Conformational Dynamics and Stability of U-Shaped and S-Shaped Amyloid β Assemblies

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
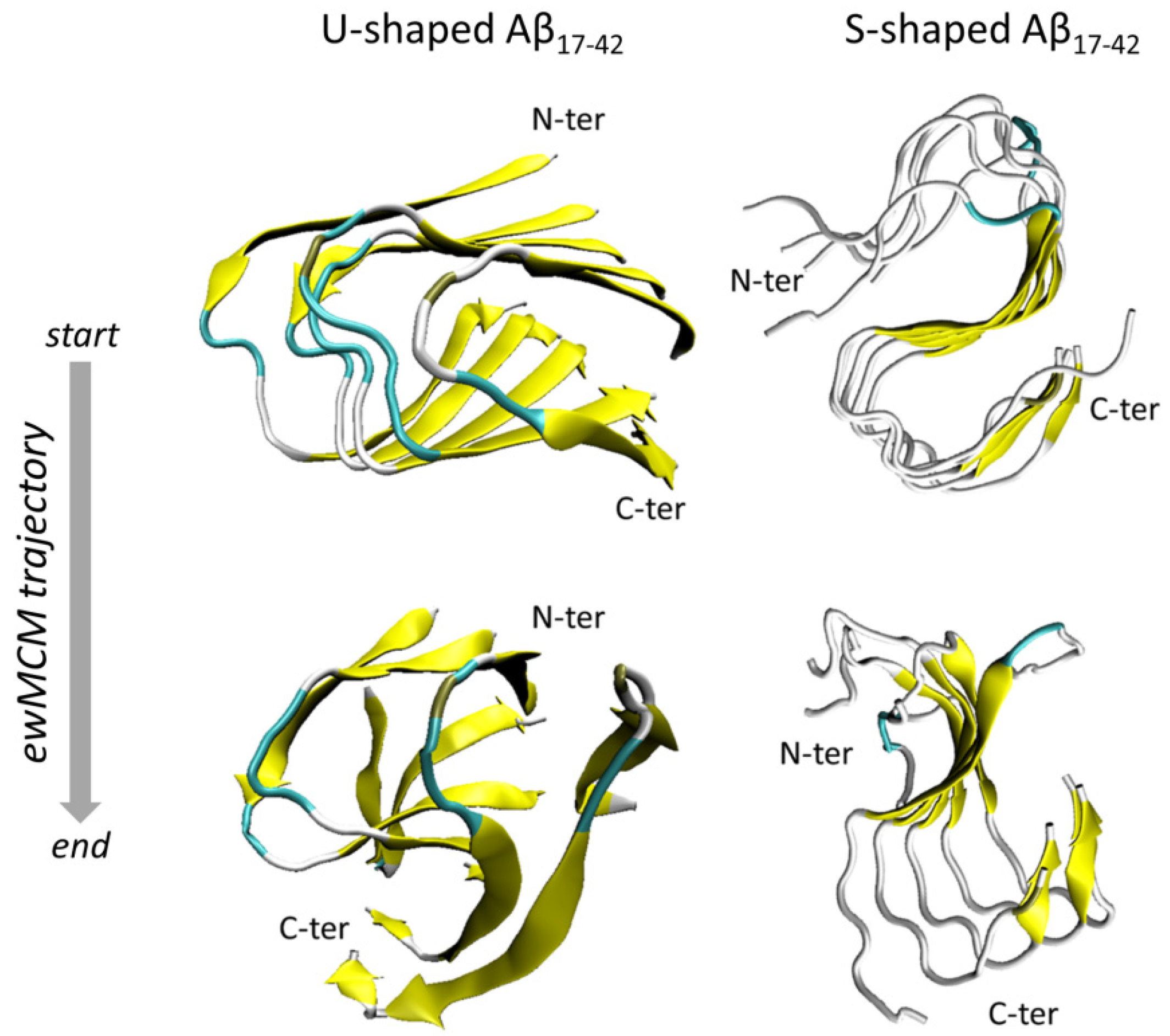{kind=link}
Abstract
:1. Introduction
2. Results
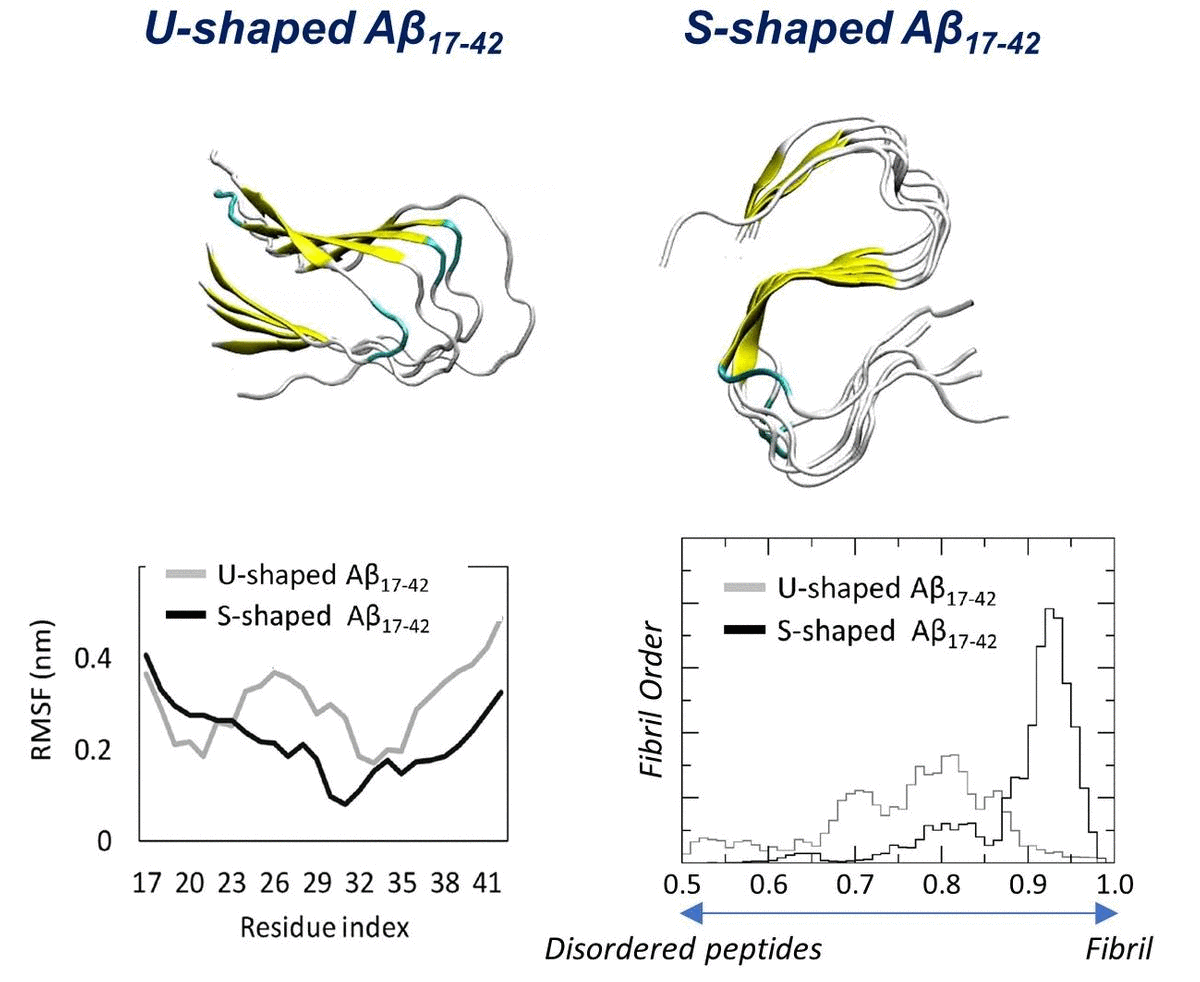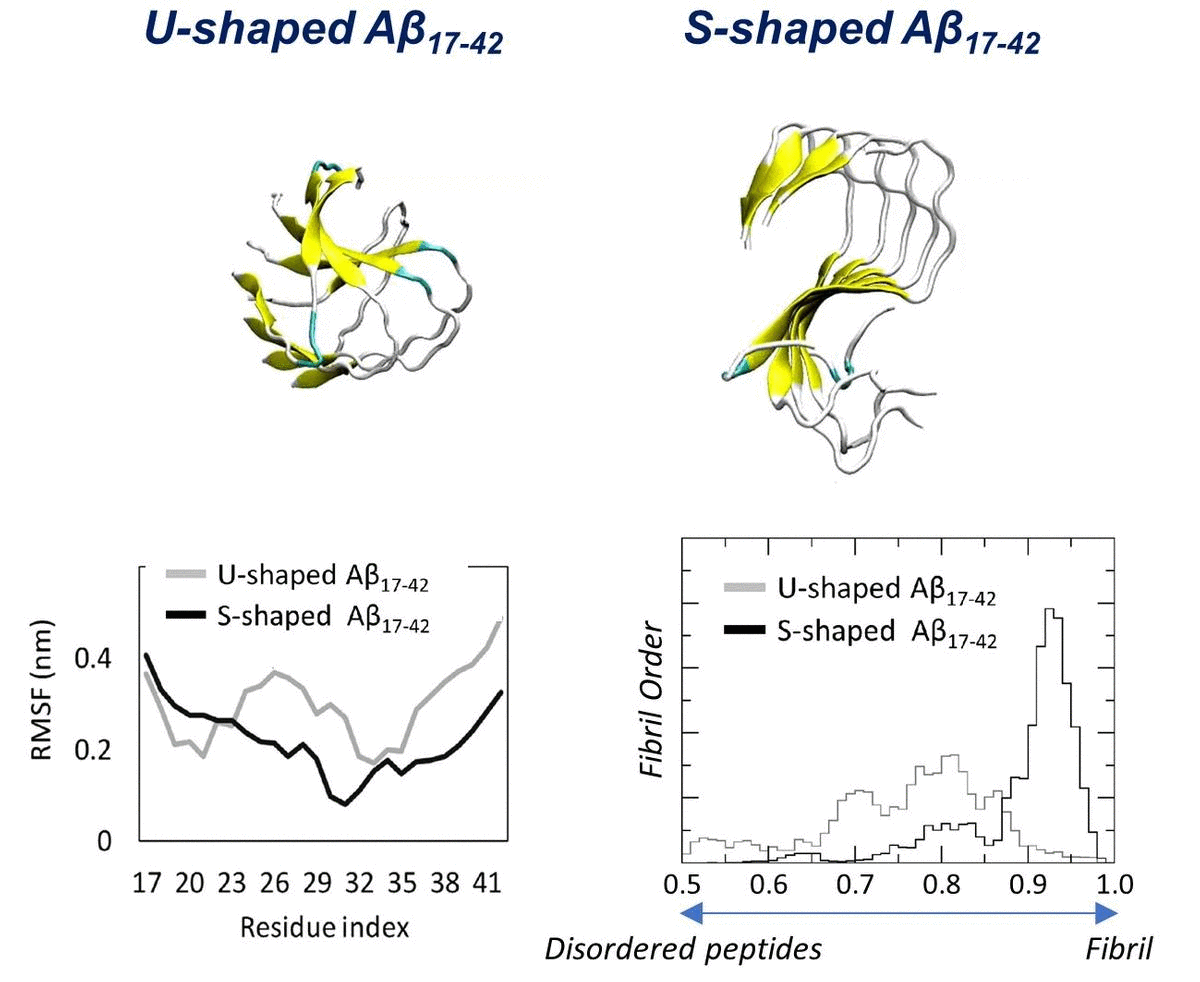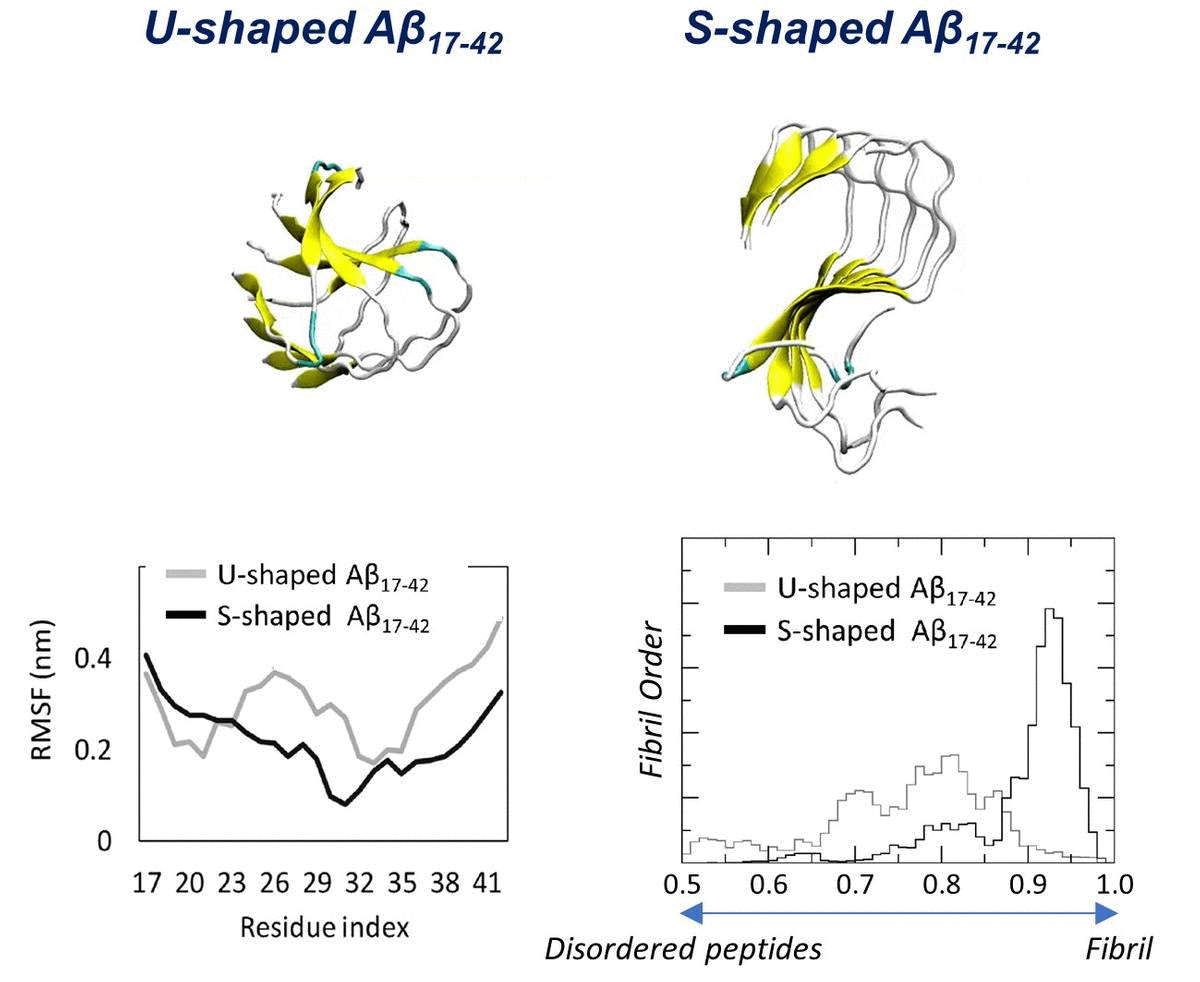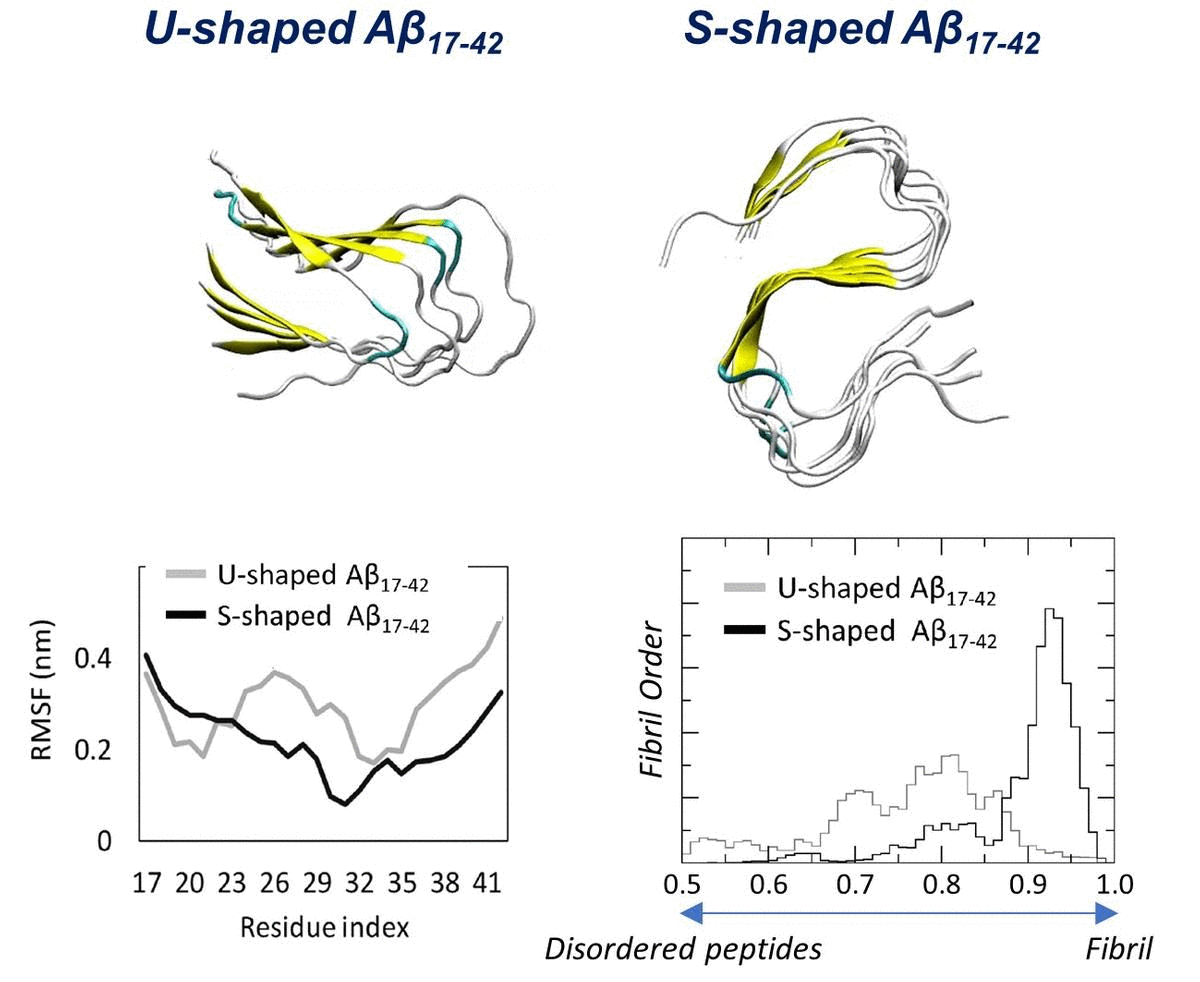
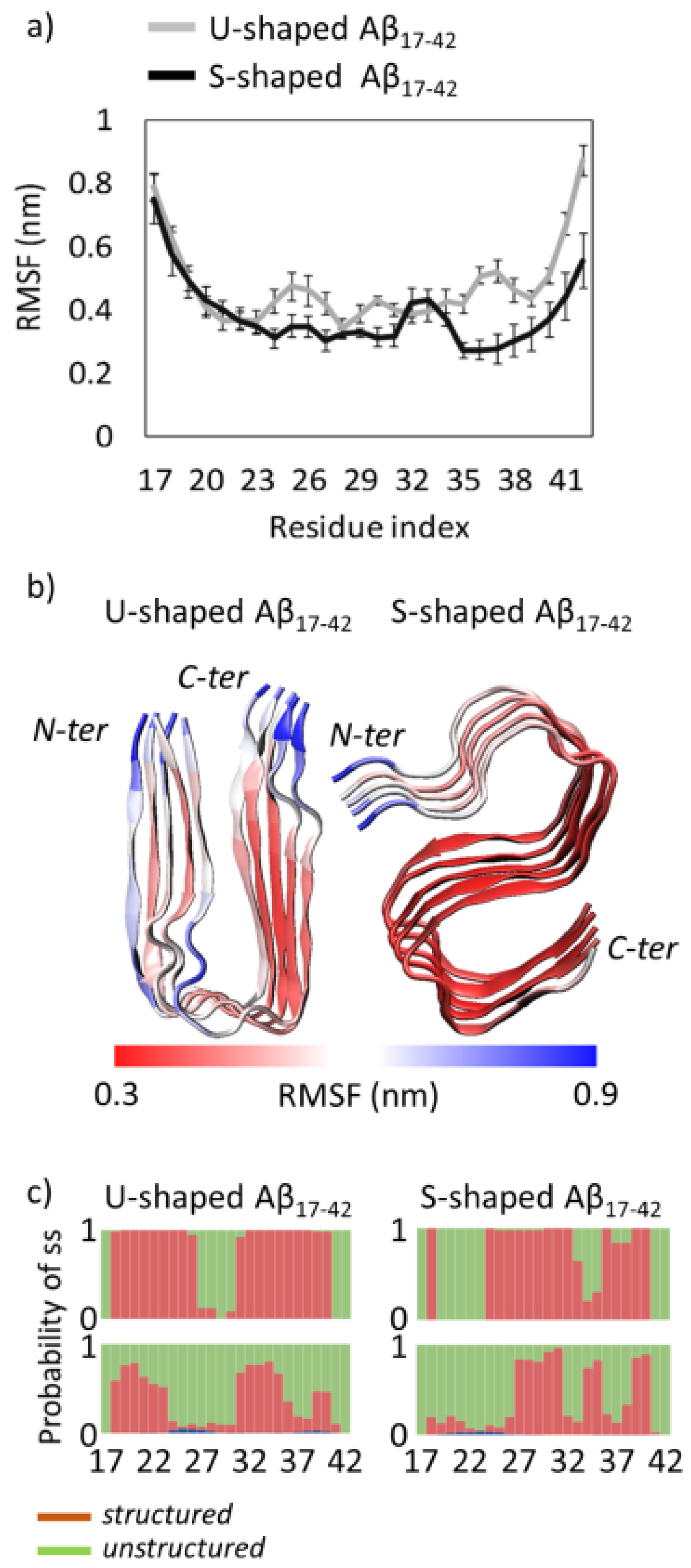
2.1. Characterization of the Aβ Conformational Arrangements
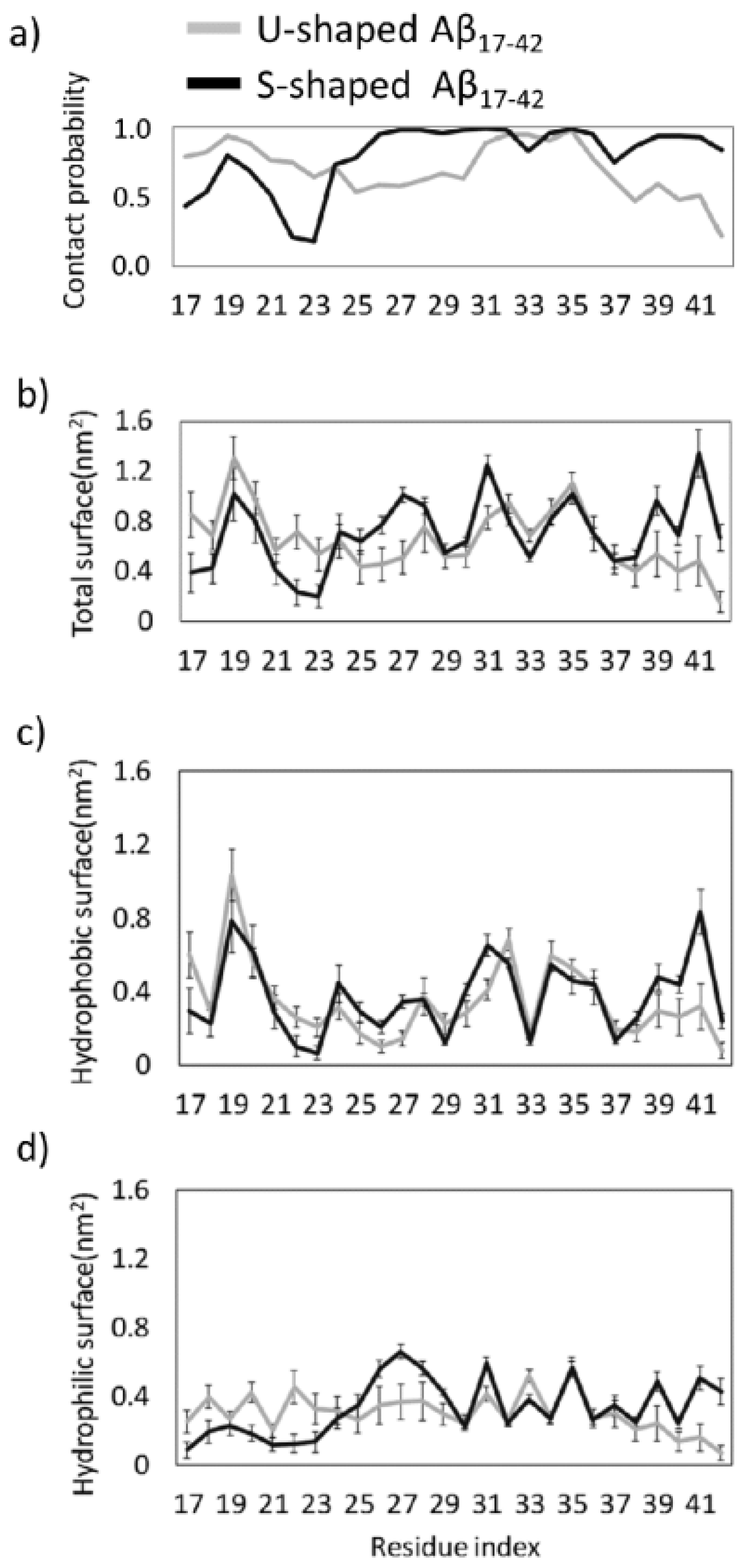
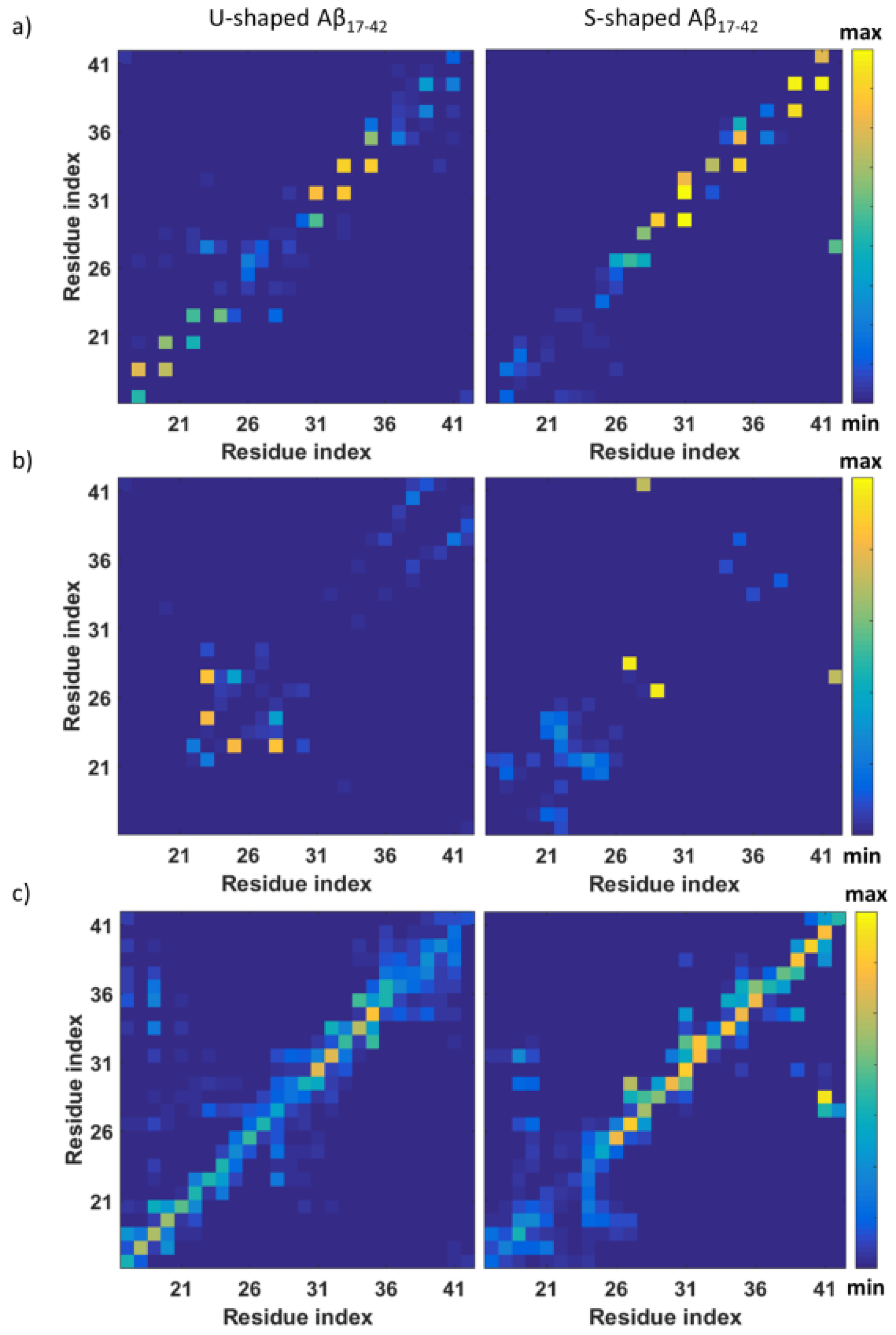
2.2. Characterization of the Aβ Interatomic Interactions
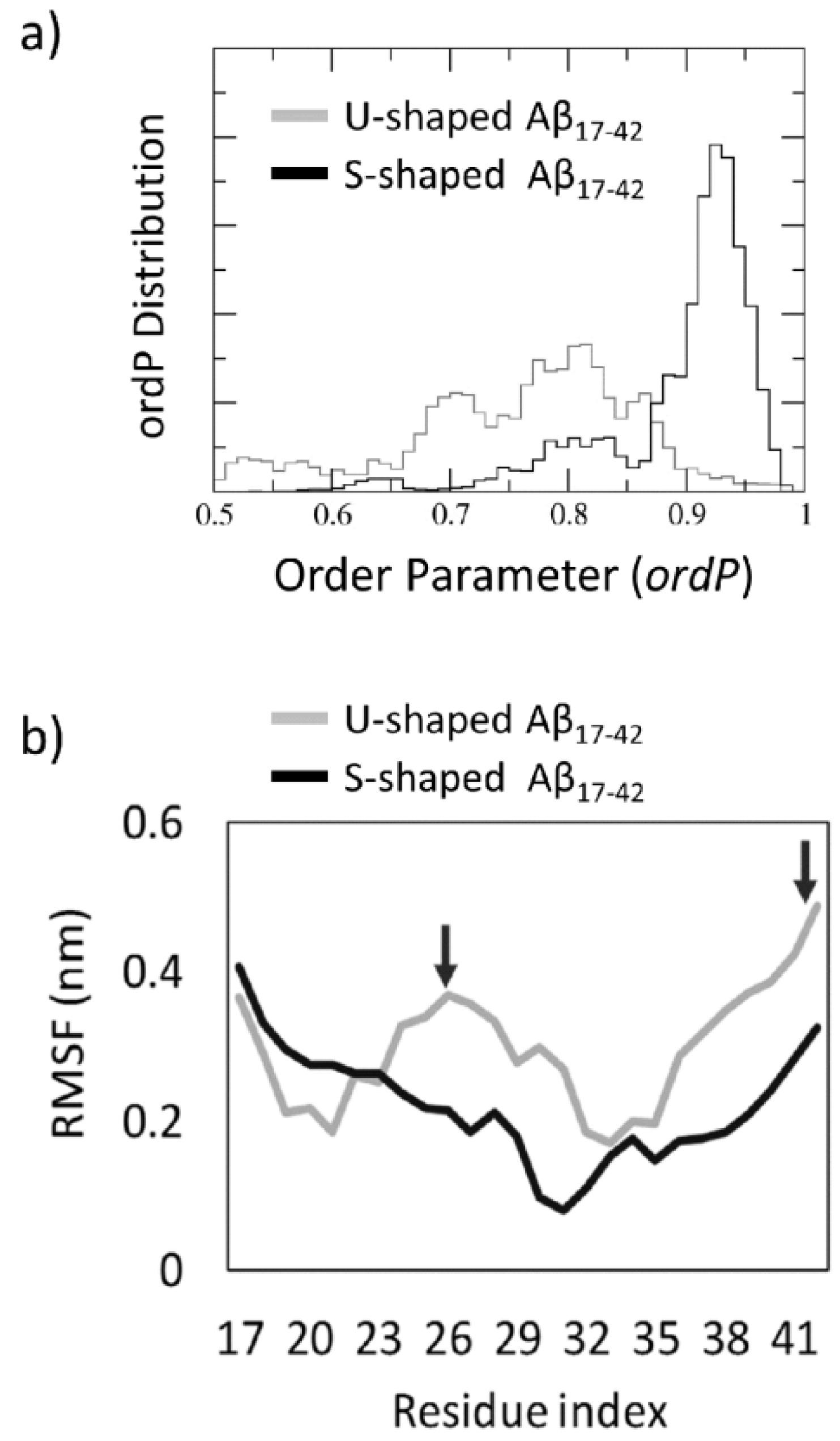
2.3. Order Parameter and Functional Mode Analysis
3. Discussion
4. Material and Methods
4.1. Replica Exchange Molecular Dynamics (REMD)
4.2. Order Parameter and Functional Mode Analysis (FMA)
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dobson, C.M. The structural basis of protein folding and its links with human disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2001, 356, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Horwich, A. Protein aggregation in disease: A role for folding intermediates forming specific multimeric interactions. J. Clin. Investig. 2002, 110, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.Y.; Pepys, M.B. Amyloidosis. Histopathology 1994, 25, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.W. The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr. Opin. Struct. Biol. 1998, 8, 101–106. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M.C. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed]
- Monsonego, A.; Monsonego, A.; Zota, V.; Zota, V.; Karni, A.; Karni, A.; Krieger, J.I.; Krieger, J.I.; Bar-or, A.; Bar-or, A.; et al. Increased T cell reactivity to amyloid. J. Clin. Investig. 2003, 112, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s Disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, J.T.; Lansbury, P.T. Seeding “one-dimensional crystallization” of amyloid: A pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 1993, 73, 1055–1058. [Google Scholar] [CrossRef]
- Cummings, J.L. Alzheimer’s Disease. N. Engl. J. Med. 2004, 351, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Schütz, A.K.; Vagt, T.; Huber, M.; Ovchinnikova, O.Y.; Cadalbert, R.; Wall, J.; Güntert, P.; Böckmann, A.; Glockshuber, R.; Meier, B.H. Atomic-Resolution Three-Dimensional Structure of Amyloid β Fibrils Bearing the Osaka Mutation. Angew. Chem. Int. Ed. 2015, 54, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Petkova, A.T.; Ishii, Y.; Balbach, J.J.; Antzutkin, O.N.; Leapman, R.D.; Delaglio, F.; Tycko, R. A structural model for Alzheimer’s-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. USA 2002, 99, 16742–16747. [Google Scholar] [CrossRef] [PubMed]
- Luhrs, T.; Ritter, C.; Adrian, M.; Riek-Loher, D.; Bohrmann, B.; Dobeli, H.; Schubert, D.; Riek, R. 3D structure of Alzheimer’s amyloid-(1-42) fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 17342–17347. [Google Scholar] [CrossRef] [PubMed]
- Petkova, A.T.; Yau, W.-M.; Tycko, R. Experimental Constraints on Quaternary Structure in Alzheimer’s β-Amyloid Fibrils. Biochemistry 2006, 45, 498–512. [Google Scholar] [CrossRef] [PubMed]
- Paravastu, A.K.; Leapman, R.D.; Yau, W.-M.; Tycko, R. Molecular structural basis for polymorphism in Alzheimer’s β-amyloid fibrils. Proc. Natl. Acad. Sci. USA 2008, 105, 18349–18354. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.-X.; Qiang, W.; Yau, W.-M.; Schwieters, C.D.; Meredith, S.C.; Tycko, R. Molecular Structure of β-Amyloid Fibrils in Alzheimer’s Disease Brain Tissue. Cell 2013, 154, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Gremer, L.; Schölzel, D.; Schenk, C.; Reinartz, E.; Labahn, J.; Ravelli, R.B.G.; Tusche, M.; Lopez-Iglesias, C.; Hoyer, W.; Heise, H.; et al. Fibril structure of amyloid-β(1–42) by cryo–electron microscopy. Science 2017, 358, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Ma, B.; McElheny, D.; Parthasarathy, S.; Long, F.; Hoshi, M.; Nussinov, R.; Ishii, Y. Aβ(1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat. Struct. Mol. Biol. 2015, 22, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Tran, J.; Jiang, L.; Guo, Z. A new structural model of Alzheimer’s Aβ42 fibrils based on electron paramagnetic resonance data and Rosetta modeling. J. Struct. Biol. 2016, 194, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Colvin, M.T.; Silvers, R.; Ni, Q.Z.; Can, T.V.; Sergeyev, I.; Rosay, M.; Donovan, K.J.; Michael, B.; Wall, J.; Linse, S.; et al. Atomic Resolution Structure of Monomorphic Aβ42 Amyloid Fibrils. J. Am. Chem. Soc. 2016, 138, 9663–9674. [Google Scholar] [CrossRef] [PubMed]
- Wälti, M.A.; Ravotti, F.; Arai, H.; Glabe, C.G.; Wall, J.S.; Böckmann, A.; Güntert, P.; Meier, B.H.; Riek, R. Atomic-resolution structure of a disease-relevant Aβ(1–42) amyloid fibril. Proc. Natl. Acad. Sci. USA 2016, 113, E4976–E4984. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Rohou, A.; Lasker, K.; Yadav, J.K.; Schiene-Fischer, C.; Fändrich, M.; Grigorieff, N. Peptide dimer structure in an Aβ(1–42) fibril visualized with cryo-EM. Proc. Natl. Acad. Sci. USA 2015, 112, 11858–11863. [Google Scholar] [CrossRef] [PubMed]
- Xi, W.; Hansmann, U.H.E. Ring-like N-fold Models of Aβ42 fibrils. Sci. Rep. 2017, 7, 6588. [Google Scholar] [CrossRef] [PubMed]
- Xi, W.; Wang, W.; Abbott, G.; Hansmann, U.H.E. Stability of a Recently Found Triple-β-Stranded Aβ1-42 Fibril Motif. J. Phys. Chem. B 2016, 120, 4548–4557. [Google Scholar] [CrossRef] [PubMed]
- Grasso, G.; Tuszynski, J.A.; Morbiducci, U.; Licandro, G.; Danani, A.; Deriu, M.A. Thermodynamic and kinetic stability of the Josephin Domain closed arrangement: Evidences from replica exchange molecular dynamics. Biol. Direct 2017, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Grasso, G.; Deriu, M.A.; Tuszynski, J.A.; Gallo, D.; Morbiducci, U.; Danani, A. Conformational fluctuations of the AXH monomer of Ataxin-1. Proteins Struct. Funct. Bioinform. 2016, 84, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Deriu, M.A.M.A.; Grasso, G.; Tuszynski, J.A.J.A.; Gallo, D.; Morbiducci, U.; Danani, A. Josephin Domain Structural Conformations Explored by Metadynamics in Essential Coordinates. PLoS Comput. Biol. 2016, 12, e1004699. [Google Scholar] [CrossRef] [PubMed]
- Janaszewska, A.; Klajnert-Maculewicz, B.; Marcinkowska, M.; Duchnowicz, P.; Appelhans, D.; Grasso, G.; Deriu, M.A.; Danani, A.; Cangiotti, M.; Ottaviani, M.F. Multivalent interacting glycodendrimer to prevent amyloid-peptide fibril formation induced by Cu(II): A multidisciplinary approach. Nano Res. 2018, 11, 1204–1226. [Google Scholar] [CrossRef]
- Grasso, G.; Morbiducci, U.; Massai, D.; Tuszynki, J.; Danani, A.; Deriu, M. Destabilizing the AXH Tetramer by Mutations: Mechanisms and Potential Antiaggregation Strategies. Biophys. J. 2018, 114, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Berhanu, W.M.; Hansmann, U.H.E. Structure and Dynamics of Amyloid-β Segmental Polymorphisms. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Alred, E.J.; Scheele, E.G.; Berhanu, W.M.; Hansmann, U.H.E. Stability of Iowa mutant and wild type Aβ-peptide aggregates. J. Chem. Phys. 2014, 175101, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cheon, M.; Hall, C.K.; Chang, I. Structural Conversion of Aβ17–42 Peptides from Disordered Oligomers to U-Shape Protofilaments via Multiple Kinetic Pathways. PLoS Comput. Biol. 2015, 11, e1004258. [Google Scholar] [CrossRef] [PubMed]
- Sunde, M.; Serpell, L.C.; Bartlam, M.; Fraser, P.E.; Pepys, M.B.; Blake, C.C. Common core structure of amyloid fibrils by synchrotron X-ray diffraction 1 1Edited by F. E. Cohen. J. Mol. Biol. 1997, 273, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Inouye, H.; Fraser, P.E.; Kirschner, D.A. Structure of beta-crystallite assemblies formed by Alzheimer beta-amyloid protein analogues: Analysis by X-ray diffraction. Biophys. J. 1993, 64, 502–519. [Google Scholar] [CrossRef]
- Ngo, S.T.; Hung, H.M.; Truong, D.T.; Nguyen, M.T. Replica exchange molecular dynamics study of the truncated amyloid beta (11–40) trimer in solution. Phys. Chem. Chem. Phys. 2017, 19, 1909–1919. [Google Scholar] [CrossRef] [PubMed]
- Tarus, B.; Straub, J.E.; Thirumalai, D. Dynamics of Asp23−Lys28 Salt-Bridge Formation in Aβ 10-35 Monomers. J. Am. Chem. Soc. 2006, 128, 16159–16168. [Google Scholar] [CrossRef] [PubMed]
- Masman, M.F.; Eisel, U.L.M.; Csizmadia, I.G.; Penke, B.; Enriz, R.D.; Marrink, S.J.; Luiten, P.G.M. In Silico Study of Full-Length Amyloid β1−42 Tri- and Penta-Oligomers in Solution. J. Phys. Chem. B 2009, 113, 11710–11719. [Google Scholar] [CrossRef] [PubMed]
- Khatua, P.; Sinha, S.K.; Bandyopadhyay, S. Size-Dependent Conformational Features of Aβ17–42 Protofilaments from Molecular Simulation Studies. J. Chem. Inf. Model. 2017, 57, 2378–2392. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Jang, H.; Ma, B.; Tsai, C.-J.; Nussinov, R. Modeling the Alzheimer Aβ17-42 Fibril Architecture: Tight Intermolecular Sheet-Sheet Association and Intramolecular Hydrated Cavities. Biophys. J. 2007, 93, 3046–3057. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.-M.; Gu, R.-X.; Wang, Y.-J.; Pi, Y.-L.; Zhang, Y.-H.; Xu, Q.; Wei, D.-Q. Destabilization of Alzheimer’s Aβ42 Protofibrils with a Novel Drug Candidate wgx-50 by Molecular Dynamics Simulations. J. Phys. Chem. B 2015, 119, 11196–11202. [Google Scholar] [CrossRef] [PubMed]
- Lemkul, J.A.; Bevan, D.R. Destabilizing Alzheimer’s Aβ42 Protofibrils with Morin: Mechanistic Insights from Molecular Dynamics Simulations. Biochemistry 2010, 49, 3935–3946. [Google Scholar] [CrossRef] [PubMed]
- Shuaib, S.; Saini, R.K.; Goyal, D.; Goyal, B. Insights into the Inhibitory Mechanism of Dicyanovinyl-Substituted J147 Derivative against Aβ42 Aggregation and Protofibril Destabilization: A Molecular Dynamics Simulation Study. ChemistrySelect 2017, 2, 1645–1657. [Google Scholar] [CrossRef]
- Kumar, A.; Srivastava, S.; Tripathi, S.; Kumar Singh, S.; Srikrishna, S.; Sharma, A. Molecular insight into amyloid oligomer destabilizing mechanism of flavonoid derivative 2-(4′ benzyloxyphenyl)-3-hydroxy-chromen-4-one through docking and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2015, 1102, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 14101. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Denschlag, R.; Lingenheil, M.; Tavan, P. Optimal temperature ladders in replica exchange simulations. Chem. Phys. Lett. 2009, 473, 193–195. [Google Scholar] [CrossRef]
- Lingenheil, M.; Denschlag, R.; Mathias, G.; Tavan, P. Efficiency of exchange schemes in replica exchange. Chem. Phys. Lett. 2009, 478, 80–84. [Google Scholar] [CrossRef]
- Grasso, G.; Deriu, M.A.; Patrulea, V.; Borchard, G.; Möller, M.; Danani, A. Free energy landscape of siRNA-polycation complexation: Elucidating the effect of molecular geometry, polymer flexibility, and charge neutralization. PLoS ONE 2017, 12, e0186816. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Hub, J.S.; de Groot, B.L. Detection of Functional Modes in Protein Dynamics. PLoS Comput. Biol. 2009, 5, e1000480. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grasso, G.; Rebella, M.; Muscat, S.; Morbiducci, U.; Tuszynski, J.; Danani, A.; Deriu, M.A. Conformational Dynamics and Stability of U-Shaped and S-Shaped Amyloid β Assemblies. Int. J. Mol. Sci. 2018, 19, 571. https://doi.org/10.3390/ijms19020571
Grasso G, Rebella M, Muscat S, Morbiducci U, Tuszynski J, Danani A, Deriu MA. Conformational Dynamics and Stability of U-Shaped and S-Shaped Amyloid β Assemblies. International Journal of Molecular Sciences. 2018; 19(2):571. https://doi.org/10.3390/ijms19020571
Chicago/Turabian StyleGrasso, Gianvito, Martina Rebella, Stefano Muscat, Umberto Morbiducci, Jack Tuszynski, Andrea Danani, and Marco A. Deriu. 2018. "Conformational Dynamics and Stability of U-Shaped and S-Shaped Amyloid β Assemblies" International Journal of Molecular Sciences 19, no. 2: 571. https://doi.org/10.3390/ijms19020571