Catalpol Inhibits Ischemia-Induced Premyelinating Oligodendrocyte Damage through Regulation of Intercellular Calcium Homeostasis via Na+/Ca2+ Exchanger 3


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
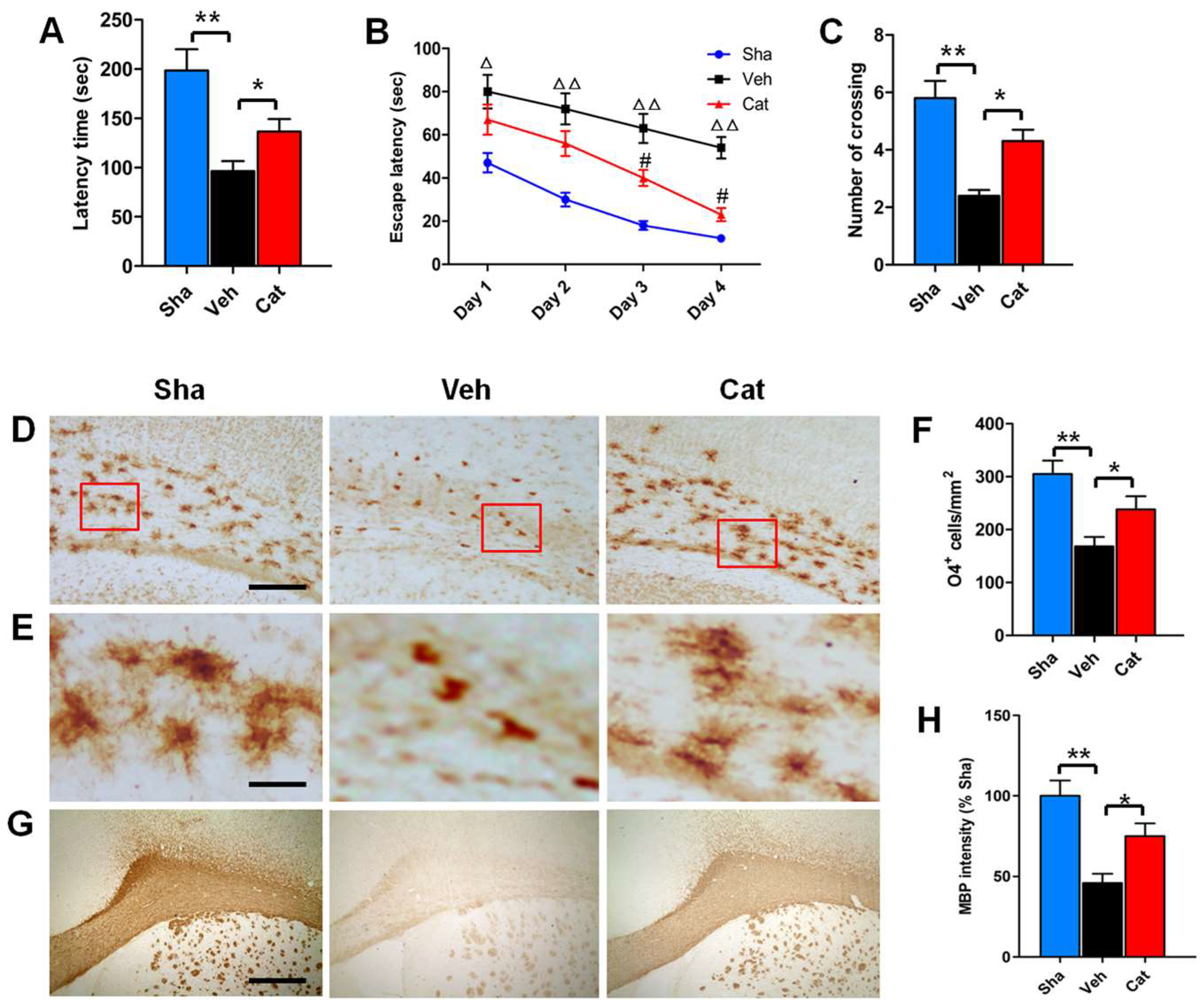
2.1. Catalpol Improves Motor and Memory Deficits of Rats after Neonatal Hypoxia–Ischemia
2.2. Catalpol Prevents PreOL Damage and Myelination Deficit after Hypoxia–Ischemia
2.3. Catalpol Improves Survival and Restores Maturation of PreOLs under OGD
2.4. Catalpol Inhibits Intracellular Calcium Elevation of PreOLs under OGD
2.5. Catalpol Enhances NCX3 Expression in PreOLs under OGD
2.6. Catalpol Protects PreOLs against OGD by Upregulating NCX3
2.7. Catalpol Reduces Mitochondrion-Mediated Activation of the ERK1/2 Pathway in PreOLs under OGD
3. Discussion
4. Materials and Methods
4.1. Neonatal Hypoxia–Ischemia Model and Drug Administration
4.2. Rotarod Test
4.3. Morris Water Maze Test
4.4. Immunohistochemistry
4.5. Oligodendrocyte Progenitor Cell Culture.
4.6. OGD Model and Drug Treatment
4.7. MTT Assay
4.8. Lactate Dehydrogenase Assay
4.9. Terminal Deoxynucleotidyl Transferase-Mediated Dutp-Biotin Nick End Labeling Assay (TUNEL) Staining
4.10. Immunofluorescent Staining
4.11. Confocal Ca2+ Imaging
4.12. Quantitative RT-PCR
4.13. Western Blot Analysis
4.14. Mitochondrial Membrane Potential Assay
4.15. Determination of Reactive Oxygen Species (ROS) Generation
4.16. Determination of Malondialdehyde Levels
4.17. Statistical Analysis
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| PreOL | Premyelinating oligodendrocyte |
| OL | Oligodendrocyte |
| OPC | Oligodendrocyte progenitor cell |
| NCX3 | Na+/Ca2+ exchanger 3 |
| OGD | Oxygen-glucose deprivation |
| ERK1/2 | Extracellular signal-regulated kinase 1/2 |
| PARP-1 | Poly-ADP-ribose polymerase-1 |
| KB-R7943 | 2-[2-[4-(4-nitrobenzyloxy)phenyl]ethyl]isothiourea |
| MBP | Myelin basic protein |
| ROS | Reactive oxygen species |
| MDA | Malondialdehyde |
References
- Volpe, J.J. Brain injury in premature infants: A complex amalgam of destructive and developmental disturbances. Lancet Neurol. 2009, 8, 110–124. [Google Scholar] [CrossRef]
- Back, S.A.; Luo, N.L.; Borenstein, N.S.; Levine, J.M.; Volpe, J.J.; Kinney, H.C. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J. Neurosci. 2001, 21, 1302–1312. [Google Scholar] [CrossRef] [PubMed]
- Mifsud, G.; Zammit, C.; Muscat, R.; Giovanni, G.D.; Valentino, M. Oligodendrocyte pathophysiology and treatment strategies in cerebral ischemia. CNS. Neurosci. Ther. 2014, 20, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Back, S.A.; Miller, S.P. Brain injury in premature neonates: A primary cerebral dysmaturation disorder? Ann. Neurol. 2014, 75, 469–486. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.P.; Li, X.Y.; Zhou, B.; Shen, W.H.; Zhang, Z.J.; Xu, T.L.; Duan, S. Ca2+ signaling evoked by activation of Na+ channels and Na+/Ca2+ exchangers is required for GABA-induced NG2 cell migration. J. Cell Biol. 2009, 186, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Soliven, B. Calcium signalling in cells of oligodendroglial lineage. Microsc. Res. Tech. 2001, 52, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Domercq, M.; Perez-Samartin, A.; Aparicio, D.; Alberdi, E.; Pampliega, O.; Matute, C. P2X7 receptors mediate ischemic damage to oligodendrocytes. Glia 2010, 58, 730–740. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.Y.; Cai, W.Q.; Chen, P.H.; Deng, Q.Y.; Zhao, C.M. Downregulation of P2X7 receptor expression in at oligodendrocyte precursor cells after hypoxia ischemia. Glia 2009, 57, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Jeon, D.; Chu, K.; Jung, K.H.; Kim, M.; Yoon, B.W.; Lee, C.J.; Oh, U.; Shin, H.S. Na+/Ca2+ exchanger 2 is neuroprotective by exporting Ca2+ during a transient focal cerebral ischemia in the mouse. Cell Calcium 2008, 43, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, P.; Cuomo, O.; Pignataro, G.; Boscia, F.; Sirabella, R.; Pannaccione, A.; Secondo, A.; Scorziello, A.; Adornetto, A.; Gala, R.; et al. Targeted disruption of Na+/Ca2+ exchanger 3 (NCX3) gene leads to a worsening of ischemic brain damage. J. Neurosci. 2008, 28, 1179–1184. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Lao, A.J.; Katsura, M.; Caputo, A.; Schweizer, F.E.; Sokolow, S. Involvement of the sodium-calcium exchanger 3 (NCX3) in ziram-induced calcium dysregulation and toxicity. Neurotoxicology 2014, 45, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, L.; Pignataro, G.; Boscia, F.; Sirabella, R.; Formisano, L.; Saggese, M.; Cuomo, O.; Gala, R.; Secondo, A.; Viggiano, D.; et al. Ncx1, ncx2, and ncx3 gene product expression and function in neuronal anoxia and brain ischemia. Ann. N. Y. Acad. Sci. 2007, 1099, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Pignataro, G.; Gala, R.; Cuomo, O.; Tortiglione, A.; Giaccio, L.; Castaldo, P.; Sirabella, R.; Matrone, C.; Canitano, A.; Amoroso, S.; et al. Two sodium/calcium exchanger gene products, NCX1 and NCX3, play a major role in the development of permanent focal cerebral ischemia. Stroke 2004, 35, 2566–2570. [Google Scholar] [CrossRef] [PubMed]
- Brustovetsky, T.; Bolshakov, A.; Brustovetsky, N. Calpain activation and Na+/Ca2+ exchanger degradation occur downstream of calcium deregulation in hippocampal neurons exposed to excitotoxic glutamate. J. Neurosci. Res. 2010, 88, 1317–1328. [Google Scholar] [CrossRef] [PubMed]
- Boscia, F.; D’Avanzo, C.; Pannaccione, A.; Secondo, A.; Casamassa, A.; Formisano, L.; Guida, N.; Sokolow, S.; Herchuelz, A.; Annunziato, L. Silencing or knocking out the Na+/Ca2+ exchanger-3 (NCX3) impairs oligodendrocyte differentiation. Cell Death Differ. 2012, 19, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Casamassa, A.; Rocca, C.L.; Sokolow, S.; Herchuelz, A.; Matarese, G.; Annunziato, L.; Boscia, F. Ncx3 gene ablation impairs oligodendrocyte precursor response and increases susceptibility to experimental autoimmune encephalomyelitis. Glia 2016, 64, 1124–1137. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Wu, X.Y.; Cai, Q.Y.; Wang, Y.; Xiao, L.; Tian, Y.P.; Li, H.L. Lead poisoning disturbs oligodendrocytes differentiation involved in decreased expression of NCX3 inducing intracellular calcium overload. Int. J. Mol. Sci. 2015, 16, 19096–19110. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.; Wang, X.B.; Chen, L.; Hao, S.; An, L.J.; Jiang, B.; Guo, L. Catalpol protects mesencephalic neurons against MPTP induced neurotoxicity via attenuation of mitochondrial dysfunction and MAO-B activity. Toxicol. In Vitro 2008, 22, 1883–1889. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.M.; Zhang, R.; Wu, P.P.; Xia, Z.Q.; Hu, Y.E. Memory defect induced by beta-amyloid plus glutamate receptor agonist is alleviated by catalpol and donepezil through different mechanisms. Brain Res. 2012, 1441, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Bao, Y.; Jiang, B.; Wang, Z.; Liu, Y.; Zhang, C.; An, L. Catalpol protects primary cultured astrocytes from in vitro ischemia-induced damage. Int. J. Dev. Neurosci. 2008, 26, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; An, L.J.; Duan, Y.L.; Li, Y.C.; Jiang, B. Catalpol protects rat pheochromocytoma cells against oxygen and glucose deprivation-induced injury. Neurol. Res. 2008, 30, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.Y.; Chen, X.S.; Zhan, X.L.; Yao, Z.X. Protective effects of catalpol on oligodendrocyte death and myelin breakdown in a rat model of chronic cerebral hypoperfusion. Neurosci. Lett. 2011, 497, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.Y.; Yao, Z.X.; Li, H.L. Catalpol promotes oligodendrocyte survival and oligodendrocyte progenitor differentiation via the Akt signaling pathway in rats with chronic cerebral hypoperfusion. Brain Res. 2014, 1560, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.Y.; Ma, T.; Li, C.R.; Tian, Y.P.; Li, H.L. Catalpol protects pre-myelinating oligodendrocytes against ischemia-induced oxidative injury through ERK1/2 signaling pathway. Int. J. Biol. Sci. 2016, 12, 1415–1426. [Google Scholar] [CrossRef] [PubMed]
- Misumi, S.; Ueda, Y.; Nishigaki, R.; Suzuki, M.; Ishida, A.; Jung, C.G.; Hida, H. Dysfunction in motor coordination in neonatal white matter injury model without apparent neuron loss. Cell Transplant. 2016, 25, 1381–1393. [Google Scholar] [CrossRef] [PubMed]
- Ziemka-Nalecz, M.; Jaworska, J.; Sypecka, J.; Polowy, R.; Filipkowski, R.K.; Zalewska, T. Sodium butyrate, a histone deacetylase inhibitor, exhibits neuroprotective/neurogenic effects in a rat model of neonatal hypoxia-ischemia. Mol. Neurobiol. 2017, 54, 5300–5318. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Rosenberg, P.A.; Volpe, J.J.; Jensen, F.E. Calcium-permeable AMPA/kainate receptors mediate toxicity and preconditioning by oxygen-glucose deprivation in oligodendrocyte precursors. Proc. Natl. Acad. Sci. USA 2003, 100, 6801–6806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwamoto, T. Forefront of Na+/Ca2+ exchanger studies: Molecular pharmacology of Na+/Ca2+ exchange inhibitors. J. Pharmacol. Sci. 2004, 96, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Domercq, M.; Alberdi, E.; Sánchez-Gómez, M.V.; Ariz, U.; Pérez-Samartín, A.; Matute, C. Dual-specific phosphatase-6 (Dusp6) and ERK mediate AMPA receptor-induced oligodendrocyte death. J. Biol. Chem. 2011, 286, 11825–11836. [Google Scholar] [CrossRef] [PubMed]
- Volpe, J.J.; Kinney, H.C.; Jensen, F.E.; Rosenberg, P.A. The developing oligodendrocyte: Key cellular target in brain injury in the premature infant. Int. J. Dev. Neurosci. 2011, 29, 423–440. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, A.; Matute, C.; Alberdi, E. Intracellular Ca2+ release through ryanodine receptors contributes to AMPA receptor-mediated mitochondrial dysfunction and ER stress in oligodendrocytes. Cell Death Dis. 2010, 1, e54. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Ibarretxe, G.; Sánchez-Gómez, M.V.; Campos-Esparza, M.R.; Alberdi, E.; Matute, C. Differential oxidative stress in oligodendrocytes and neurons after excitotoxic insults and protection by natural polyphenols. Glia 2006, 53, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, P.; Cantile, M.; Cuomo, O.; Secondo, A.; Pannaccione, A.; Ambrosino, P.; Pignataro, G.; Fiorino, F.; Severino, B.; Gatta, E.; et al. Neurounina-1, a novel compound that increases Na+/Ca2+ exchanger activity, effectively protects against stroke damage. Mol. Pharmacol. 2013, 83, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Boscia, F.; Gala, R.; Pignataro, G.; de Bartolomeis, A.; Cicale, M.; Ambesi-Impiombato, A.; di Renzo, G.; Annunziato, L. Permanent focal brain ischemia induces isoform-dependent changes in the pattern of Na+/Ca2+ exchanger gene expression in the ischemic core, periinfarct area, and intact brain regions. J. Cereb. Blood Flow Metab. 2006, 26, 502–517. [Google Scholar] [CrossRef] [PubMed]
- Kip, S.N.; Strehler, E.E. Rapid downregulation of NCX and PMCA in hippocampal neurons following H2O2 oxidative stress. Ann. N.Y. Acad. Sci. 2007, 1099, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Bano, D.; Young, K.W.; Guerin, C.J.; Lefeuvre, R.; Rothwell, N.J.; Naldini, L.; Rizzuto, R.; Carafoli, E.; Nicotera, P. Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell 2005, 120, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Volpe, J.J. Neurology of the newborn, 5th ed.; Elsevier: Philadelphia, PA, USA, 2008. [Google Scholar]
- Wright, J.; Zhang, G.; Yu, T.S.; Kernie, S. Age-related changes in the oligodendrocyte progenitor pool influence brain remodeling after injury. Dev. Neurosci. 2010, 32, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Elitt, C.M.; Rosenberg, P.A. The challenge of understanding cerebral white matter injury in the premature infant. Neuroscience 2014, 276, 216–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segovia, K.; McClure, M.; Moravec, M.; Luo, N.L.; Wan, Y.; Gong, X.; Riddle, A.; Craig, A.; Struve, J.; Sherman, L.S.; et al. Arrested oligodendrocyte lineage maturation in chronic perinatal white matter injury. Ann. Neurol. 2008, 63, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Buser, J.R.; Maire, J.; Riddle, A.; Gong, X.; Nguyen, T.; Nelson, K.; Luo, N.L.; Ren, J.; Struve, J.; Sherman, L.S.; et al. Arrested preoligodendrocyte maturation contributes myelination failure in premature infants. Ann. Neurol. 2012, 71, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Back, S.A.; Rosenberg, P.A. Pathophysiology of glia in perinatal white matter injury. Glia 2014, 62, 1790–1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friess, M.; Hammann, J.; Unichenko, P.; Luhmann, H.J.; White, R.; Kirischuk, S. Intracellular ion signaling influences myelin basic protein synthesis in oligodendrocyte precursor cells. Cell Calcium 2016, 60, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Dinicola, S.; Mariggiò, M.A.; Morabito, C.; Guarnieri, S.; Cucina, A.; Pasqualato, A.; D’Anselmi, F.; Proietti, S.; Coluccia, P.; Bizzarri, M. Grape seed extract triggers apoptosis in Caco-2 human colon cancer cells through reactive oxygen species and calcium increase: Extracellular signal-regulated kinase involvement. Br. J. Nutr. 2013, 110, 797–809. [Google Scholar] [CrossRef] [PubMed]
- Ulbrich, F.; Kaufmann, K.B.; Coburn, M.; Lagrèze, W.A.; Roesslein, M.; Biermann, J.; Buerkle, H.; Loop, T.; Goebel, U. Neuroprotective effects of argon are mediated via an ERK-1/2 dependent regulation of heme-oxygenase-1 in retinal ganglion cells. J. Neurochem. 2015, 134, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Domercq, M.; Mato, S.; Soria, F.N.; Sánchez-gómez, M.V.; Alberdi, E.; Matute, C. Zn2+-induced ERK activation mediates PARP-1-dependent ischemic-reoxygenation damage to oligodendrocytes. Glia 2013, 61, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Armon, M. PARP-1 activation in the ERK signaling pathway. Trends Pharmacol. Sci. 2007, 28, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.X.; Tang, S.; Huang, W.W.; Liu, X.M.; Li, G.H.; Chi, H.; Zhu, M.; Tang, J. Protection of the brain following cerebral ischemia through the attenuation of PARP-1-induced neurovascular unit damage in rats. Brain Res. 2015, 1624, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Veto, S.; Acs, P.; Bauer, J.; Lassmann, H.; Berente, Z.; Setalo, G., Jr.; Borgulya, G.; Sumegi, B.; Komoly, S.; Gallyas, F., Jr.; et al. Inhibiting poly-(ADP-ribose) polymerase: A potential therapy against oligodendrocyte death. Brain 2010, 133, 822–834. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Ohlsson, M.; Warner, E.A.; Loo, K.K.; Hoang, T.X.; Voskuhl, R.R.; Havton, L.A. Glial reactions and degeneration of myelinated processes in spinal cord gray matter in chronic experimental autoimmune encephalomyelitis. Neuroscience 2008, 156, 586–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.; Lin, S.; Fan, L.W.; Pang, Y.; Rhodes, P.G. Minocycline alleviates hypoxic–ischemic injury to developing oligodendrocytes in the neonatal rat brain. Neuroscience 2006, 137, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Mei, F.; Li, N.; Wang, H.; Li, X.; Kong, J.; Xiao, L. Haloperidol promotes proliferation but inhibits differentiation in rat oligodendrocyte progenitor cell cultures. Biochem. Cell Biol. 2010, 88, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Zha, Y.Y.; Yang, B.; Tang, M.L.; Guo, Q.C.; Chen, J.T.; Wen, L.P.; Wang, M. Concentration-dependent effects of fullerenol on cultured hippocampal neuron viability. Int. J. Nanomedicine 2012, 7, 3099–3109. [Google Scholar] [PubMed]
- Yan, S.; Zhang, H.; Wang, J.; Zheng, F.; Dai, J. Perfluorooctanoic acid exposure induces endoplasmic reticulum stress inthe liver andits effects are amelioratedby 4-phenylbutyrate. Free Radic. Biol. Med. 2015, 87, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Yuan, X.; Jia, T.; Liu, C.; Ni, Z.; Qin, Z.; Yuan, Y. Polymeric prodrug of bufalin for increasing solubility and stability: Synthesis and anticancer study in vitro and in vivo. Int. J. Pharm. 2016, 506, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wu, W.Y.; Huang, H.; Li, W.Z.; Chen, H.Q.; Yin, Y.Y. Biochanin A protects against lipopolysaccharide-induced damage of dopaminergic neurons both in vivo and in vitro via inhibition of microglial activation. Neurotox. Res. 2016, 30, 486–498. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Bai, Y.; Zhang, C.; Zhang, X.; Zhang, Y.X.; Chen, J.; Xiong, L.; Shi, M.; Zhao, G. Cinepazide maleate protects PC12 cells against oxygen-glucose deprivation-induced injury. Neurol. Sci. 2014, 35, 875–881. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, Q.; Ma, T.; Tian, Y.; Li, C.; Li, H. Catalpol Inhibits Ischemia-Induced Premyelinating Oligodendrocyte Damage through Regulation of Intercellular Calcium Homeostasis via Na+/Ca2+ Exchanger 3. Int. J. Mol. Sci. 2018, 19, 1925. https://doi.org/10.3390/ijms19071925
Cai Q, Ma T, Tian Y, Li C, Li H. Catalpol Inhibits Ischemia-Induced Premyelinating Oligodendrocyte Damage through Regulation of Intercellular Calcium Homeostasis via Na+/Ca2+ Exchanger 3. International Journal of Molecular Sciences. 2018; 19(7):1925. https://doi.org/10.3390/ijms19071925
Chicago/Turabian StyleCai, Qiyan, Teng Ma, Yanping Tian, Chengren Li, and Hongli Li. 2018. "Catalpol Inhibits Ischemia-Induced Premyelinating Oligodendrocyte Damage through Regulation of Intercellular Calcium Homeostasis via Na+/Ca2+ Exchanger 3" International Journal of Molecular Sciences 19, no. 7: 1925. https://doi.org/10.3390/ijms19071925