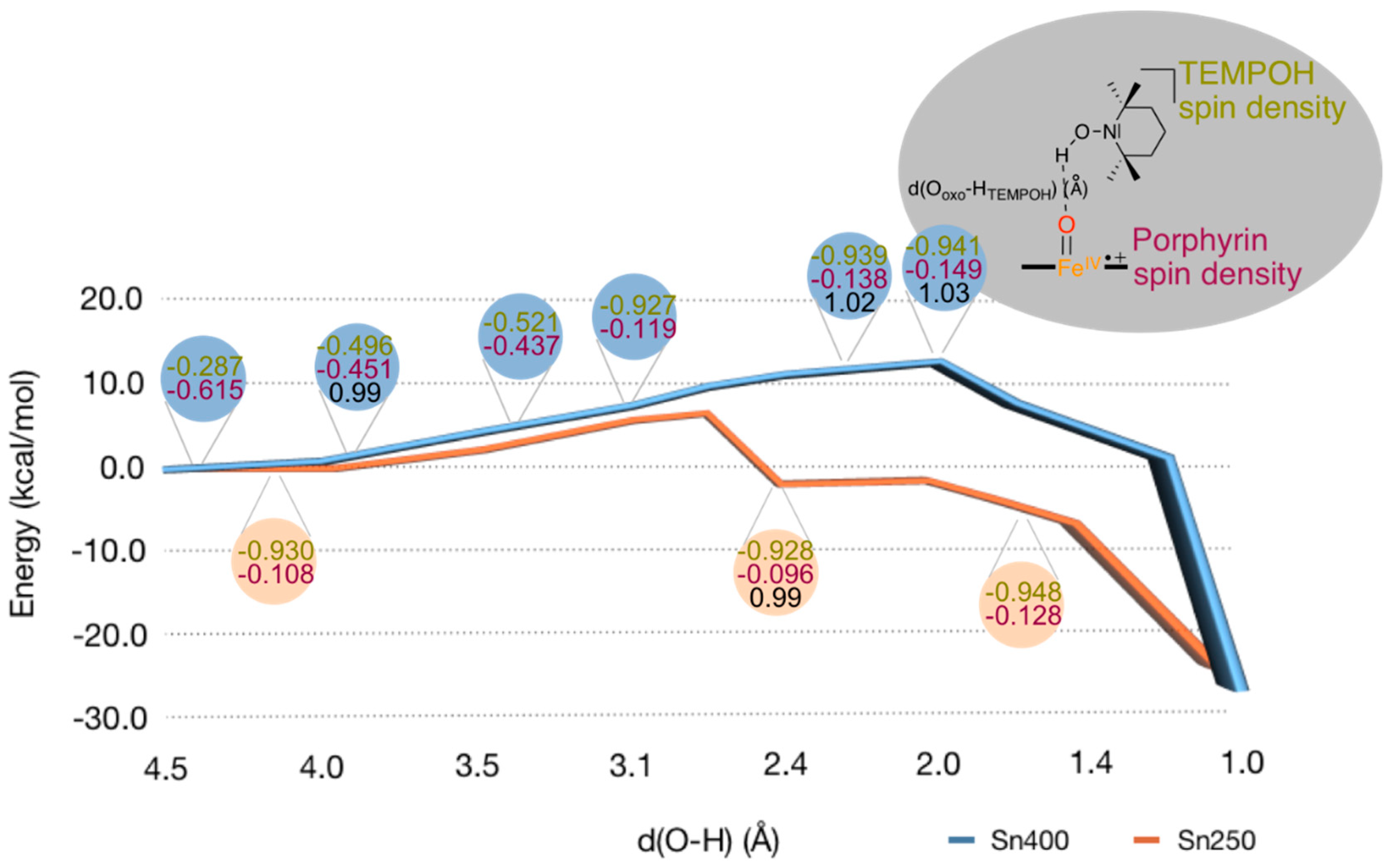
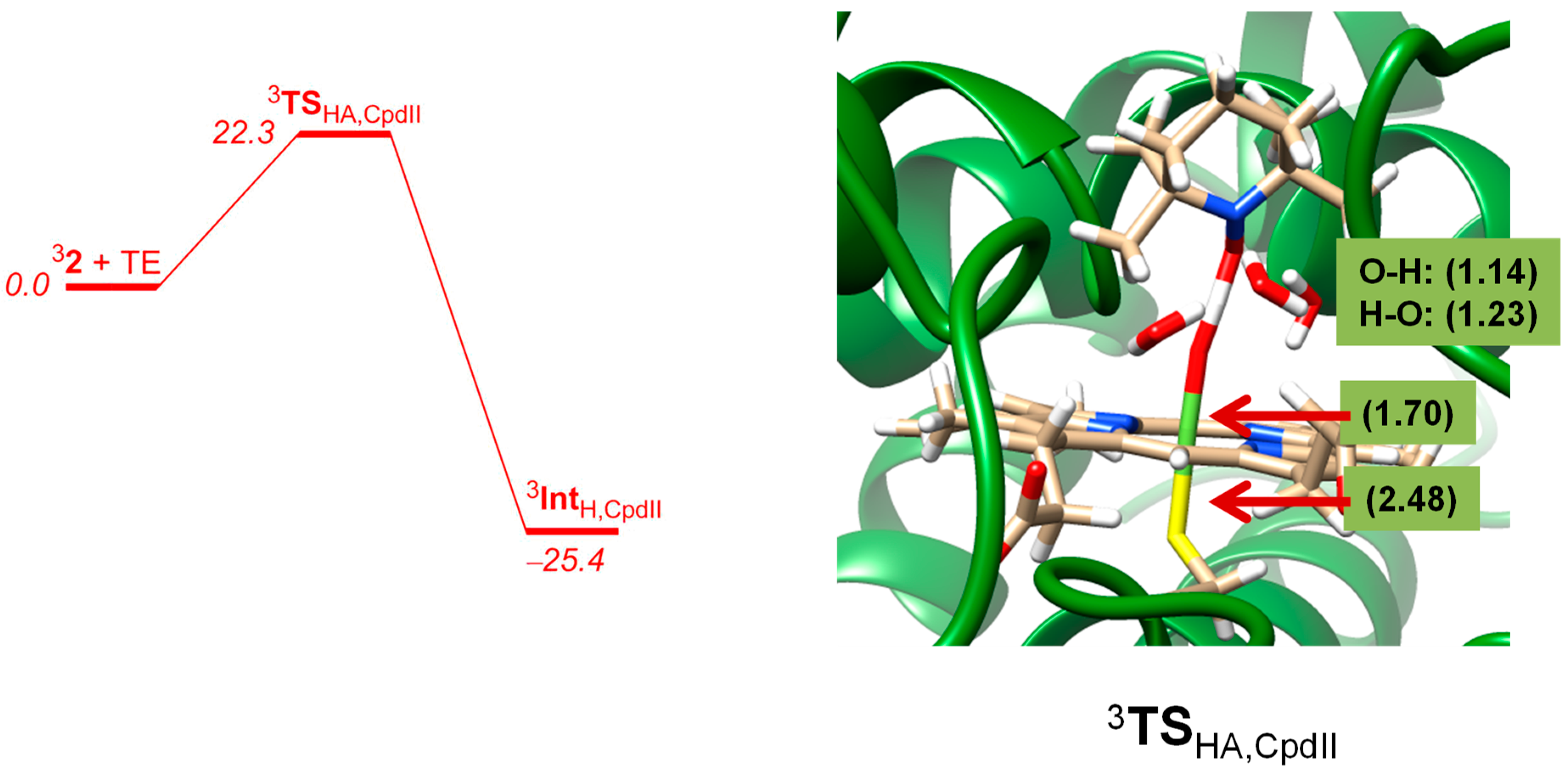
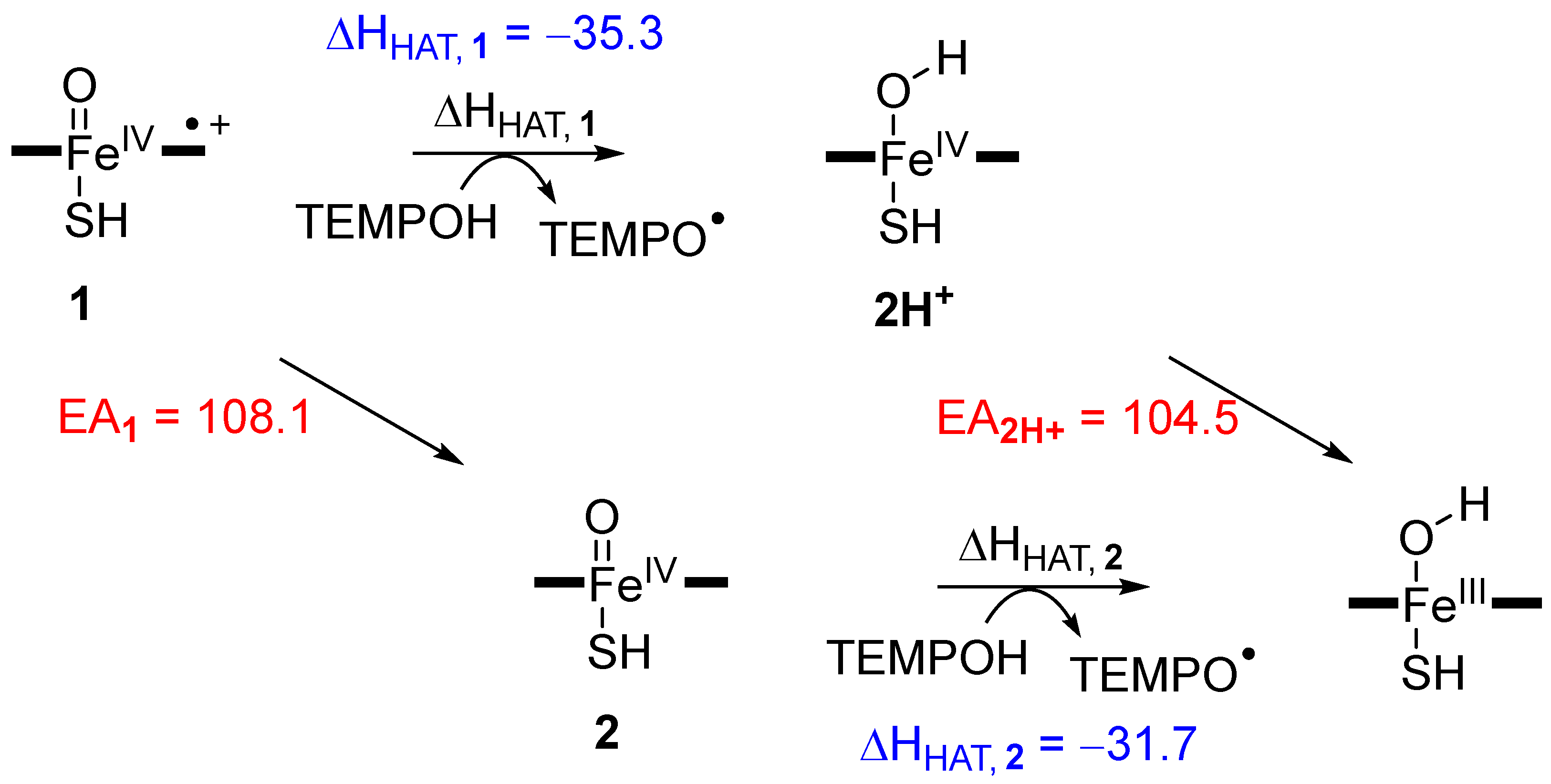
2. Results
2.1. DFT on Model Complexes
2.2. QM/MM Studies
3. Discussion
4. Materials and Methods
4.1. DFT Model Complexes
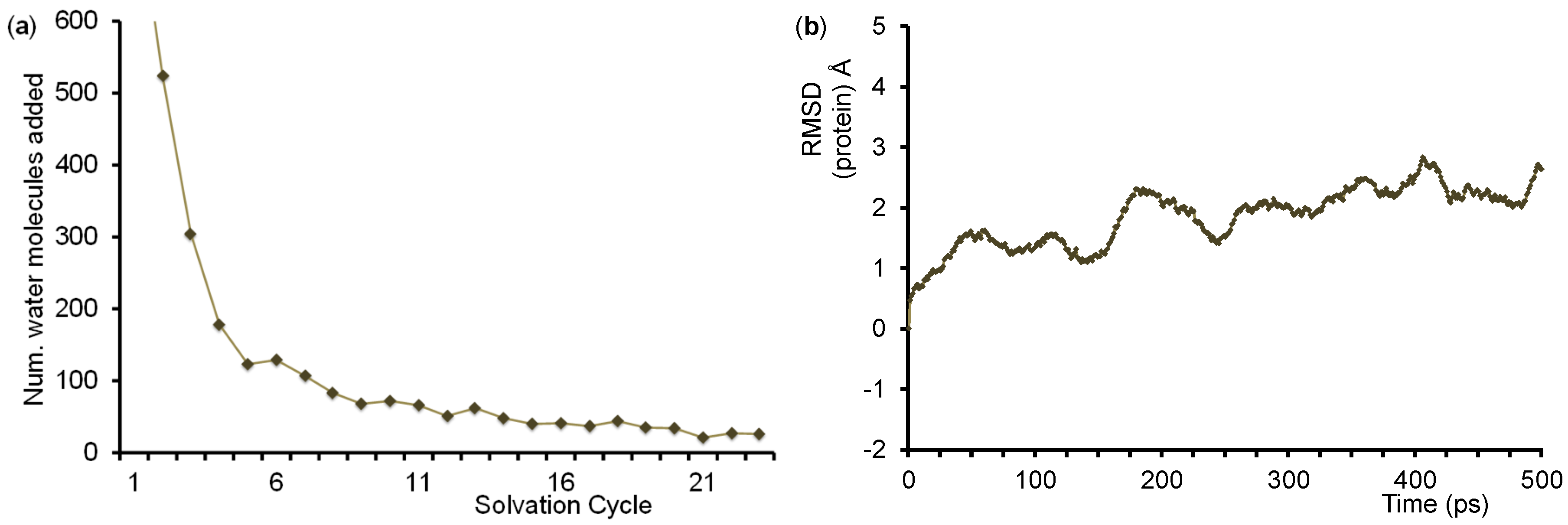
4.2. QM/MM Set Up
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sono, M.; Roach, M.P.; Coulter, E.D.; Dawson, J.H. Heme-containing oxygenases. Chem. Rev. 1996, 96, 2841–2887. [Google Scholar] [CrossRef] [PubMed]
- Ortiz de Montellano, P.R. (Ed.) Cytochrome P450: Structure, Mechanism, and Biochemistry, 3rd ed.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2004. [Google Scholar]
- Meunier, B.; de Visser, S.P.; Shaik, S. Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef] [PubMed]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schlichting, I. Structure and chemistry of cytochrome P450. Chem. Rev. 2005, 105, 2253–2277. [Google Scholar] [CrossRef] [PubMed]
- Munro, A.W.; Girvan, H.M.; McLean, K.J. Variations on a (t)heme—novel mechanisms, redox partners and catalytic functions in the cytochrome P450 superfamily. Nat. Prod. Rep. 2007, 24, 585–609. [Google Scholar] [CrossRef] [PubMed]
- Ortiz de Montellano, P.R. Hydrocarbon hydroxylation by cytochrome P450 enzymes. Chem. Rev. 2010, 110, 932–948. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Guengerich, F.P. Reaction of trichloroethylene and trichloroethylene oxide with cytochrome P450 enzymes: Inactivation and sites of modification. Chem. Res. Toxicol. 2001, 14, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Grogan, G. Cytochromes P450: Exploiting diversity and enabling application as biocatalysts. Curr. Opin. Chem. Biol. 2011, 15, 241–248. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Kumar, D. (Eds.) Iron-Containing Enzymes: Versatile Catalysts of Hydroxylation Reactions in Nature; Royal Society of Chemistry Publishing: Cambridge, UK, 2011; ISBN 978-1-84973-181-2. [Google Scholar]
- Groves, J.T. The bioinorganic chemistry of iron in oxygenases and supramolecular assemblies. Proc. Natl. Acad. Sci. USA 2003, 100, 3569–3574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, Y.; Nakajima, H.; Ueno, T. Reactivities of oxo and peroxo intermediates studied by hemoprotein mutants. Acc. Chem. Res. 2007, 40, 554–562. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Nam, W. High-valent iron-oxo porphyrins in oxygenation reactions. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific Publishing Co.: Hackensack, NJ, USA, 2010; Chapter 44; pp. 85–140. ISBN 13-978-981-4307-23-9. [Google Scholar]
- Huang, X.; Groves, J.T. Oxygen activation and radical transformations in heme proteins and metalloporphyrins. Chem. Rev. 2018, 118, 2491–2553. [Google Scholar] [CrossRef] [PubMed]
- Davydov, R.; Makris, T.M.; Kofman, V.; Werst, D.E.; Sligar, S.G.; Hoffman, B.M. Hydroxylation of camphor by reduced oxy-cytochrome P450cam: Mechanistic implications of EPR and ENDOR studies of catalytic intermediates in native and mutant enzymes. J. Am. Chem. Soc. 2001, 123, 1403–1415. [Google Scholar] [CrossRef] [PubMed]
- Gelb, M.H.; Heimbrook, D.C.; Malkonen, P.; Sligar, S.G. Stereochemistry and deuterium isotope effects in camphor hydroxylation by the cytochrome P450cam monoxygenase system. Biochemistry 1982, 21, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Makris, T.M.; von Koenig, K.; Schlichting, I.; Sligar, S.G. Alteration of P450 distal pocket solvent leads to impaired proton delivery and changes in heme geometry. Biochemistry 2007, 46, 14129–14140. [Google Scholar] [CrossRef] [PubMed]
- Poulos, T.L.; Finzel, B.C.; Howard, A.J. High-resolution crystal structure of cytochrome P450cam. J. Mol. Biol. 1987, 195, 687–700. [Google Scholar] [CrossRef]
- Cong, Z.; Shoji, O.; Kasai, C.; Kawakami, N.; Sugimoto, H.; Shiro, Y.; Watanabe, Y. Activation of Wild-Type Cytochrome P450BM3 by the Next Generation of Decoy Molecules: Enhanced Hydroxylation of Gaseous Alkanes and Crystallographic Evidence. ACS Catal. 2015, 5, 150–156. [Google Scholar] [CrossRef]
- Butler, C.R.; Ogilvie, K.; Martinez-Alsina, L.; Barreiro, G.; Beck, E.M.; Nolan, C.E.; Atchison, K.; Benvenuti, E.; Buzon, L.; Doran, S.; et al. Aminomethyl-derived beta secretase (BACE1) inhibitors: Engaging Gly230 without an anilide functionality. J. Med. Chem. 2017, 60, 386–402. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Hirao, H.; de Visser, S.P.; Zheng, J.; Wang, D.; Thiel, W.; Shaik, S. New features in the catalytic cycle of cytochrome P450 during the formation of Compound I from Compound 0. J. Phys. Chem. B 2005, 109, 19946–19951. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Kumar, D.; de Visser, S.P.; Altun, A.; Thiel, W. Theoretical perspective on the structure and mechanism of cytochrome P450 enzymes. Chem. Rev. 2005, 105, 2279–2328. [Google Scholar] [CrossRef] [PubMed]
- Balding, P.R.; Porro, C.S.; McLean, K.J.; Sutcliffe, M.J.; Maréchal, J.-D.; Munro, A.W.; de Visser, S.P. How do azoles inhibit cytochrome P450 enzymes? A density functional study. J. Phys. Chem. A 2008, 112, 12911–12918. [Google Scholar] [CrossRef] [PubMed]
- Davydov, R.; Perera, R.; Jin, S.; Yang, T.-C.; Bryson, T.A.; Sono, M.; Dawson, J.H.; Hoffman, B.M. Substrate modulation of the properties and reactivity of the oxy-ferrous and hydroperoxo-ferric intermediates of cytochrome P450cam as shown by cryoreduction-EPR/ENDOR spectroscopy. J. Am. Chem. Soc. 2005, 127, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Rittle, J.; Green, M.T. Cytochrome P450 Compound I: Capture, characterization, and C-H bond activation kinetics. Science 2010, 330, 933–937. [Google Scholar] [CrossRef] [PubMed]
- Ogliaro, F.; Harris, N.; Cohen, S.; Filatov, M.; de Visser, S.P.; Shaik, S. A model “rebound” mechanism of hydroxylation by cytochrome P450: Stepwise and effectively concerted pathways, and their reactivity patterns. J. Am. Chem. Soc. 2000, 122, 8977–8989. [Google Scholar] [CrossRef]
- Kamachi, T.; Yoshizawa, K. A Theoretical study on the mechanism of camphor hydroxylation by Compound I of cytochrome P450. J. Am. Chem. Soc. 2003, 125, 4652–4661. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, Y.; Han, K. Recent density functional theory model calculations of drug metabolism by cytochrome P450. Coord. Chem. Rev. 2012, 256, 1137–1150. [Google Scholar] [CrossRef] [Green Version]
- Vaz, A.D.N.; Pernecky, S.J.; Raner, G.M.; Coon, M.J. Peroxo-iron and oxenoid-iron species as alternative oxygenating agents in cytochrome P450-catalyzed reactions: Switching by Threonine-302 to Alanine mutagenesis of cytochrome P450 2B4. Proc. Natl. Acad. Sci. USA 1996, 93, 4644–4648. [Google Scholar] [CrossRef] [PubMed]
- Ogliaro, F.; de Visser, S.P.; Cohen, S.; Sharma, P.K.; Shaik, S. Searching for the second oxidant in the catalytic cycle of cytochrome P450: A theoretical investigation of the iron(III)-hydroperoxo species and its epoxidation pathways. J. Am. Chem. Soc. 2002, 124, 2806–2817. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.K.; de Visser, S.P.; Shaik, S. Can a single oxidant with two spin states masquerade as two different oxidants? A study of the sulfoxidation mechanism by cytochrome P450. J. Am. Chem. Soc. 2003, 125, 8698–8699. [Google Scholar] [CrossRef] [PubMed]
- Porro, C.S.; Sutcliffe, M.J.; de Visser, S.P. Quantum mechanics/molecular mechanics studies on the sulfoxidation of dimethyl sulfide by Compound I and Compound 0 of Cytochrome P450: Which is the better oxidant? J. Phys. Chem. A 2009, 113, 11635–11642. [Google Scholar] [CrossRef] [PubMed]
- Song, W.J.; Ryu, Y.O.; Song, R.; Nam, W. Oxoiron(IV) porphyrin pi-cation radical complexes with a chameleon behavior in cytochrome P450 model reactions. J. Biol. Inorg. Chem. 2005, 10, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Fertinger, C.; Hessenauer-Ilicheva, N.; Franke, A.; van Eldik, R. Direct comparison of the reactivity of model complexes for Compounds 0, I, and II in oxygenation, hydrogen-abstraction, and hydride-transfer processes. Chem. Eur. J. 2009, 15, 13435–13440. [Google Scholar] [CrossRef] [PubMed]
- Fertinger, C.; Franke, A.; van Eldik, R. Mechanistic insight from thermal activation parameters for oxygenation reactions of different substrates with biomimetic iron porphyrin models for compounds I and II. J. Biol. Inorg. Chem. 2012, 17, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Franke, A.; Brindell, M.; Oszajca, M.; Zahl, A.; van Eldik, R. Combined experimental and theoretical study on the reactivity of Compounds I and II in horseradish peroxidase biomimetics. Chem. Eur. J. 2014, 20, 14437–14450. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-X.; Postils, V.; Sun, W.; Faponle, A.S.; Solà, M.; Wang, Y.; Nam, W.; de Visser, S.P. Reactivity patterns of (protonated) Compound II and Compound I of Cytochrome P450: Which is the better oxidant? Chem. Eur. J. 2017, 23, 6406–6418. [Google Scholar] [CrossRef] [PubMed]
- Oszajca, M.; Franke, A.; Drzewiecka-Matuszek, A.; Brindell, M.; Stochel, G.; van Eldik, R. Temperature and pressure effects on C–H abstraction reactions involving compound I and II mimics in aqueous solution. Inorg. Chem. 2014, 53, 2848–2857. [Google Scholar] [CrossRef] [PubMed]
- Mallick, D.; Shaik, S. Kinetic isotope effect probes the reactive spin state, as well as the geometric feature and constitution of the transition state during H-abstraction by heme Compound II complexes. J. Am. Chem. Soc. 2017, 139, 11451–11459. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Tan, L.S. Is the bound substrate in nitric oxide synthase protonated or neutral and what is the active oxidant that performs substrate hydroxylation? J. Am. Chem. Soc. 2008, 130, 12961–12974. [Google Scholar] [CrossRef] [PubMed]
- İşci, Ü.; Faponle, A.S.; Afanasiev, P.; Albrieux, F.; Briois, V.; Ahsen, V.; Dumoulin, F.; Sorokin, A.B.; de Visser, S.P. Site-selective formation of an iron(IV)-oxo species at the more electron-rich iron atom of heteroleptic μ-nitrido diiron phthalocyanines. Chem. Sci. 2015, 6, 5063–5075. [Google Scholar] [CrossRef]
- De Visser, S.P.; Kumar, D.; Neumann, R.; Shaik, S. Computer-generated high-valent iron-oxo and manganese-oxo species with polyoxometalate ligands: How do they compare with the iron-oxo active species of heme enzymes? Angew. Chem. Int. Ed. 2004, 43, 5661–5665. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P. Trends in substrate hydroxylation reactions by heme and nonheme iron(IV)-oxo oxidants give correlations between intrinsic properties of the oxidant with barrier height. J. Am. Chem. Soc. 2010, 132, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P. Propene activation by the oxo-iron active species of taurine/α-ketoglutarate dioxygenase (TauD) enzyme. How does the catalysis compare to heme-enzymes? J. Am. Chem. Soc. 2006, 128, 9813–9824. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P. What factors influence the ratio of C–H hydroxylation versus C=C epoxidation by a nonheme cytochrome P450 biomimetic? J. Am. Chem. Soc. 2006, 128, 15809–15818. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Latifi, R.; Kumar, S.; Rybak-Akimova, E.V.; Sainna, M.A.; de Visser, S.P. Rationalization of the barrier height for para-Z-styrene epoxidation by iron(IV)-oxo porphyrins with variable axial ligands. Inorg. Chem. 2013, 52, 7968–7979. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Guan, J.; Liu, H.; Huang, X. Analysis of an alternative to the H-atom abstraction mechanism in methane C-H bond activation by nonheme iron(IV)-oxo oxidants. Dalton Trans. 2013, 42, 10260–10270. [Google Scholar] [CrossRef] [PubMed]
- Quesne, M.G.; Senthilnathan, D.; Singh, D.; Kumar, D.; Maldivi, P.; Sorokin, A.B.; de Visser, S.P. Origin of the enhanced reactivity of μ-nitrido-bridged diiron(IV)-oxo porphyrinoid complexes over cytochrome P450 Compound I. ACS Catal. 2016, 6, 2230–2243. [Google Scholar] [CrossRef]
- De Visser, S.P.; Kumar, D.; Cohen, S.; Shacham, R.; Shaik, S. A predictive pattern of computed barriers for C–H hydroxylation by Compound I of cytochrome P450. J. Am. Chem. Soc. 2004, 126, 8362–8363. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Kumar, D.; de Visser, S.P. A valence bond modeling of trends in hydrogen abstraction barriers and transition states of hydroxylation reactions catalyzed by cytochrome P450 enzymes. J. Am. Chem. Soc. 2008, 130, 10128–10140. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Tahsini, L.; de Visser, S.P.; Kang, H.Y.; Kim, S.J.; Nam, W. The effect of porphyrin ligands on the regioselective dehydrogenation versus epoxidation of olefins by oxoiron(IV) mimics of cytochrome P450. J. Phys. Chem. A 2009, 113, 11713–11722. [Google Scholar] [CrossRef] [PubMed]
- Bernasconi, L.; Baerends, E.J. The EDTA complex of oxidoiron(IV) as realisation of an optimal ligand environment for high activity of FeO2+. Eur. J. Inorg. Chem. 2008, 1672–1681. [Google Scholar] [CrossRef]
- Ye, S.; Geng, C.-Y.; Shaik, S.; Neese, F. Electronic structure analysis of multistate reactivity in transition metal catalyzed reactions: The case of C–H bond activation by non-heme iron(IV)–oxo cores. Phys. Chem. Chem. Phys. 2013, 15, 8017–8030. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Latifi, R. Carbon dioxide, a waste product in the catalytic cycle of α-ketoglutarate dependent halogenases prevents the formation of hydroxylated by-products. J. Phys. Chem. B 2009, 113, 12–14. [Google Scholar] [CrossRef] [PubMed]
- Pratter, S.M.; Konstantinovics, C.; DiGiuro, C.L.M.; Leitner, E.; Kumar, D.; de Visser, S.P.; Grogan, G.; Straganz, G.D. Inversion of enantio-selectivity of a mononuclear non-heme iron(II)-dependent hydroxylase by tuning the interplay of metal-center geometry and protein structure. Angew. Chem. Int. Ed. 2013, 52, 9677–9681. [Google Scholar] [CrossRef] [PubMed]
- Timmins, A.; Saint-André, M.; de Visser, S.P. Understanding how prolyl-4-hydroxylase structure steers a ferryl oxidant toward scission of a strong C–H bond. J. Am. Chem. Soc. 2017, 139, 9855–9866. [Google Scholar] [CrossRef] [PubMed]
- Timmins, A.; de Visser, S.P. How are substrate binding and catalysis affected by mutating Glu127 and Arg161 in prolyl-4-hydroxylase? A QM/MM and MD study. Front. Chem. 2017, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Ogliaro, F.; Sharma, P.K.; Shaik, S. Hydrogen bonding modulates the selectivity of enzymatic oxidation by P450: A chameleon oxidant behavior of Compound I. Angew. Chem. Int. Ed. 2002, 41, 1947–1951. [Google Scholar] [CrossRef]
- Kumar, D.; de Visser, S.P.; Sharma, P.K.; Hirao, H.; Shaik, S. Sulfoxidation mechanisms catalyzed by cytochrome P450 and horseradish peroxidase models: Spin selection induced by the ligand. Biochemistry 2005, 44, 8148–8158. [Google Scholar] [CrossRef] [PubMed]
- Sahu, S.; Widger, L.R.; Quesne, M.G.; de Visser, S.P.; Matsumura, H.; Moënne-Loccoz, P.; Siegler, M.A.; Goldberg, D.P. Secondary coordination sphere influence on the reactivity of nonheme iron(II) complexes: An experimental and DFT approach. J. Am. Chem. Soc. 2013, 135, 10590–10593. [Google Scholar] [CrossRef] [PubMed]
- Morozov, A.N.; Pardillo, A.D.; Chatfield, D.C. Chloroperoxidase-catalyzed epoxidation of cis-β-methylstyrene: NH–S hydrogen bonds and proximal helix dipole change the catalytic mechanism and significantly lower the reaction barrier. J. Phys. Chem. B 2015, 119, 14350–14363. [Google Scholar] [CrossRef] [PubMed]
- Morozov, A.N.; Chatfield, D.C. How the proximal pocket may influence the enantiospecificities of chloroperoxidase-catalyzed epoxidations of olefins. Int. J. Mol. Sci. 2016, 17, 1297. [Google Scholar] [CrossRef] [PubMed]
- Quesne, M.G.; Borowski, T.; de Visser, S.P. Quantum mechanics/molecular mechanics modelling of enzymatic processes: Caveats and breakthroughs. Chem. Eur. J. 2016, 22, 2562–2581. [Google Scholar] [CrossRef] [PubMed]
- Faponle, A.S.; Seebeck, F.P.; de Visser, S.P. Sulfoxide synthase versus cysteine dioxygenase reactivity in a nonheme iron enzyme. J. Am. Chem. Soc. 2017, 139, 9259–9270. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Li, H.; Poulos, T.L. Structural basis for effect or control and redox partner recognition in cytochrome P450. Science 2013, 340, 1227–1230. [Google Scholar] [CrossRef] [PubMed]
- Schöneboom, J.C.; Lin, H.; Reuter, N.; Thiel, W.; Cohen, S.; Ogliaro, F.; Shaik, S. The elusive oxidant species of cytochrome P450 enzymes: Characterization by combined quantum mechanical/molecular mechanical (QM/MM) calculations. J. Am. Chem. Soc. 2002, 124, 8142–8151. [Google Scholar] [CrossRef] [PubMed]
- Bathelt, C.M.; Zurek, J.; Mulholland, A.J.; Harvey, J.N. Electronic structure of compound I in human isoforms of cytochrome P450 from QM/MM modeling. J. Am. Chem. Soc. 2005, 127, 12900–12908. [Google Scholar] [CrossRef] [PubMed]
- Cantú Reinhard, F.G.; de Visser, S.P. Oxygen atom transfer using an iron(IV)-oxo embedded in a tetracyclic N-heterocyclic carbene system: How does the reactivity compare to Cytochrome P450 Compound I? Chem. Eur. J. 2017, 23, 2935–2944. [Google Scholar] [CrossRef] [PubMed]
- Schlichting, I.; Berendzen, J.; Chu, K.; Stock, A.M.; Maves, S.A.; Benson, D.E.; Sweet, B.M.; Ringe, D.; Petsko, G.A.; Sligar, S.G. The catalytic pathway of cytochrome P450cam at atomic resolution. Science 2000, 287, 1615–1622. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Ogliaro, F.; Sharma, P.K.; Shaik, S. What factors affect the regioselectivity of oxidation by cytochrome P450? A DFT study of allylic hydroxylation and double bond epoxidation in a model reaction. J. Am. Chem. Soc. 2002, 124, 11809–11826. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.K.; de Visser, S.P.; Ogliaro, F.; Shaik, S. Is the ruthenium analogue of Compound I of cytochrome P450 an efficient oxidant? A theoretical investigation of the methane hydroxylation reaction. J. Am. Chem. Soc. 2003, 125, 2291–2300. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P. Substitution of hydrogen by deuterium changes the regioselectivity of ethylbenzene hydroxylation by an oxo-iron-porphyrin catalyst. Chem. Eur. J. 2006, 12, 8168–8177. [Google Scholar] [CrossRef] [PubMed]
- Latifi, R.; Bagherzadeh, M.; de Visser, S.P. Origin of the correlation of the rate constant of substrate hydroxylation by nonheme iron(IV)-oxo complexes with the bond-dissociation energy of the C–H bond of the substrate. Chem. Eur. J. 2009, 15, 6651–6662. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Faponle, A.S.; Quesne, M.G.; Sainna, M.A.; Zhang, J.; Franke, A.; Kumar, D.; van Eldik, R.; Liu, W.; de Visser, S.P. Drug metabolism by cytochrome P450 enzymes: What distinguishes the pathways leading to substrate hydroxylation over desaturation? Chem. Eur. J. 2015, 21, 9083–9092. [Google Scholar] [CrossRef] [PubMed]
- Latifi, R.; Sainna, M.A.; Rybak-Akimova, E.V.; de Visser, S.P. Does hydrogen bonding-donation to manganese(IV)-oxo and iron(IV)-oxo oxidants affect the oxygen atom transfer ability? A computational study. Chem. Eur. J. 2013, 19, 4058–4068. [Google Scholar] [CrossRef] [PubMed]
- Bordwell, F.G.; Cheng, J.-P. Substituent effects on the stabilities of phenoxyl radicals and the acidities of phenoxyl radical cations. J. Am. Chem. Soc. 1991, 113, 1736–1743. [Google Scholar] [CrossRef]
- Mayer, J.M. Hydrogen atom abstraction by metal-oxo complexes: Understanding the analogy with organic radical reactions. Acc. Chem. Res. 1998, 31, 441–450. [Google Scholar] [CrossRef]
- Barman, P.; Upadhyay, P.; Faponle, A.S.; Kumar, J.; Nag, S.S.; Kumar, D.; Sastri, C.V.; de Visser, S.P. Deformylation reaction by a nonheme manganese(III)-peroxo complex via initial hydrogen atom abstraction. Angew. Chem. Int. Ed. 2016, 55, 11091–11095. [Google Scholar] [CrossRef] [PubMed]
- Cantú Reinhard, F.G.; Barman, P.; Mukherjee, G.; Kumar, J.; Kumar, D.; Kumar, D.; Sastri, C.V.; de Visser, S.P. Keto-enol tautomerization triggers an electrophilic aldehyde deformylation reaction by a nonheme manganese(III)-peroxo complex. J. Am. Chem. Soc. 2017, 139, 18328–18338. [Google Scholar] [CrossRef] [PubMed]
- Latifi, R.; Valentine, J.S.; Nam, W.; de Visser, S.P. Predictive studies of H-atom abstraction reactions by an iron(IV)-oxo corrole cation radical oxidant. Chem. Commun. 2012, 48, 3491–3493. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Sastry, G.N.; de Visser, S.P. Axial ligand effect on the rate constant of aromatic hydroxylation by iron(IV)-oxo complexes mimicking cytochrome P450 enzymes. J. Phys. Chem. B 2012, 116, 718–730. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Sastry, G.N.; de Visser, S.P. Effect of the axial ligand on substrate sulfoxidation mediated by iron(IV)-oxo porphyrin cation radical oxidants. Chem. Eur. J. 2011, 17, 6196–6205. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Karamzadeh, B.; Sastry, G.N.; de Visser, S.P. What factors influence the rate constant of substrate epoxidation by Compound I of cytochrome P450 and analogous iron(IV)-oxo oxidants. J. Am. Chem. Soc. 2010, 132, 7656–7667. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Ortega, A.; Quesne, M.G.; Bui, S.; Heuts, D.P.H.M.; Steiner, R.A.; Heyes, D.J.; de Visser, S.P.; Scrutton, N.S. Origin of the proton-transfer step in the cofactor-free 1-H-3-hydroxy-4-oxoquinaldine 2,4-dioxygenase: Effect of the basicity of an active site His residue. J. Biol. Chem. 2014, 289, 8620–8632. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Ortega, A.; Quesne, M.G.; Bui, S.; Heyes, D.J.; Steiner, R.A.; Scrutton, N.S.; de Visser, S.P. Catalytic mechanism of cofactor-free dioxygenases and how they circumvent spin-forbidden oxygenation of their substrates. J. Am. Chem. Soc. 2015, 137, 7474–7487. [Google Scholar] [CrossRef] [PubMed]
- Faponle, A.S.; Quesne, M.G.; de Visser, S.P. Origin of the regioselective fatty-acid hydroxylation versus decarboxylation by a cytochrome P450 peroxygenase: What drives the reaction to biofuel production? Chem. Eur. J. 2016, 22, 5478–5483. [Google Scholar] [CrossRef] [PubMed]
- Fellner, M.; Siakkou, E.; Faponle, A.S.; Tchesnokov, E.P.; de Visser, S.P.; Wilbanks, S.M.; Jameson, G.N.L. Influence of cysteine 164 on active site structure in rat cysteine dioxygenase. J. Biol. Inorg. Chem. 2016, 21, 501–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dakhili, S.Y.T.; Caslin, S.A.; Faponle, A.S.; Quayle, P.; de Visser, S.P.; Wong, L.S. Recombinant silicateins as model biocatalysts in organosiloxane chemistry. Proc. Natl. Acad. Sci. USA 2017, 114, 5285–5291. [Google Scholar] [CrossRef] [PubMed]
- Cantú Reinhard, F.G.; de Visser, S.P. Biodegradation of cosmetics products: A computational study of Cytochrome P450 metabolism of phthalates. Inorganics 2017, 5, 77. [Google Scholar] [CrossRef]
- Godfrey, E.; Porro, C.S.; de Visser, S.P. Comparative quantum mechanics/molecular mechanics (QM/MM) and density functional theory calculations on the oxo-iron species of taurine/α-ketoglutarate dioxygenase. J. Phys. Chem. A 2008, 112, 2464–2468. [Google Scholar] [CrossRef] [PubMed]
- Quesne, M.G.; Latifi, R.; Gonzalez-Ovalle, L.E.; Kumar, D.; de Visser, S.P. Quantum mechanics/molecular mechanics study on the oxygen binding and substrate hydroxylation step in AlkB repair enzymes. Chem. Eur. J. 2014, 20, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Thiel, W.; de Visser, S.P. Theoretical study on the mechanism of the oxygen activation process in cysteine dioxygenase enzymes. J. Am. Chem. Soc. 2011, 133, 3869–3882. [Google Scholar] [CrossRef] [PubMed]
- Prokop, K.A.; de Visser, S.P.; Goldberg, D.P. Unprecedented rate enhancements of hydrogen-atom transfer to a manganese(V)-oxo corrolazine complex. Angew. Chem. Int. Ed. 2010, 49, 5091–5095. [Google Scholar] [CrossRef] [PubMed]
- Vardhaman, A.K.; Sastri, C.V.; Kumar, D.; de Visser, S.P. Nonheme ferric hydroperoxo intermediates are efficient oxidants of bromide oxidation. Chem. Commun. 2011, 47, 11044–11046. [Google Scholar] [CrossRef] [PubMed]
- Vardhaman, A.K.; Barman, P.; Kumar, S.; Sastri, C.V.; Kumar, D.; de Visser, S.P. Comparison of the reactivity of nonheme iron(IV)-oxo versus iron(IV)-imido complexes: Which is the better oxidant? Angew. Chem. Int. Ed. 2013, 52, 12288–12292. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Faponle, A.S.; Barman, P.; Vardhaman, A.K.; Sastri, C.V.; Kumar, D.; de Visser, S.P. Long-range electron transfer triggers mechanistic differences between iron(IV)-oxo and iron(IV)-imido oxidants. J. Am. Chem. Soc. 2014, 136, 17102–17115. [Google Scholar] [CrossRef] [PubMed]
- Sainna, M.A.; Kumar, S.; Kumar, D.; Fornarini, S.; Crestoni, M.E.; de Visser, S.P. A comprehensive test set of epoxidation rate constants by iron(IV)-oxo porphyrin complexes. Chem. Sci. 2015, 6, 1516–1529. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Quesne, M.G.; Neu, H.M.; Cantú Reinhard, F.G.; Goldberg, D.P.; de Visser, S.P. Singlet versus triplet reactivity in an Mn(V)-Oxo species: Testing theoretical predictions against experimental evidence. J. Am. Chem. Soc. 2016, 138, 12375–12386. [Google Scholar] [CrossRef] [PubMed]
- Barman, P.; Faponle, A.S.; Vardhaman, A.K.; Angelone, D.; Löhr, A.-M.; Browne, W.R.; Comba, P.; Sastri, C.V.; de Visser, S.P. Influence of ligand architecture in tuning reaction bifurcation pathways for chlorite oxidation by nonheme iron complexes. Inorg. Chem. 2016, 55, 10170–10181. [Google Scholar] [CrossRef] [PubMed]
- Cantú Reinhard, F.G.; Sainna, M.A.; Upadhyay, P.; Balan, G.A.; Kumar, D.; Fornarini, S.; Crestoni, M.E.; de Visser, S.P. A systematic account on aromatic hydroxylation by a cytochrome P450 model Compound I: A low-pressure mass spectrometry and computational study. Chem. Eur. J. 2016, 22, 18608–18619. [Google Scholar] [CrossRef] [PubMed]
- Cantú Reinhard, F.G.; Faponle, A.S.; de Visser, S.P. Substrate sulfoxidation by an iron(IV)-oxo complex: Benchmarking computationally calculated barrier heights to experiment. J. Phys. Chem. A 2016, 120, 9805–9814. [Google Scholar] [CrossRef] [PubMed]
- Cantú Reinhard, F.G.; Fornarini, S.; Crestoni, M.E.; de Visser, S.P. Hydrogen atom versus hydride transfer in cytochrome P450 oxidations: A combined mass spectrometry and computational study. Eur. J. Inorg. Chem. 2018, 2018, 1854–1865. [Google Scholar] [CrossRef]
- Fowler, N.J.; Blanford, C.F.; Warwicker, J.; de Visser, S.P. Prediction of reduction potentials of copper proteins with continuum electrostatics and density functional theory. Chem. Eur. J. 2017, 23, 15436–15445. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, K.A.; Malhotra, A.; Balan, G.A.; Timmins, A.; de Visser, S.P. Nitrogen reduction to ammonia on a biomimetic mononuclear iron center: Insights into the nitrogenase enzyme. Chem. Eur. J. 2018, 24, 5293–5302. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–272. [Google Scholar] [CrossRef]
- Sainna, M.A.; Sil, D.; Sahoo, D.; Martin, B.; Rath, S.P.; Comba, P.; de Visser, S.P. Spin state ordering in hydroxo-bridged diiron(III)bisporphyrin complexes. Inorg. Chem. 2015, 54, 1919–1930. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P. What affects the quartet-doublet energy splitting in peroxidase enzymes? J. Phys. Chem. A 2005, 109, 11050–11057. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Ogliaro, F.; Harris, N.; Shaik, S. Multi-state epoxidation of ethene by cytochrome P450: A quantum chemical study. J. Am. Chem. Soc. 2001, 123, 3037–3047. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Shaik, S.; Sharma, P.K.; Kumar, D.; Thiel, W. Active species of horseradish peroxidase (HRP) and cytochrome P450: Two electronic chameleons. J. Am. Chem. Soc. 2003, 125, 15779–15788. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.T.; Sligar, S.G. Control of heme protein redox potential and reduction rate: Linear free energy relation between potential and ferric spin state equilibrium. J. Am. Chem. Soc. 1985, 107, 5018–5019. [Google Scholar] [CrossRef]
- Ogliaro, F.; Cohen, S.; de Visser, S.P.; Shaik, S. Medium polarization and hydrogen bonding effects on Compound I of cytochrome P450: What kind of a radical is it really? J. Am. Chem. Soc. 2000, 122, 12892–12893. [Google Scholar] [CrossRef]
- Ogliaro, F.; de Visser, S.P.; Cohen, S.; Kaneti, J.; Shaik, S. The experimentally elusive oxidant of cytochrome P450: A theoretical “trapping” defining more closely the “real” species. ChemBioChem 2001, 2, 848–851. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Czodrowski, P.; Li, H.; Nielsen, J.E.; Jensen, J.H.; Klebe, G.; Baker, N.A. PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucl. Acids Res. 2007, 35, 522–525. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.; Karplus, M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Schafer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Sherwood, P.; de Vries, A.H.; Guest, M.F.; Schreckenbach, G.; Catlow, C.R.A.; French, S.A.; Sokol, A.A.; Bromley, S.T.; Thiel, W.; Turner, A.J.; et al. QUASI: A general purpose implementation of the QM/MM approach and its application to problems in catalysis. J. Mol. Struct. Theochem. 2003, 632, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Smith, W.; Forester, T.R. DL_POLY_2.0: A general-purpose parallel molecular dynamics simulation package. J. Mol. Graph. 1996, 14, 136–141. [Google Scholar] [CrossRef]
- Bakowies, D.; Thiel, W. Hybrid models for combined quantum mechanical and molecular mechanical approaches. J. Phys. Chem. 1996, 100, 10580–10594. [Google Scholar] [CrossRef]
- Hitzenberger, M.; Hofer, T.S. Probing the range of applicability of structure- and energy-adjusted QM/MM link bonds. J. Comput. Chem. 2015, 36, 1929–1939. [Google Scholar] [CrossRef] [PubMed]












© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Postils, V.; Saint-André, M.; Timmins, A.; Li, X.-X.; Wang, Y.; Luis, J.M.; Solà, M.; De Visser, S.P. Quantum Mechanics/Molecular Mechanics Studies on the Relative Reactivities of Compound I and II in Cytochrome P450 Enzymes. Int. J. Mol. Sci. 2018, 19, 1974. https://doi.org/10.3390/ijms19071974
Postils V, Saint-André M, Timmins A, Li X-X, Wang Y, Luis JM, Solà M, De Visser SP. Quantum Mechanics/Molecular Mechanics Studies on the Relative Reactivities of Compound I and II in Cytochrome P450 Enzymes. International Journal of Molecular Sciences. 2018; 19(7):1974. https://doi.org/10.3390/ijms19071974
Chicago/Turabian StylePostils, Verònica, Maud Saint-André, Amy Timmins, Xiao-Xi Li, Yong Wang, Josep M. Luis, Miquel Solà, and Sam P. De Visser. 2018. "Quantum Mechanics/Molecular Mechanics Studies on the Relative Reactivities of Compound I and II in Cytochrome P450 Enzymes" International Journal of Molecular Sciences 19, no. 7: 1974. https://doi.org/10.3390/ijms19071974