Neuroimmune Tau Mechanisms: Their Role in the Progression of Neuronal Degeneration

{kind=link}
Abstract
:1. Tau Protein the Context of Alzheimer’s Disease
2. Molecular and Structural Aspects of Tauopathies and AD
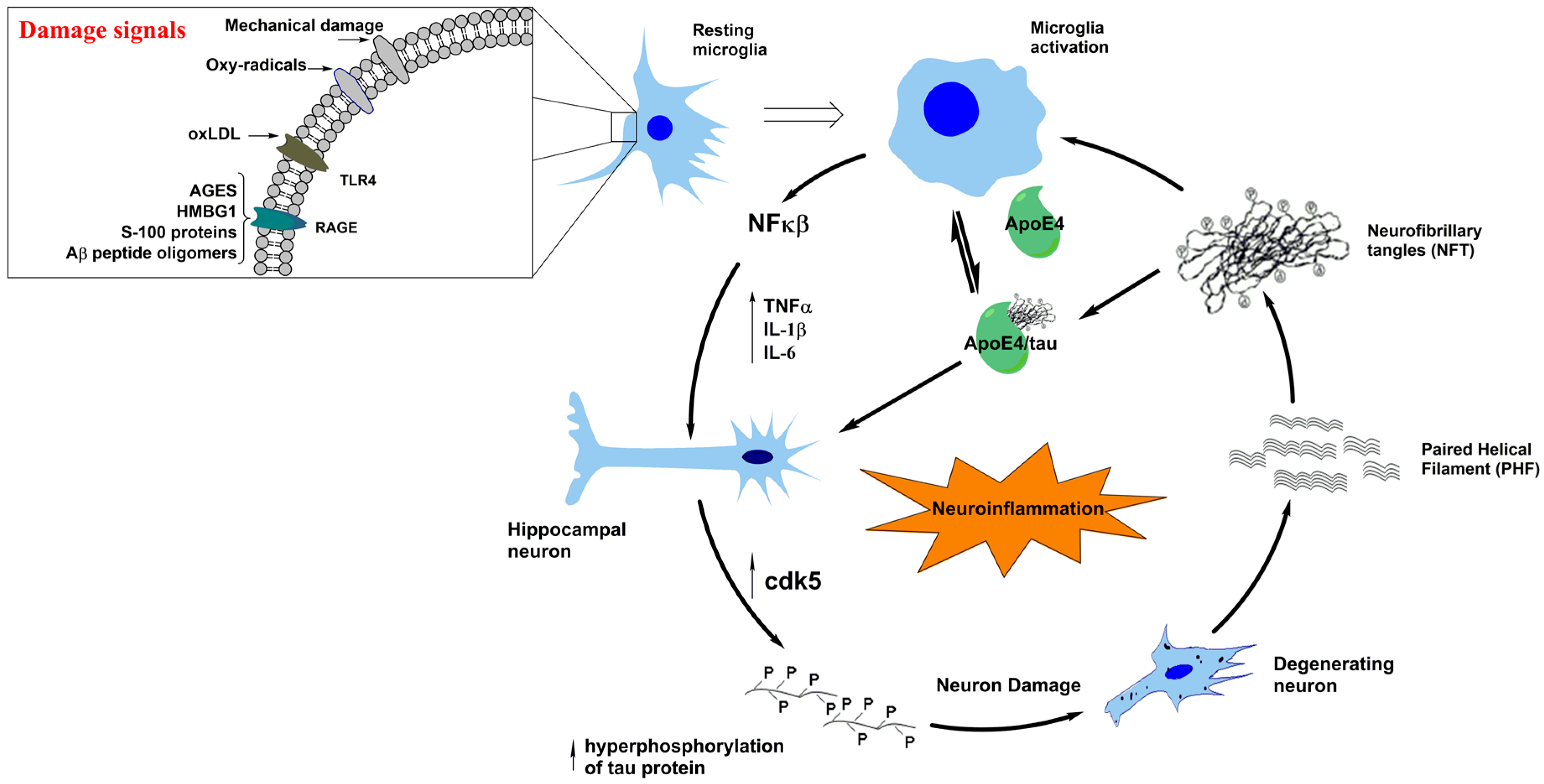
3. Neuroinflammation in Tauopathies
4. A Typical Tauopathy: Frontotemporal Dementia (FTD)
5. Molecular Interactions and the Links between Tauopathies and Parkinson’s Disease
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| WHO | World Health Organization |
| NFT | Neurofibrillary tangles |
| PHF | Paired helical filaments |
| SP | Senile plaques |
| Aβ | Amyloid-β |
| APP | Amyloid precursor protein |
| MAP | Microtubule-associated proteins |
| NFκB | Nuclear factor κB |
| CNS | Central nervous system |
| EEG | Electroencephalogram |
| NMDA | N-methyl-d-aspartate |
| CDK5 | Cyclin-dependent kinase 5 |
| GSK3-β | Glycogen synthase kinase 3-β |
| JNK | c-Jun-N-terminal kinase |
| Akt | Serine/threonine kinase |
| LTD | Long-term depression |
| LTP | Long-term potentiation |
| ERK | Extracellular signal-regulated kinases |
| MEMRI | Manganese-enhanced magnetic resonance imaging |
| PET | Positron-emission tomography |
| PSP | Supranuclear palsy |
| FTD | Frontotemporal dementia |
| bvFTD | Behavioural variant of Frontotemporal dementia |
| nfFTD | Nonfluent variant of Frontotemporal dementia |
| MNFTD | Motoneuron disorder |
| PSP | Progressive supranuclear palsy |
| svFTD | Semantic variant of Frontotemporal dementia |
| CBS | Corticobasal syndromes |
| FTLD | Frontotemporal lobar degeneration |
| TDP | Transactive response of DNA-binding protein |
| FET | FUS, EWS, and TAF15 protein family |
| UPS | Ubiquitin Proteasome System |
| PiD | Picks disease |
| GGT | Globular glial tauopathies |
| AGD | Argyrophilic grain disease |
| C9ORF72 | Chromosome 9 open reading frame 72 |
| TNF-α | Tumor necrosis factor α |
| TGF-β | Transforming growth factor β |
| IL | Interleukin |
| COX2 | Cyclooxygenase-2 |
| TSPO | Translocator protein |
| CSF | Cerebrospinal fluid |
| 5-HT | Serotonin |
| SSRIs | Selective serotonin reuptake inhibitors |
| PD | Parkinson’s disease |
| α-syn | α-Synuclein |
| LB | Lewy Bodies |
| NAC | Non-amyloid component |
| PDD | Parkinson’s disease with dementia |
| DLB | Lewy body dementia |
| MSA | Multiple system atrophy |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| A53T SNCA | Ala53Thr synuclein α |
| PKA | Protein Kinase A |
| FIDA | Fluorescence intensity distribution analysis |
References
- Bettens, K.; Sleegers, K.; Van Broeckhoven, C. Current status on Alzheimer disease molecular genetics: From past, to present, to future. Hum. Mol. Genet. 2010, 19, R4–R11. [Google Scholar] [CrossRef] [PubMed]
- Maccioni, R.B. Introductory remarks. Molecular, biological and clinical aspects of Alzheimer’s disease. Arch. Med. Res. 2012, 43, 593–594. [Google Scholar] [CrossRef] [PubMed]
- Maccioni, R.B.; Munoz, J.P.; Barbeito, L. The molecular bases of Alzheimer’s disease and other neurodegenerative disorders. Arch. Med. Res. 2001, 32, 367–381. [Google Scholar] [CrossRef]
- Farias, G.; Cornejo, A.; Jimenez, J.; Guzman, L.; Maccioni, R.B. Mechanisms of tau self-aggregation and neurotoxicity. Curr. Alzheimer Res. 2011, 8, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Martinez, L.; Farias, G.A.; Maccioni, R.B. Tau Oligomers as Potential Targets for Alzheimer’s Diagnosis and Novel Drugs. Front. Neurol. 2013, 4, 167. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Schraen-Maschke, S.; Richard, F.; Fievet, N.; Rouaud, O.; Berr, C.; Dartigues, J.F.; Tzourio, C.; Alpérovitch, A.; Buée, L.; et al. Association of plasma amyloid β with risk of dementia: The prospective Three-City Study. Neurology 2009, 73, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, D.W.; Hwo, S.Y.; Kirschner, M.W. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J. Mol. Biol. 1977, 116, 207–225. [Google Scholar] [CrossRef]
- Seeds, N.W.; Maccioni, R.B. Proteins from morphologically differentiated neuroblastoma cells promote tubulin polymerization. J. Cell Biol. 1978, 76, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, J.A.; Rojo, L.; Kuljis, R.O.; Maccioni, R.B. The damage signals hypothesis of Alzheimer’s disease pathogenesis. J. Alzheimer Dis. 2008, 14, 329–333. [Google Scholar] [CrossRef]
- Maccioni, R.B.; Rojo, L.E.; Fernandez, J.A.; Kuljis, R.O. The role of neuroimmunomodulation in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2009, 1153, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Morales, I.; Farias, G.; Maccioni, R.B. Neuroimmunomodulation in the pathogenesis of Alzheimer’s disease. Neuroimmunomodulation 2010, 17, 202–204. [Google Scholar] [CrossRef] [PubMed]
- Morales, I.; Guzman-Martinez, L.; Cerda-Troncoso, C.; Farias, G.A.; Maccioni, R.B. Neuroinflammation in the pathogenesis of Alzheimer’s disease. A rational framework for the search of novel therapeutic approaches. Front. Cell. Neurosci. 2014, 8, 112. [Google Scholar] [CrossRef] [PubMed]
- Rojo, L.E.; Fernandez, J.A.; Maccioni, A.A.; Jimenez, J.M.; Maccioni, R.B. Neuroinflammation: Implications for the pathogenesis and molecular diagnosis of Alzheimer’s disease. Arch. Med. Res. 2008, 39, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Morales, I.; Jimenez, J.M.; Mancilla, M.; Maccioni, R.B. Tau oligomers and fibrils induce activation of microglial cells. J. Alzheimer Dis. 2013, 37, 849–856. [Google Scholar] [CrossRef]
- Martin, L.; Latypova, X.; Wilson, C.M.; Magnaudeix, A.; Perrin, M.L.; Yardin, C.; Terro, F. Tau protein kinases: Involvement in Alzheimer’s disease. Ageing Res. Rev. 2013, 12, 289–309. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.L.; Wang, C.; Jiang, T.; Tan, L.; Xing, A.; Yu, J.T. The Role of Cdk5 in Alzheimer’s Disease. Mol. Neurobiol. 2016, 53, 4328–4342. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Sasamoto, K.; Nagai, J.; Yamazaki, Y.; Saito, K.; Goshima, Y.; Inoue, T.; Ohshima, T. Phosphorylation of CRMP2 by Cdk5 Regulates Dendritic Spine Development of Cortical Neuron in the Mouse Hippocampus. Neural Plast. 2016, 2016, 6790743. [Google Scholar] [CrossRef] [PubMed]
- Hanger, D.P.; Noble, W. Functional implications of glycogen synthase kinase-3-mediated tau phosphorylation. Int. J. Alzheimers Dis. 2011, 2011, 352805. [Google Scholar] [CrossRef] [PubMed]
- Meunier, C.N.; Cancela, J.M.; Fossier, P. Lack of GSK3-β activation and modulation of synaptic plasticity by dopamine in 5-HT1A-receptor KO mice. Neuropharmacology 2017, 113 Pt A, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Maixner, D.W.; Weng, H.R. The role of glycogen synthase kinase 3 β in neuroinflammation and pain. J. Pharm. Pharmacol. 2013, 1, 1. [Google Scholar] [CrossRef]
- Lee, H.G.; Ogawa, O.; Zhu, X.; O’Neill, M.J.; Petersen, R.B.; Castellani, R.J.; Ghanbari, H.; Perry, G.; Smith, M.A. Aberrant expression of metabotropic glutamate receptor 2 in the vulnerable neurons of Alzheimer’s disease. Acta Neuropathol. 2004, 107, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Florenzano, F.; Veronica, C.; Ciasca, G.; Ciotti, M.T.; Pittaluga, A.; Olivero, G.; Feligioni, M.; Iannuzzi, F.; Latina, V.; Maria Sciacca, M.F.; et al. Extracellular truncated tau causes early presynaptic dysfunction associated with Alzheimer’s disease and other tauopathies. Oncotarget 2017. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; McInnes, J.; Wierda, K.; Holt, M.; Herrmann, A.G.; Jackson, R.J.; Wang, Y.-C.; Swerts, J.; Beyens, J.; Miskiewicz, K.; et al. Tau association with synaptic vesicles causes presynaptic dysfunction. Nat. Commun. 2017, 8, 15295. [Google Scholar] [CrossRef] [PubMed]
- Swanson, E.; Breckenridge, L.; McMahon, L.; Som, S.; McConnell, I.; Bloom, G.S. Extracellular Tau Oligomers Induce Invasion of Endogenous Tau into the Somatodendritic Compartment and Axonal Transport Dysfunction. J. Alzheimer Dis. 2017, 58, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, S.N.; Ingram, A.; Cloyd, R.A.; Meier, S.E.; Miller, E.; Lyons, D.; Nation, G.K.; Mechas, E.; Weiss, B.; Lanzillotta, C.; et al. Identification of changes in neuronal function as a consequence of aging and tauopathic neurodegeneration using a novel and sensitive magnetic resonance imaging approach. Neurobiol. Aging 2017, 56, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Kepe, V.; Bordelon, Y.; Boxer, A.; Huang, S.C.; Liu, J.; Thiede, F.C.; Mazziotta, J.C.; Mendez, M.F.; Donoghue, N.; Small, G.W.; et al. PET imaging of neuropathology in tauopathies: Progressive supranuclear palsy. J. Alzheimer Dis. 2013, 36, 145–153. [Google Scholar] [CrossRef]
- Andrade, V.; Cortes, N.; Guzmán-Martínez, L.; Maccioni, R.B. An overview of the links between behavioral disorders and Alzheimer’ disease. JSM Alzheimer’s Dis. Relat. Dement. 2017, 4, 1031. [Google Scholar]
- Bellucci, A.; Westwood, A.J.; Ingram, E.; Casamenti, F.; Goedert, M.; Spillantini, M.G. Induction of inflammatory mediators and microglial activation in mice transgenic for mutant human P301S tau protein. Am. J. Pathol. 2004, 165, 1643–1652. [Google Scholar] [CrossRef]
- Bellucci, A.; Bugiani, O.; Ghetti, B.; Spillantini, M.G. Presence of reactive microglia and neuroinflammatory mediators in a case of frontotemporal dementia with P301S mutation. Neurodegener. Dis. 2011, 8, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Sjogren, M.; Folkesson, S.; Blennow, K.; Tarkowski, E. Increased intrathecal inflammatory activity in frontotemporal dementia: Pathophysiological implications. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Wes, P.D.; Easton, A.; Corradi, J.; Barten, D.M.; Devidze, N.; DeCarr, L.B.; Truong, A.; He, A.; Barrezueta, N.X.; Polson, C.; et al. Tau overexpression impacts a neuroinflammation gene expression network perturbed in Alzheimer’s disease. PLoS ONE 2014, 9, e106050. [Google Scholar] [CrossRef] [PubMed]
- Cagnin, A.; Rossor, M.; Sampson, E.L.; Mackinnon, T.; Banati, R.B. In vivo detection of microglial activation in frontotemporal dementia. Ann. Neurol. 2004, 56, 894–897. [Google Scholar] [CrossRef] [PubMed]
- Venneti, S.; Wiley, C.A.; Kofler, J. Imaging microglial activation during neuroinflammation and Alzheimer’s disease. J. Neuroimmune Pharmacol. 2009, 4, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Mapping neuroinflammation in frontotemporal dementia with molecular PET imaging. J. Neuroinflamm. 2015, 12, 108. [Google Scholar] [CrossRef] [PubMed]
- Neary, D.; Snowden, J.S.; Gustafson, L.; Passant, U.; Stuss, D.; Black, S.; Freedman, M.; Kertesz, A.; Robert, P.H.; Albert, M.; et al. Frontotemporal lobar degeneration: A consensus on clinical diagnostic criteria. Neurology 1998, 51, 1546–1554. [Google Scholar] [CrossRef] [PubMed]
- Olney, N.T.; Spina, S.; Miller, B.L. Frontotemporal Dementia. Neurol. Clin. 2017, 35, 339–374. [Google Scholar] [CrossRef] [PubMed]
- Hughes, L.E.; Rittman, T.; Regenthal, R.; Robbins, T.W.; Rowe, J.B. Improving response inhibition systems in frontotemporal dementia with citalopram. Brain J. Neurol. 2015, 138 Pt 7, 1961–1975. [Google Scholar] [CrossRef] [PubMed]
- Knopman, D.S.; Roberts, R.O. Estimating the number of persons with frontotemporal lobar degeneration in the US population. J. Mol. Neurosci. 2011, 45, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Coyle-Gilchrist, I.T.; Dick, K.M.; Patterson, K.; Vazquez Rodriquez, P.; Wehmann, E.; Wilcox, A.; Lansdall, C.J.; Dawson, K.E.; Wiggins, J.; Mead, S.; et al. Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology 2016, 86, 1736–1743. [Google Scholar] [CrossRef] [PubMed]
- Hodges, J.R.; Davies, R.; Xuereb, J.; Kril, J.; Halliday, G. Survival in frontotemporal dementia. Neurology 2003, 61, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Onyike, C.U.; Diehl-Schmid, J. The epidemiology of frontotemporal dementia. Int. Rev. Psychiatry 2013, 25, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Ratnavalli, E.; Brayne, C.; Dawson, K.; Hodges, J.R. The prevalence of frontotemporal dementia. Neurology 2002, 58, 1615–1621. [Google Scholar] [CrossRef] [PubMed]
- Rascovsky, K.; Hodges, J.R.; Knopman, D.; Mendez, M.F.; Kramer, J.H.; Neuhaus, J.; van Swieten, J.C.; Seelaar, H.; Dopper, E.G.; Onyike, C.U.; et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain J. Neurol. 2011, 134 Pt 9, 2456–2477. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2015, 17, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.J.; Cairns, N.J.; Grossman, M.; McMillan, C.T.; Lee, E.B.; Van Deerlin, V.M.; Lee, V.M.; Trojanowski, J.Q. Frontotemporal lobar degeneration: Defining phenotypic diversity through personalized medicine. Acta Neuropathol. 2015, 129, 469–491. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Neumann, M.; Bigio, E.H.; Cairns, N.J.; Alafuzoff, I.; Kril, J.; Kovacs, G.G.; Ghetti, B.; Halliday, G.; Holm, I.E.; et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: An update. Acta Neuropathol. 2010, 119, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sieben, A.; Van Langenhove, T.; Engelborghs, S.; Martin, J.J.; Boon, P.; Cras, P.; De Deyn, P.P.; Santens, P.; Van Broeckhoven, C.; Cruts, M. The genetics and neuropathology of frontotemporal lobar degeneration. Acta Neuropathol. 2012, 124, 353–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghetti, B.; Oblak, A.L.; Boeve, B.F.; Johnson, K.A.; Dickerson, B.C.; Goedert, M. Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: A chameleon for neuropathology and neuroimaging. Neuropathol. Appl. Neurobiol. 2015, 41, 24–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bevan Jones, W.R.; Cope, T.E.; Passamonti, L.; Fryer, T.D.; Hong, Y.T.; Aigbirhio, F.; Kril, J.J.; Forrest, S.L.; Allinson, K.; Coles, J.P.; et al. [18F]AV-1451 PET in behavioral variant frontotemporal dementia due to MAPT mutation. Ann. Clin. Transl. Neurol. 2016, 3, 940–947. [Google Scholar] [CrossRef] [PubMed]
- Ishiki, A.; Harada, R.; Okamura, N.; Tomita, N.; Rowe, C.C.; Villemagne, V.L.; Yanai, K.; Kudo, Y.; Arai, H.; Furumoto, S.; et al. Tau imaging with [18F]THK-5351 in progressive supranuclear palsy. Eur. J. Neurol. 2017, 24, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Ono, M.; Sahara, N.; Kumata, K.; Ji, B.; Ni, R.; Koga, S.; Dickson, D.W.; Trojanowski, J.Q.; Lee, V.M.; Yoshida, M.; et al. Distinct binding of PET ligands PBB3 and AV-1451 to tau fibril strains in neurodegenerative tauopathies. Brain J. Neurol. 2017, 140, 764–780. [Google Scholar] [CrossRef] [PubMed]
- Meeter, L.H.; Kaat, L.D.; Rohrer, J.D.; van Swieten, J.C. Imaging and fluid biomarkers in frontotemporal dementia. Nat. Rev. Neurol. 2017, 13, 406–419. [Google Scholar] [CrossRef] [PubMed]
- Struyfs, H.; Van Broeck, B.; Timmers, M.; Fransen, E.; Sleegers, K.; Van Broeckhoven, C.; De Deyn, P.P.; Streffer, J.R.; Mercken, M.; Engelborghs, S. Diagnostic Accuracy of Cerebrospinal Fluid Amyloid-β Isoforms for Early and Differential Dementia Diagnosis. J. Alzheimer Dis. 2015, 45, 813–822. [Google Scholar] [CrossRef]
- Bastin, C.; Feyers, D.; Souchay, C.; Guillaume, B.; Pepin, J.L.; Lemaire, C.; Degueldre, C.; Collette, F.; Salmon, E. Frontal and posterior cingulate metabolic impairment in the behavioral variant of frontotemporal dementia with impaired autonoetic consciousness. Hum. Brain Mapp. 2012, 33, 1268–1278. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, N.; Black, S.E.; Chow, T.; Cappell, J.; Tang-Wai, D.F.; Lanctot, K.L. Serotonergic function and treatment of behavioral and psychological symptoms of frontotemporal dementia. Am. J. Geriatr. Psychiatry 2012, 20, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Lebert, F.; Stekke, W.; Hasenbroekx, C.; Pasquier, F. Frontotemporal dementia: A randomised, controlled trial with trazodone. Dement. Geriatr. Cogn. Disord. 2004, 17, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Solfrizzi, V.; Seripa, D.; Imbimbo, B.P.; Lozupone, M.; Santamato, A.; Zecca, C.; Barulli, M.R.; Bellomo, A.; Pilotto, A.; et al. Tau-Centric Targets and Drugs in Clinical Development for the Treatment of Alzheimer’s Disease. BioMed Res. Int. 2016, 2016, 3245935. [Google Scholar] [CrossRef] [PubMed]
- Munoz, Y.; Carrasco, C.M.; Campos, J.D.; Aguirre, P.; Nunez, M.T. Parkinson’s Disease: The Mitochondria-Iron Link. Parkinson’s Dis. 2016, 2016, 7049108. [Google Scholar] [CrossRef]
- Forno, L.S. Neuropathology of Parkinson’s disease. J. Neuropathol. Exp. Neurol. 1996, 55, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Neuropathology, biochemistry, and biophysics of α-synuclein aggregation. J. Neurochem. 2007, 103, 17–37. [Google Scholar] [CrossRef] [PubMed]
- Maarouf, C.L.; Beach, T.G.; Adler, C.H.; Shill, H.A.; Sabbagh, M.N.; Wu, T.; Walker, D.G.; Kokjohn, T.A.; Roher, A.E.; Consortium, A.P.D. Cerebrospinal fluid biomarkers of neuropathologically diagnosed Parkinson’s disease subjects. Neurol. Res. 2012, 34, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.; Beal, M.F. Parkinson’s disease. Hum. Mol. Genet. 2007, R183–R194. [Google Scholar] [CrossRef] [PubMed]
- McKeith, I.G.; Dickson, D.W.; Lowe, J.; Emre, M.; O’Brien, J.T.; Feldman, H.; Cummings, J.; Duda, J.E.; Lippa, C.; Perry, E.K.; et al. Diagnosis and management of dementia with Lewy bodies: Third report of the DLB Consortium. Neurology 2005, 65, 1863–1872. [Google Scholar] [CrossRef] [PubMed]
- Golde, T.E.; Miller, V.M. Proteinopathy-induced neuronal senescence: A hypothesis for brain failure in Alzheimer’s and other neurodegenerative diseases. Alzheimer’s Res. Ther. 2009, 1, 5. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically disordered proteins and their environment: Effects of strong denaturants, temperature, pH, counter ions, membranes, binding partners, osmolytes, and macromolecular crowding. Protein J. 2009, 28, 305–325. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Basic mechanisms of neurodegeneration: A critical update. J. Cell. Mol. Med. 2010, 14, 457–487. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. The neuropathologic substrate of Parkinson disease dementia. Acta Neuropathol. 2010, 119, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Botond, G.; Budka, H. Protein coding of neurodegenerative dementias: The neuropathological basis of biomarker diagnostics. Acta Neuropathol. 2010, 119, 389–408. [Google Scholar] [CrossRef] [PubMed]
- Arai, Y.; Yamazaki, M.; Mori, O.; Muramatsu, H.; Asano, G.; Katayama, Y. Α-synuclein-positive structures in cases with sporadic Alzheimer’s disease: Morphology and its relationship to tau aggregation. Brain Res. 2001, 888, 287–296. [Google Scholar] [CrossRef]
- Iseki, E.; Togo, T.; Suzuki, K.; Katsuse, O.; Marui, W.; de Silva, R.; Lees, A.; Yamamoto, T.; Kosaka, K. Dementia with Lewy bodies from the perspective of tauopathy. Acta Neuropathol. 2003, 105, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.A.; Li, J.L.; Li, Y.; Wilson, R.S.; Kordower, J.H.; Bennett, D.A. Substantia nigra tangles are related to gait impairment in older persons. Ann. Neurol. 2006, 59, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Joachim, C.L.; Morris, J.H.; Kosik, K.S.; Selkoe, D.J. Tau antisera recognize neurofibrillary tangles in a range of neurodegenerative disorders. Ann. Neurol. 1987, 22, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Wills, J.; Jones, J.; Haggerty, T.; Duka, V.; Joyce, J.N.; Sidhu, A. Elevated tauopathy and α-synuclein pathology in postmortem Parkinson’s disease brains with and without dementia. Exp. Neurol. 2010, 225, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Haggerty, T.; Credle, J.; Rodriguez, O.; Wills, J.; Oaks, A.W.; Masliah, E.; Sidhu, A. Hyperphosphorylated Tau in an α-synuclein-overexpressing transgenic model of Parkinson’s disease. Eur. J. Neurosci. 2011, 33, 1598–1610. [Google Scholar] [CrossRef] [PubMed]
- Duka, T.; Sidhu, A. The neurotoxin, MPP+, induces hyperphosphorylation of Tau, in the presence of α-Synuclein, in SH-SY5Y neuroblastoma cells. Neurotox. Res. 2006, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Duka, T.; Rusnak, M.; Drolet, R.E.; Duka, V.; Wersinger, C.; Goudreau, J.L.; Sidhu, A. Α-synuclein induces hyperphosphorylation of Tau in the MPTP model of parkinsonism. FASEB J. 2006, 20, 2302–2312. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Interaction between α-synuclein and tau in Parkinson’s disease comment on Wills et al.: Elevated tauopathy and α-synuclein pathology in postmortem Parkinson’s disease brains with and without dementia. Exp. Neurol. 2010; 225: 210–218. Exp. Neurol. 2011, 227, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Kraybill, M.L.; Larson, E.B.; Tsuang, D.W.; Teri, L.; McCormick, W.C.; Bowen, J.D.; Kukull, W.A.; Leverenz, J.B.; Cherrier, M.M. Cognitive differences in dementia patients with autopsy-verified, A.D.; Lewy body pathology, or both. Neurology 2005, 64, 2069–2073. [Google Scholar] [CrossRef] [PubMed]
- Langlais, P.J.; Thal, L.; Hansen, L.; Galasko, D.; Alford, M.; Masliah, E. Neurotransmitters in basal ganglia and cortex of Alzheimer’s disease with and without Lewy bodies. Neurology 1993, 43, 1927–1934. [Google Scholar] [CrossRef] [PubMed]
- Olichney, J.M.; Galasko, D.; Salmon, D.P.; Hofstetter, C.R.; Hansen, L.A.; Katzman, R.; Thal, L.J. Cognitive decline is faster in Lewy body variant than in Alzheimer’s disease. Neurology 1998, 51, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Forman, M.S.; Higuchi, M.; Golbe, L.I.; Graves, C.L.; Kotzbauer, P.T.; Trojanowski, J.Q.; Lee, V.M. Initiation and synergistic fibrillization of tau and α-synuclein. Science 2003, 300, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.M.; Giasson, B.I.; Trojanowski, J.Q. More than just two peas in a pod: Common amyloidogenic properties of tau and α-synuclein in neurodegenerative diseases. Trends Neurosci. 2004, 27, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.H.; Hager, H.; Nielsen, M.S.; Hojrup, P.; Gliemann, J.; Jakes, R. α-synuclein binds to Tau and stimulates the protein kinase A-catalyzed tau phosphorylation of serine residues 262 and 356. J. Biol. Chem. 1999, 274, 25481–25489. [Google Scholar] [CrossRef] [PubMed]
- Muntane, G.; Dalfo, E.; Martinez, A.; Ferrer, I. Phosphorylation of tau and α-synuclein in synaptic-enriched fractions of the frontal cortex in Alzheimer’s disease, and in Parkinson’s disease and related α-synucleinopathies. Neuroscience 2008, 152, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Frasier, M.; Walzer, M.; McCarthy, L.; Magnuson, D.; Lee, J.M.; Haas, C.; Kahle, P.; Wolozin, B. Tau phosphorylation increases in symptomatic mice overexpressing A30P α-synuclein. Exp. Neurol. 2005, 192, 274–287. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.Y.; Crary, J.F.; Rao, C.; Sacktor, T.C.; Mirra, S.S. Atypical protein kinase C in neurodegenerative disease II: PKCiota/lambda in tauopathies and α-synucleinopathies. J. Neuropathol. Exp. Neurol. 2006, 65, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Riedel, M.; Goldbaum, O.; Richter-Landsberg, C. α-Synuclein promotes the recruitment of tau to protein inclusions in oligodendroglial cells: Effects of oxidative and proteolytic stress. J. Mol. Neurosci. 2009, 39, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Duka, T.; Duka, V.; Joyce, J.N.; Sidhu, A. Α-Synuclein contributes to GSK-3β-catalyzed Tau phosphorylation in Parkinson’s disease models. FASEB J. 2009, 23, 2820–2830. [Google Scholar] [CrossRef] [PubMed]
- Ciaccioli, G.; Martins, A.; Rodrigues, C.; Vieira, H.; Calado, P. A powerful yeast model to investigate the synergistic interaction of α-synuclein and tau in neurodegeneration. PLoS ONE 2013, 8, e55848. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, F.; Suzuki, M.; Shimada, N.; Kagiya, G.; Ohta, E.; Tamura, K.; Maruyama, H.; Ichikawa, T. Stimulatory effect of α-synuclein on the tau-phosphorylation by GSK-3β. FEBS J. 2011, 278, 4895–4904. [Google Scholar] [CrossRef] [PubMed]
- Wills, J.; Credle, J.; Haggerty, T.; Lee, J.H.; Oaks, A.W.; Sidhu, A. Tauopathic changes in the striatum of A53T α-synuclein mutant mouse model of Parkinson’s disease. PLoS ONE 2011, 6, e17953. [Google Scholar] [CrossRef] [PubMed]
- Kaul, T.; Credle, J.; Haggerty, T.; Oaks, A.W.; Masliah, E.; Sidhu, A. Region-specific tauopathy and synucleinopathy in brain of the α-synuclein overexpressing mouse model of Parkinson’s disease. BMC Neurosci. 2011, 12, 79. [Google Scholar] [CrossRef] [PubMed]
- Oaks, A.W.; Frankfurt, M.; Finkelstein, D.I.; Sidhu, A. Age-dependent effects of A53T α-synuclein on behavior and dopaminergic function. PLoS ONE 2013, 8, e60378. [Google Scholar] [CrossRef] [PubMed]
- Nubling, G.; Bader, B.; Levin, J.; Hildebrandt, J.; Kretzschmar, H.; Giese, A. Synergistic influence of phosphorylation and metal ions on tau oligomer formation and coaggregation with α-synuclein at the single molecule level. Mol. Neurodegener. 2012, 7, 35. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R.; Spillantini, M.G.; Hasegawa, M.; Smith, M.J.; Crowther, R.A. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 1996, 383, 550–553. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cortés, N.; Andrade, V.; Guzmán-Martínez, L.; Estrella, M.; Maccioni, R.B. Neuroimmune Tau Mechanisms: Their Role in the Progression of Neuronal Degeneration. Int. J. Mol. Sci. 2018, 19, 956. https://doi.org/10.3390/ijms19040956
Cortés N, Andrade V, Guzmán-Martínez L, Estrella M, Maccioni RB. Neuroimmune Tau Mechanisms: Their Role in the Progression of Neuronal Degeneration. International Journal of Molecular Sciences. 2018; 19(4):956. https://doi.org/10.3390/ijms19040956
Chicago/Turabian StyleCortés, Nicole, Víctor Andrade, Leonardo Guzmán-Martínez, Matías Estrella, and Ricardo B. Maccioni. 2018. "Neuroimmune Tau Mechanisms: Their Role in the Progression of Neuronal Degeneration" International Journal of Molecular Sciences 19, no. 4: 956. https://doi.org/10.3390/ijms19040956