Mast Cells: Key Contributors to Cardiac Fibrosis


Kolling Institute for Medical Research, University of Sydney, St Leonards 2065, Australia
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(1), 231; https://doi.org/10.3390/ijms19010231
Submission received: 27 November 2017
/
Revised: 21 December 2017
/
Accepted: 22 December 2017
/
Published: 12 January 2018
(This article belongs to the Special Issue Extracellular Matrix in Development and Disease)
Abstract
:Historically, increased numbers of mast cells have been associated with fibrosis in numerous cardiac pathologies, implicating mast cells in the development of cardiac fibrosis. Subsequently, several approaches have been utilised to demonstrate a causal role for mast cells in animal models of cardiac fibrosis including mast cell stabilising compounds, rodents deficient in mast cells, and inhibition of the actions of mast cell-specific proteases such as chymase and tryptase. Whilst most evidence supports a pro-fibrotic role for mast cells, there is evidence that in some settings these cells can oppose fibrosis. A major gap in our current understanding of cardiac mast cell function is identification of the stimuli that activate these cells causing them to promote a pro-fibrotic environment. This review will present the evidence linking mast cells to cardiac fibrosis, as well as discuss the major questions that remain in understanding how mast cells contribute to cardiac fibrosis.
1. Introduction
Mast cells (MCs) are non-circulating immune cells that develop only once bone marrow-derived precursors have reached their target tissue. These tissue MCs then go through several stages of maturation driven primarily by the c-kit ligand, stem cell factor, with the final MC phenotype being highly dependent on the microenvironment in which they reside. There is now a substantial amount of evidence supporting a role for MCs in cardiac remodeling and heart failure [1]. In fact, MC numbers increase in the heart both in humans and in experimental animals in a wide variety of cardiac pathologies including myocardial infarction (MI) [2,3,4], hypertension [5,6,7], transplantation [8], myocarditis [9], volume overload [10,11], and in the failing heart [12]. Many of the effects of cardiac MCs involve regulation of the extracellular matrix (ECM), whether it be inducing ECM degradation or promoting increased ECM synthesis. The latter characterises cardiac fibrosis, which is a characteristic of almost all cardiac pathologies and is the focus of this review article. Cardiac MCs can influence fibrosis by direct effects on fibroblasts, as well as indirect effects, both brought about by the many proteases, cytokines, growth factors, and other products manufactured by these multi-faceted cells. This review article will discuss the evidence supporting a role for MCs in cardiac fibrosis by presenting studies that have utilised MC stabilizing compounds, rodents deficient in MCs, and specific inhibitors of MC proteases. This review will also discuss important unanswered questions in the field, including the elusive mediators that activate cardiac MCs causing them to promote fibrosis.
2. Studies Associating Mast Cells with Cardiac Fibrosis
MCs were first linked to cardiac fibrosis more than 50 years ago with the observation that these cells were increased in human hearts with endocardial fibrosis [13]. Since then, there have been numerous other observations that have associated MCs with cardiac fibrosis arising from multiple etiologies. In 1988, increased MCs were found to be associated with areas of fibrosis in biopsies obtained from 92 human diseased hearts by Turlington et al. [14]. In 1989, Olivetti et al. [6] observed an increased number of MCs in the right ventricle (RV) of rats following constriction of the pulmonary artery, a technique that results in RV fibrosis. Subsequently, Li et al. [8] reported that MCs were increased in human hearts following transplantation and that MC number correlated with fibrosis (r = 0.63). Strengthening this relationship was the observation that patients with high numbers of MCs at two weeks post-transplantation showed a 17% increase in fibrosis by week 3, whilst those patients with lesser numbers of MCs had only a 3.5% increase in fibrosis. Perhaps not surprisingly, patients in the high MC group also scored higher on the rejection scale.
Levels of the MC-specific amine, histamine, were reported to be elevated in experimental Chagas’ disease induced by infection of mice with Trypanosoma cruzi virus [15], with MCs in these mice appearing in areas of fibrosis [16]. Further, MC degranulation occurs soon after infection of mice with experimental myocarditis induced by coxsackievirus [17]. MC density also increases [18] in myocarditis and very strongly correlates with collagen volume fraction (r = 0.946) [19].
MCs were also linked to fibrosis in the hypertensive left ventricle (LV) when Panizo et al. [5] observed an increase in MC density in the LV of spontaneously hypertensive rats (SHR) that strongly correlated with collagen volume fraction (r = 0.87). Shiota et al. [7] also reported increased MC densities across the lifespan of the SHR. Even in stenotic aortic valves, MCs contained increased cathepsin G, which correlated with expression levels of collagen I and III [20]. More recently, Luitel et al. [21] confirmed in mice the earlier findings of Olivetti [6] in rats that MC density and degranulation increase in the RV following constriction of the pulmonary artery. Whilst these studies clearly show a strong association between MCs and fibrosis in the heart from varying etiologies, these associations do not establish causality. These studies are summarized in Table 1.
3. Evidence for the Causal Involvement of Mast Cells in Cardiac Fibrosis
3.1. Studies with Mast Cell Stabilizers
MC stabilizers prevent the release of MC mediators (e.g., histamine). This stabilization may ultimately involve blocking calcium channels, without which MC granules cannot fuse to the cell membrane and be exuded. Several studies have used this approach to examine the role of MCs in cardiac fibrosis. Palaniyandi et al. [19] demonstrated that the MC stabilizer disodium cromoglycate (also known as cromolyn) could dramatically reduce fibrosis in a model of myocarditis, whereby rats were injected with porcine cardiac myosin emulsified with complete freund’s adjuvant with Mycobacterium tuberculosis H37RA. A subsequent study confirmed the anti-fibrotic effect of cromolyn in myocarditis in rats [22]. We provided the first causal evidence that MCs play a role in cardiac fibrosis in the hypertensive heart [23]. SHR were treated with the MC stabilizing compound nedocromil (30 mg/kg/day) from 8 weeks of age (prior to the development of fibrosis) through to 24 weeks of age. This resulted in complete prevention of fibrosis in the LV, as determined by collagen volume fraction (Figure 1A). This included the observation that MC stabilization prevented macrophage recruitment and normalized cytokine profiles (IFN-γ, IL-4, IL-6 and IL-10). Interestingly, we found that IL-10 was dramatically decreased in untreated SHR, and was returned to normal after MC inhibition. In a previous study, Palaniyandi et al. [24] had demonstrated that IL-10 inhibited acute myocarditis-induced pathological changes in the heart, and that this likely involved the inhibition of MCs since histamine levels and MC density were reduced by IL-10. Thus, IL-10 may represent an endogenous MC inhibitor, with a loss of IL-10 leaving MCs susceptible to activation stimuli. Confirming the pro-fibrotic role of MCs in the pressure overloaded heart, Kanellakis et al. [25] showed that cromolyn prevented LV fibrosis in mice with transaortic constriction. Similarly in the atria, the MC stabilizer, cromolyn, prevented fibrosis following transaortic constriction-induced pressure overload on the heart [26]. Even in STZ-induced diabetic hearts, nedocromil was able to reduce cardiac fibrosis [27]. More recently, Li et al. [28] found that nedocromil (30 mg/kg/day) prevented fibrosis from developing in rats following five weeks of transaortic constriction. Thus, the MC stabilzer studies strongly argue for a role for MCs in cardiac fibrosis. However, one must be aware of possible off target effects of these compounds, such as inhibition of sensory nerves. These studies are summarized in Table 2.
3.2. Studies with Mast Cell-Deficient Rodents
3.2.1. Types of Mast Cell-Deficient Rodents
There are several mutant mice that have been utilized as models of MC-deficiency to study MC function in vivo including KitW/W-v and KitW/W-sh mice [38,39,40,41]. These mouse strains carry spontaneous loss of function mutations at both alleles of the dominant white spotting locus (W, i.e., c-kit). This mutation exhibits a significant reduction in c-kit tyrosine kinase-dependent signalling, hence disrupting normal MC development and survival [39,42]. The different mutant alleles of c-kit reflect the variable non-MC-related effects that these mice display. The mutated W allele gives rise to truncated c-kit without the transmembrane domain, resulting in no protein expression on the cell surface [43]. Alternatively, the Wv mouse has a point mutation at the tyrosine kinase-encoding domain of c-kit. Unlike the W mouse, KitW/W-v mice still express the c-kit protein on the cell surface, although with reduced tyrosine kinase activity [43,44]. KitW/W-v mice have no detectable MCs in multiple organs by the time they reach 6 to 8 weeks of age [39]. However, due to malfunction of the c-kit protein, these mice display phenotypic abnormalities such as macrocytic anaemia, infertility, impaired melanogenesis, lack of intestinal cells of Cajal, spontaneous dermatitis, gastric ulcers and duodenum dilatation [45,46,47,48,49,50,51,52,53]. This strain has traditionally been the most popular strain used to study MC-deficiency.
The W-sash (KitW/W-sh) mutation is an inversion mutation in the transcriptional regulatory elements, upstream of the c-kit transcription start site of mouse chromosome 5, resulting in functionally impaired c-kit protein [54]. This mutation was first described 23 years ago from crossing two inbred strain mice (C3H/HEH × 101/H), although it is only fairly recently that this strain has gained popularity as a model of MC-deficiency in vivo [40,55]. Adult KitW/W-sh mice are MC-deficient at multiple anatomical sites [56], however, unlike the KitW/W-v mouse, they are fertile and not anaemic [40,57]. They also exhibit normal levels of myeloid cells, B cells, T cells, dendritic cells, and basophils [58]. Importantly, like the KitW/W-v mouse, KitW/W-sh mice can be successfully reconstituted with bone marrow derived mast cells with normal c-kit expression by adoptive transfer via intraperitoneal, intradermal or intravenous injection [55,56,58]. A comprehensive study by Grimbaldeston et al. [58] details the advantageous of KitW/W-sh mice over KitW/W-v mice.
Another mutant mouse that can be used to study MC biology is the steel-Dickie (Sld) mouse. This mutation occurs due to a 4.0 kilobase intragenic deletion and truncates the Sl coding sequence. Mast cell growth factor (MGF) is encoded by the Sl gene, hence this mutation results in soluble truncated growth factor that lacks both transmembrane and cytosplasmic domains [59]. Sld mice carry a homozygous mutation in the Sl gene as a complete deletion of Sl alleles, resulting in the complete absence of MGF and is lethal [60,61,62]. Phenotypically, Sld mice exhibit melanocytes defects, severe anaemia and sterility [63].
There is also a MC-deficient rat. The Ws/Ws rat has a 12-base deletion in the tyrosine kinase domain of c-kit, the receptor for stem cell factor. The Ws/Ws phenotype is otherwise normal except for white spotting of the skin and macrocytic anemia that is spontaneously ameliorated by 10 weeks of age [41]. Therefore, these rats do not exhibit the severe anemia seen in some MC-deficient mouse strains.
3.2.2. Pro-Fibrotic Role for Mast Cells
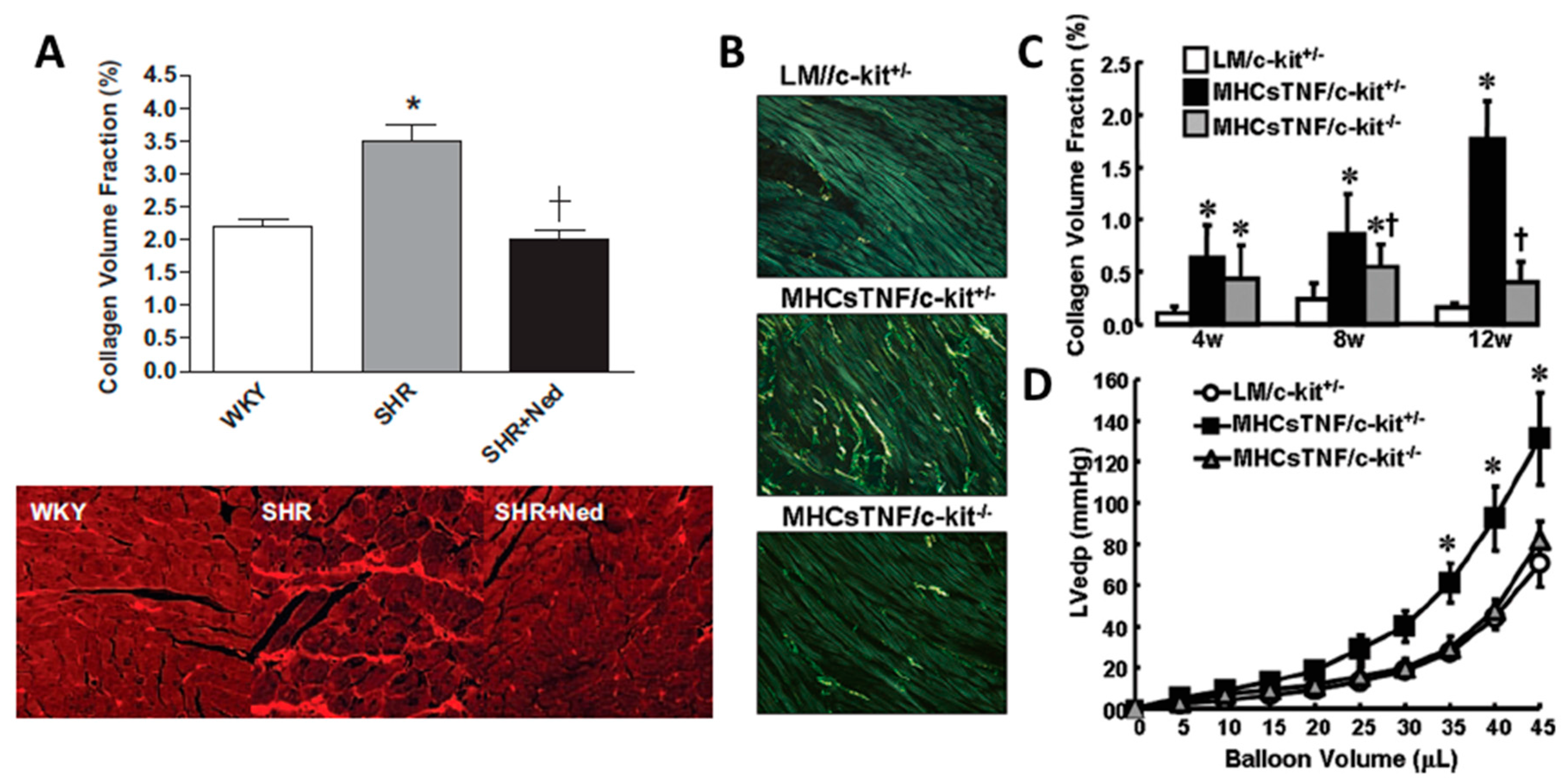
Hara et al. [29] were the first to use MC-deficient mice to evaluate cardiac fibrosis in any form. They used male KitW/Wv mice exposed to abdominal aortic banding for 15 weeks and found that whilst perivascular fibrosis occurred in WT mice with banding, collagen levels were normal in KitW/Wv mice. To definitively confirm the role of MCs in MC-deficient mice, MCs must be replaced by adoptive transfer. Unfortunately, Hara et al. were unsuccessful in their attempt to replenish MCs in KitW/Wv mice, thus, could not confirm that a lack of MCs was solely responsible for the resistance to adverse remodelling. The inability to reconstitute MCs to the hearts of these mice may have been due to beginning reconstitution just 2 days before initiating banding. Typically, it takes 6 to 8 weeks for injected MCs to reconstitute to the heart. Liao et al. [26] used the KitW/Wv mouse to investigate the contribution of MCs to atrial fibrosis following transaortic constriction. These authors reported that mice deficient in MCs were protected against atrial fibrosis, however, they also did not perform MC reconstitution experiments. In an interesting study, Zhang et al. [30] investigated the role of MCs in cardiac fibrosis in a model of TNF-α overexpression. Mice with cardiac-restricted TNF-α overexpression were crossed with KitW/W-sh mice. While fibrosis developed in the hearts of TNF-α overexpressing mice, this did not occur in MC-deficient TNF-α overexpressing hearts, indicating that MCs mediate the pro-fibrotic actions of TNF-α in this setting (Figure 1B,C). Further, the lack of MCs restored normal diastolic function as indicated by normalisation of the LV pressure volume relationship (Figure 1D). This study shows that in the setting of elevated TNF-α, MCs mediate the pro-fibrotic actions of TNF-α and that MC-mediated cardiac fibrosis contributes to diastolic dysfunction. This study offers probably the most conclusive evidence to date that MCs contribute to cardiac fibrosis, however, the caveat here is that this is an artificial up-regulation of TNF-α, which may or may not be relevant to conditions such as hypertension. These studies are summarized in Table 2.
3.2.3. Anti-Fibrotic Role of Mast Cells
Despite the evidence that MCs are pro-fibrotic in the heart, there does appear to be some exceptions. Martin Hauer-Jensen’s laboratory has used the Ws/Ws MC-deficient rat to identify some of these exceptions. In one study, Joseph et al. [31] fed Ws/Ws rats a diet high in homocysteine for 10 weeks to induce hyperhomocysteinemia that causes perivascular and interstitial cardiac fibrosis, without concomitant cardiomyocyte hypertrophy. Both forms of fibrosis were further increased in Ws/Ws rats fed homocysteine, indicating that MCs were protective in this setting. In another study on cardiac injury caused by radiation (single dose at 18 Gy), Boerma et al. [32] found that MC-competent rats had increased collagen III compared to rats deficient in MCs. However, this was not the case for collagen I. These studies are summarized in Table 2.
The reasons why MCs appear to be pro-fibrotic except in the instances of hyperhomocysteinemia and radiation are not clear. One tempting explanation is the use of MC-deficient rats in the studies indicating an anti-fibrotic role versus MC-deficient mice in the studies supporting a pro-fibrotic role. However, MC stabilisers in rats appear to be anti-fibrotic, arguing against this hypothesis. It may be that MCs are directed to take on a pro-fibrotic phenotype when the stimulus involves altered cardiovascular hemodynamics (e.g., hypertension) or infection (e.g., myocarditis), whereas remodelling not related to these types of stimuli invoke an anti-fibrotic MC phenotype. Deeper analysis of the types of mediators released by MCs in each of these settings will be required to confirm or refute this hypothesis.
4. MC products
Figure 2 indicates specific mediators released by MCs and their potential contributions to cardiac fibrosis. These mediators are discussed below.
4.1. Proteases
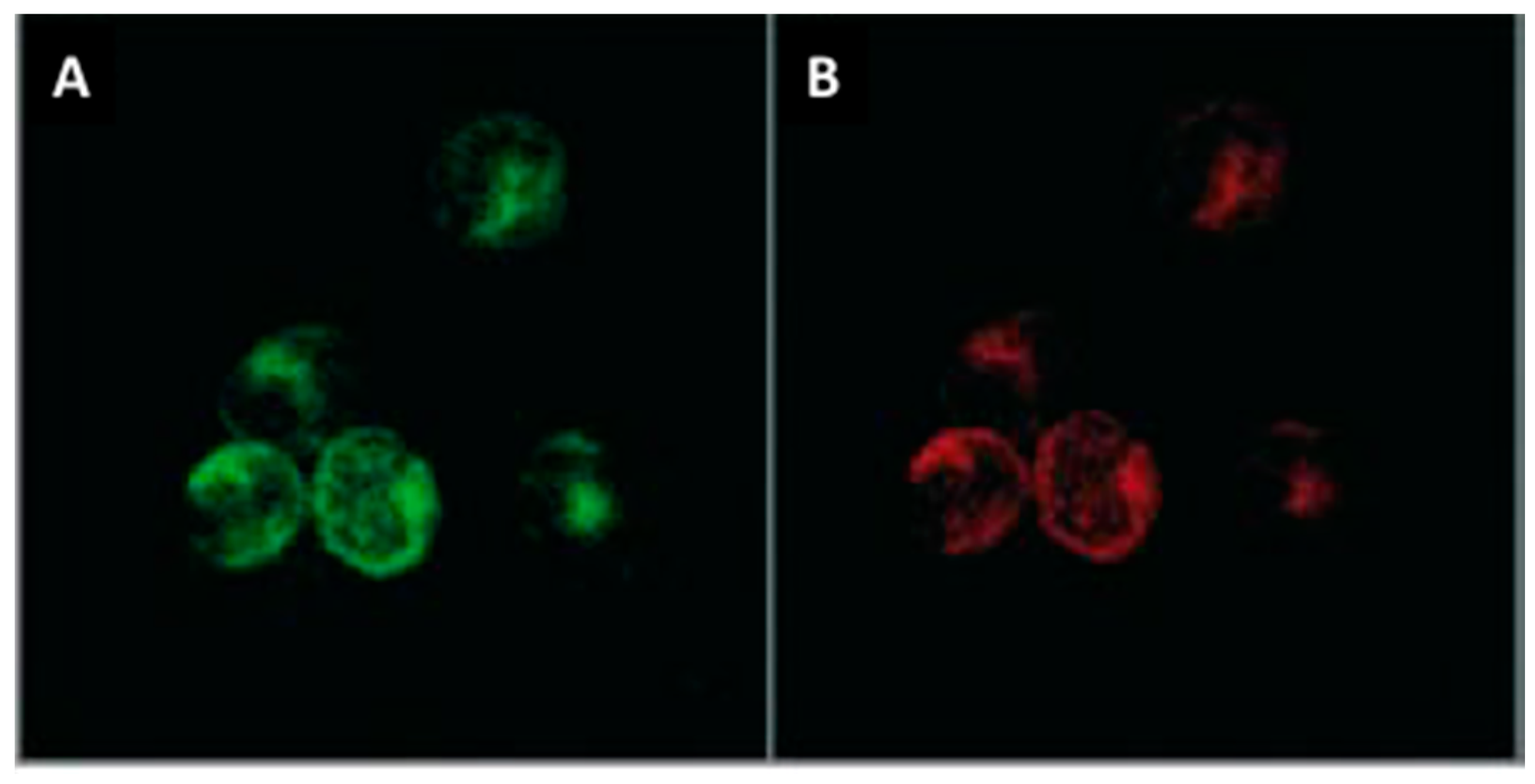
Cardiac MC phenotype has not been well studied. However, cardiac MCs, like MCs in general, store large amounts of specific proteases. Cardiac MCs fall under the connective tissue type MC phenotype since they contain both chymase and tryptase (Figure 3). On the other hand, mucosal MCs are tryptase+, but chymase-. Most of the work pertaining to cardiac MC products that contribute to fibrosis has focused on these proteases.
4.1.1. Chymase
Chymase is a chymotrypsin-like serine protease stored in the granules of MCs. In a mouse model of transaortic constriction, Hara et al. [29] observed an increase in Mcp5 in the heart, one of the mouse genes for chymase. Chymase activity in the heart was reported to increase 5.2-fold in hamsters with hypertension induced by the two-kidney, one-clip approach [64]. Similarly, there is evidence of chymase up-regulation in humans, with chymase mRNA and protein increased in patients with aortic stenosis undergoing valve replacement surgery [65].
The first study to demonstrate a causal role for chymase in cardiac fibrosis was by Matsumoto et al. [33]. In this study, heart failure was induced by rapid pacing in beagles (270 bpm, 22 days). Dogs with heart failure were treated with the chymase inhibitor SUNC8257 (10 mg/kg, orally twice a day), with chymase inhibition reducing collagen I and III mRNA levels and fibrosis as determined by picrosirius red staining (Figure 4). In the setting of MI-induced remodelling in rats, the chymase inhibitor, NK3201 reduced collagen I and III levels as well as fibrosis following 4 weeks post-MI, although it was unclear whether this analysis only included the infarct region or the entire LV [34]. Intriguingly, whilst there was a small improvement in diastolic function with chymase inhibition, as determined by E/A ratio, LV dilatation and systolic function were not improved. In an interesting study, Matsumoto et al. [35] investigated the role of chymase in cardiac remodelling caused by intermittent hypoxia mimicking sleep apnoea. Mice were placed in chambers that delivered intermittent hypoxia (30 s of 4.5% to 5.5% O2 followed by 30 s of 21% O2 for 8 h/day during the daytime) or normoxic conditions for 10 days. In addition to other remodelling parameters, perivascular fibrosis was increased by hypoxia and reduced by the chymase inhibitor, NK3201.
In the Matsumoto study in dogs, TGF-β1 mRNA was reduced by chymase inhibition whereas ACE mRNA was not. Further, Shimizu et al. [66] observed that aortic banding in male Syrian hamsters induced an increase in chymase activity and a decrease in angiotensin converting enzyme (ACE) activity, while causing cardiac fibrosis. The authors concluded that this indicated that chymase was primarily responsible for angiotensin II formation in this setting. These studies reflect the two known mechanisms likely to underlie chymases role in promoting fibrosis (i.e., angiotensin II and TGF-β1). Evidence has shown that chymase plays a role in the formation of angiotensin II in a non-canonical pathway of the renin angiotensin system (RAS). Chymase acted as an angiotensin-(1–12) converting enzyme in generating angiotensin II in SHR neonatal myocytes [67]. Chymase was also found to be responsible for this bio-conversion in the left atria of patients undergoing surgery for treatment of resistant atrial fibrillation or LV of normal patients dying from motor vehicle accidents [68,69]. Further supporting this, cardiac angiotensin II formation was reduced by the chymase inhibitor NK3201 in a mouse model of intermittent hypoxia [35]. Thus, one mechanism by which chymase exerts pro-fibrotic actions is via angiotensin II. Although, one needs to be aware of species differences in the contribution of chymase versus ACE to the cardiac angiotensin II pool. Balcells et al. [70] have noted that the human heart has dramatically more angiotensin II than other species, with dog being second, followed by mouse, rabbit, and rat. Human cardiac angiotensin II was almost entirely accounted for by chymase activity, as was also the case in the dog [70]. Approximately 25% of cardiac angiotensin II formation in the rat was attributed to chymase, whereas the contribution was less again in the rabbit and mouse. Akasu et al. [71] found similar results. They reported that angiotensin II was almost entirely accounted for by chymase in human, hamster, dog, and marmoset hearts. Pig and rabbit hearts showed ACE as the primary mechanism for angiotensin II synthesis. Interestingly, and in contrast to Balcells et al. [70], Akasu et al. [71] found that chymase was the primary determinant of angiotensin II formation in the rat heart. The mouse heart was not investigated.
The other pathway by which chymase exerts pro-fibrotic actions involves TGF-β1. At a cellular level, treatment of isolated neonatal cardiac fibroblasts with chymase (15–30 ng/mL) for 24 h resulted in cell proliferation [72]. This was accompanied by increased mRNA and protein levels of TGF-β1. Chymase up-regulation of TGF-β1 was independent of angiotensin II since blockade of AT1 and AT2 receptors did not alter TGF-β1 production. The proliferative and collagen producing effects of chymase could be reduced by a neutralising antibody to TGF-β1. The downstream mediator of TGF-β1 was Smad2/3 and not p38 or ERK pathways. In vivo, cromolyn reduced TGF-β1 levels in rats with myocarditis [19]. Shiota et al. [7] demonstrated that activated MCs were a major source of TGF-β1 and were localised to areas of fibrosis in 12 month and 20 month old SHR that were in advanced stages of LV hypertrophy and heart failure, respectively. In a mouse model of TNF-α overexpression, crossing these mice with MC-deficient mice reduced TGF-β1 levels as well TGF-β receptors in the heart [30].
4.1.2. Tryptase
Tryptase is a serine protease also stored in the granules of MCs. Fewer efforts have focused on the role of tryptase in cardiac fibrosis, even though tryptase levels increase in fibrotic hearts [23,28]. Mice with encephalomyocarditis virus-induced myocarditis show up-regulated mRNA levels of Mcp6, the gene for tryptase, 14 days after infection, which tracked with an increase in collagen I gene expression [18]. Using an in vitro approach, we demonstrated that tryptase could increase ECM synthesis by cardiac fibroblasts after 72 h [23]. Interestingly, proliferation was induced much more rapidly (24 h), suggesting that a fibroblast proliferative response might be the primary action of tryptase. In a follow-up study, we demonstrated that the effects of tryptase on cardiac fibroblasts were mediated by protease activated receptor-2 (PAR-2), which induced selective MAP kinase pathways with ERK1/2 mediating the pro-fibrotic actions of tryptase on cardiac fibroblasts, with no involvement from p38 or JNK [36]. This pathway also mediated cardiac fibroblast conversion to the myofibroblast phenotype. Critically, blockade of PAR-2 with FSLLRY (10 μg/kg/day) in SHR prevented fibrosis from occurring, independent of blood pressure. Thus, the role of tryptase appears to be more direct than is the case for chymase. In another follow-up study, we identified an autocrine/paracrine response by cardiac MCs mediating their own protease release. Inhibition of tryptase with nafamostat mesilate (5 mg/kg/day) reduced plasma chymase levels in rats with transaortic constriction [28]. To investigate this further, sections of rat LV were cultured in a novel tissue culture system and treated with tryptase. Tryptase caused the release of chymase into the media and a concomitant increase in collagen production that could be reduced by the chymase inhibitor chymostatin. These results suggest that tryptase also acts in an autocrine/paracrine manner to induce chymase release from MCs, and subsequent fibrosis.
4.2. Other Mast Cell Products
MCs release many products other than proteases that are capable of influencing the ECM. Several of these will be discussed below. However, it is important to recognize that many of these products can be produced by other cell types, and the relative contribution of the MCs to the overall pool of some of these products is unclear.
4.2.1. Histamine
The human heart contains considerable amounts of histamine (1035 ± 65 ng/g of atrial tissue) [73]. Histamine is the classic MC product mediating hypersensitivity reactions, and this is also true in the heart, which participates in anaphylaxis. However, histamine can also contribute to cardiac remodelling [74]. In the most direct assessment to date, Zeng et al. [37] performed transaortic constriction on H2 histamine receptor deficient mice (H2R−/−, Table 2). After four weeks, H2R−/− mice showed reduced cardiac fibrosis and slightly improved systolic function, indicating a role for histamine in cardiac fibrosis. The investigators performed additional studies in isolated cardiac fibroblasts and determined that both histamine and the H2R-selective agonist amthamine dihydrobromide increased protein levels of calcineurin; this was prevented by the H2R antagonist famotidine. Importantly, H2R activation also up-regulated myofibroblast conversion, fibronectin production, and procollagen I and III up-regulation at the gene level. Calcineurin was subsequently shown to mediate fibroblast proliferation, fibronectin production, and collagen gene regulation in response to H2R activation. This study clearly shows the capability of histamine to have direct effects on cardiac fibroblasts. Interestingly though, the actions of histamine may extend beyond this. Of the four known histamine receptors, three (H1, H2 and H3) are found in the heart. Due to their localisation, the actions of these receptors in the heart are extremely complex. H1R and H2R are present in the sinoatrial and atrioventricular nodes of the heart, suggesting regulation of heart rate [75]. H1R modulates cardiac autonomic nerve function [76,77]. Interestingly, histamine enables noradrenaline release in the rat heart via H2R [78]. Given the pro-fibrotic properties of noradrenaline, this raises the possibility that MC-mediated release of noradrenaline via H2R could promote fibrosis. In fact, a recent clinical study linked H2R antagonist use to a 62% reduced risk of heart failure [79]. Cardiac sensory nerves possess H3R. Our recent findings that the sensory nerve neuropeptide substance P plays a critical role in cardiac fibrosis in the hypertensive heart raises the possibility that MC histamine could be responsible for its release [80]. If this is the case it sets up an interesting feed forward mechanism since we have data indicating that substance P and its cognate receptor the neurokinin-1 receptor are responsible for the increase in MC density observed in the hypertensive heart, but neurokinin-1 receptors do not contribute to MC activation in this setting. Thus, other stimuli activate MCs, potentially resulting in histamine release and amplification of the substance P response.
4.2.2. Components of the Renin Angiotensin System
Renin is the first enzyme in the RAS, its role being to cleave angiotensinogen to angiotensin I, which in turn can be cleaved by ACE or chymase to active angiotensin II. In 2004, Roberto Levi’s group demonstrated that rat cardiac MCs contain renin [81]. Extrapolating the relevance of this finding to humans, Levi’s group also showed that the human MC line HMC-1 produced active renin that could convert angiotensinogen to angiotensin I. Subsequently, Hara et al. [82] confirmed the presence of renin mRNA in HMC-1 cells and further identified angiotensinogen mRNA in these cells. Interestingly, these authors identified pre-formed angiotensin II in HMC-1 cells that was released in response to calcitonin gene-related peptide. Although MC-derived renin contributes to ischaemia-induced arrhythmias in the heart [83], no specific evidence shows directly the role of MC-derived components of the RAS in cardiac fibrosis. However, given the known pro-fibrotic effect of angiotensin II, it is reasonable to assume that MC RAS contributes at least to some degree to cardiac fibrosis.
4.2.3. TNF-α
Immunolabelling indicates that MCs are likely the main source of TNF-α in the heart [84,85]. MC stabilizers such as ketotifen and cromoglycate prevented TNF-α release in hearts undergoing ischemia reperfusion, further supporting this supposition [86]. MC-derived TNF-α has been shown to stimulate collagen production by dermal fibroblasts [87], however, the extent to which MC-derived TNF-α contributes to cardiac fibrosis has not yet been investigated.
4.2.4. TGF-β
The role of MCs in generating TGF-β via chymase has already been discussed in Section 4.1.1, however, direct production of TGF-β by MCs could also contribute to organ fibrosis. Inhibition of TGF-β1 has been shown to mediate the pro-fibrotic effects of MCs on dermal fibroblasts in culture [87]. Although this suggests effects of MC-derived TGF-β, the alternate possibility cannot be ruled out that chymase induced activation of TGF-β produced by fibroblasts. There are no studies directly investigating the contribution of MC-derived TGF-β to cardiac fibrosis.
4.2.5. Matrix Metalloproteinases
Investigation of MCs in airways of dogs found that these cells produce matrix metalloproteinase (MMP)-2 and -9 (gelatinase A and B) [88]. Stem cell factor selectively up-regulated MMP-9 [88]. Interestingly, TGF-β opposed the actions of stem cell factor on MMP-9. The significance of MMPs is their ability to regulate the ECM, whether it be by initiating degradation, or alternatively, inducing pro-fibrotic responses [89]. While MC regulation of MMPs has been established in cardiac volume overload [10,90], this results in ECM degradation in that setting. The contribution of MCs to MMPs has not been investigated in relation to cardiac fibrosis. However, conditioned media from MCs was able to increase MMP-2 activation in neonatal cardiac fibroblasts [91]. Additionally, chymase inhibition prevented MMP-9 activation in pigs undergoing ischemia reperfusion [92].
5. Important Questions
While there is accumulating evidence that MCs contribute to cardiac fibrosis, there are several important questions that remain unanswered.
5.1. What Activates Cardiac Mast Cells?
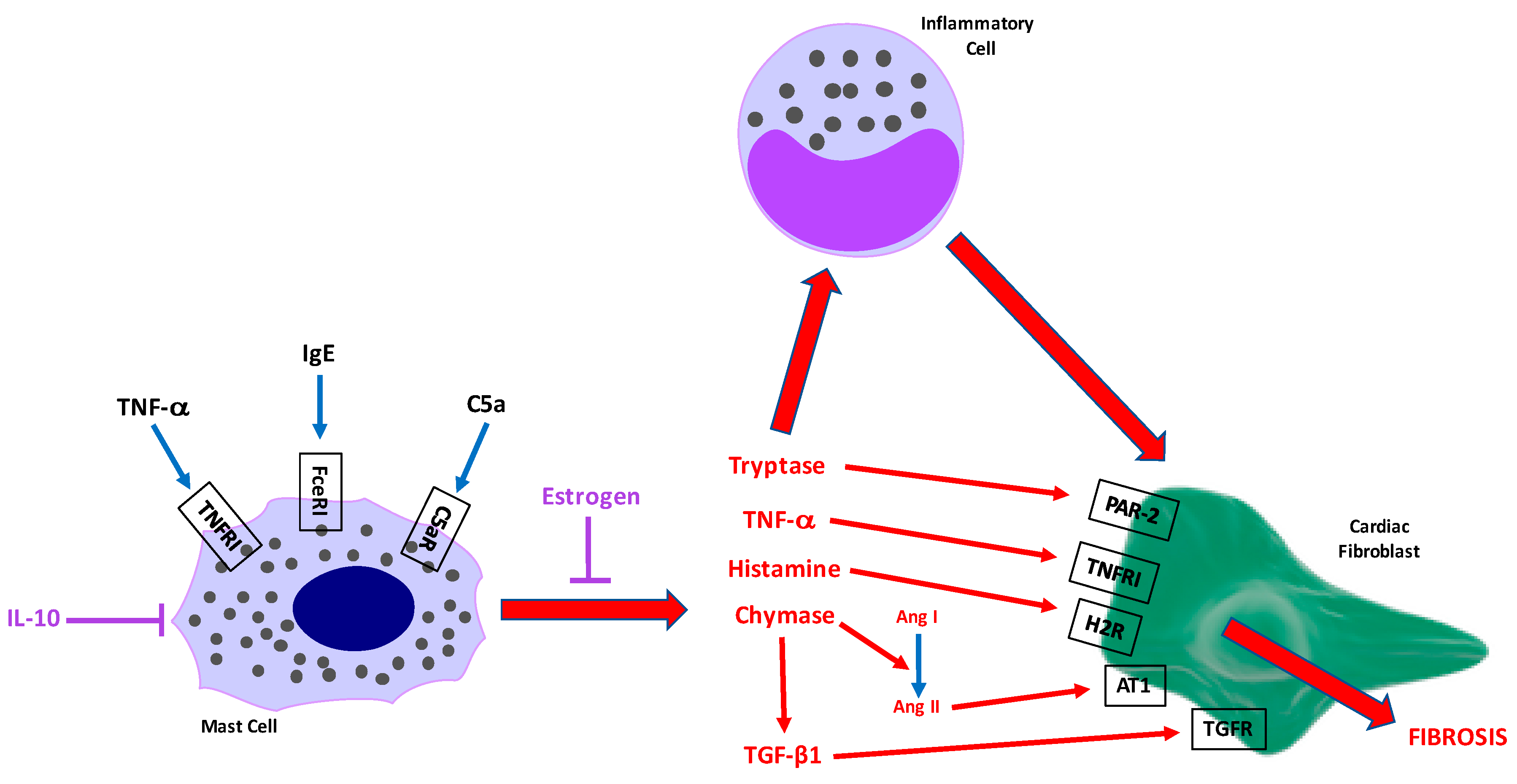
Probably the most pressing question is what are the stimuli that activate these cells causing them to promote fibrosis? There are several good candidates still to be investigated. These are depicted in Figure 2.
5.1.1. Immunoglobulin E
Like all MCs, cardiac MCs possess the IgE receptor, FcεRI. This is evidenced by their degranulation in response to antibody against FcεRI in vitro [93]. Interestingly, FcεRI activation causes release of histamine and tryptase, as well as leukotriene C4 and prostaglandin D2 by isolated human cardiac MCs [94]. As discussed earlier, MC histamine and tryptase are linked to cardiac fibrosis. Surprisingly, the role of IgE and FcεRI in causing cardiac fibrosis has not yet been investigated.
5.1.2. TNF-α
TNF-α receptor I (TNFRI) and TNF-α receptor II (TNFRII) mediate the actions of TNF-α. TnfrI−/− mice have improved cardiac remodelling responses (including fibrosis) following MI, while TnfrII−/− mice have worse fibrosis. Thus, TNFRI exacerbates remodelling leading to heart failure, whereas TNFRII has cardioprotective actions. The evidence implicating TNF-α and TNFRI comes from studies that crossed TNF-α overexpressing mice with MC-deficient mice. TNF-α overexpressing mice develop fibrosis; this fibrosis is reduced in mice lacking MCs. This indicates that MCs mediate the pro-fibrotic effects of TNF-α, implying that TNF-α plays a role in activating MCs. What is not clear from that study is whether this is direct activation of MCs by TNF-α via TNFRI or whether the activation is indirect with TNF-α up-regulating other mediators that then activate cardiac MCs. This question requires MC-specific deletion of TNFRI. Also, whether MC-derived TNF-α represents a viable treatment target is questionable given the failure of targeting TNF-α in heart failure patients.
5.1.3. Complement 5a
Patella et al., demonstrated more than 20 years ago that isolated human cardiac MCs degranulate in response to the complement factor, C5a [93]. The response to C5a was more rapid than to IgE and reached the same maximal response as IgE. However, Füreder et al. [95] subsequently reported that only 5% to 15% of human cardiac MCs that they examined possessed the C5a receptor and that human cardiac MCs did not release histamine in response to C5a. Nevertheless, coronary infusion of C5a (500 ng) in pigs undergoing MI led to an increase in coronary histamine levels indicating MC activation by C5a [96]. It will require MC-specific deletion of the C5a receptor to help resolve this question.
5.2. What Are the Specific Mechanisms by Which Mast Cells Cause Cardiac Fibrosis?
Despite clear evidence from multiple animal models that MCs are involved in cardiac fibrosis and that MC proteases mediate these actions along with possible contributions from other MC mediators, we know very little about the specifics. One example is the temporal involvement of MCs in cardiac fibrosis. Our own belief is that these cells are important in initiating fibrosis, but may not be involved continually throughout the process. This somewhat stems from our experience in MC regulation of the ECM in volume overload models where MC density increases in the first few days following initiation of volume overload, before returning to normal [90]. This suggests that MCs are important early to initiate processes that then continue throughout the remodelling process. Supporting this, MCs density is increased in the young SHR [7] and continued to be increased at 8 and 12 weeks of age, when fibrosis develops, before returning to normal levels by 16 weeks of age once fibrosis is established. Interestingly, MC density increases again around the age that heart failure develops in the SHR [7].
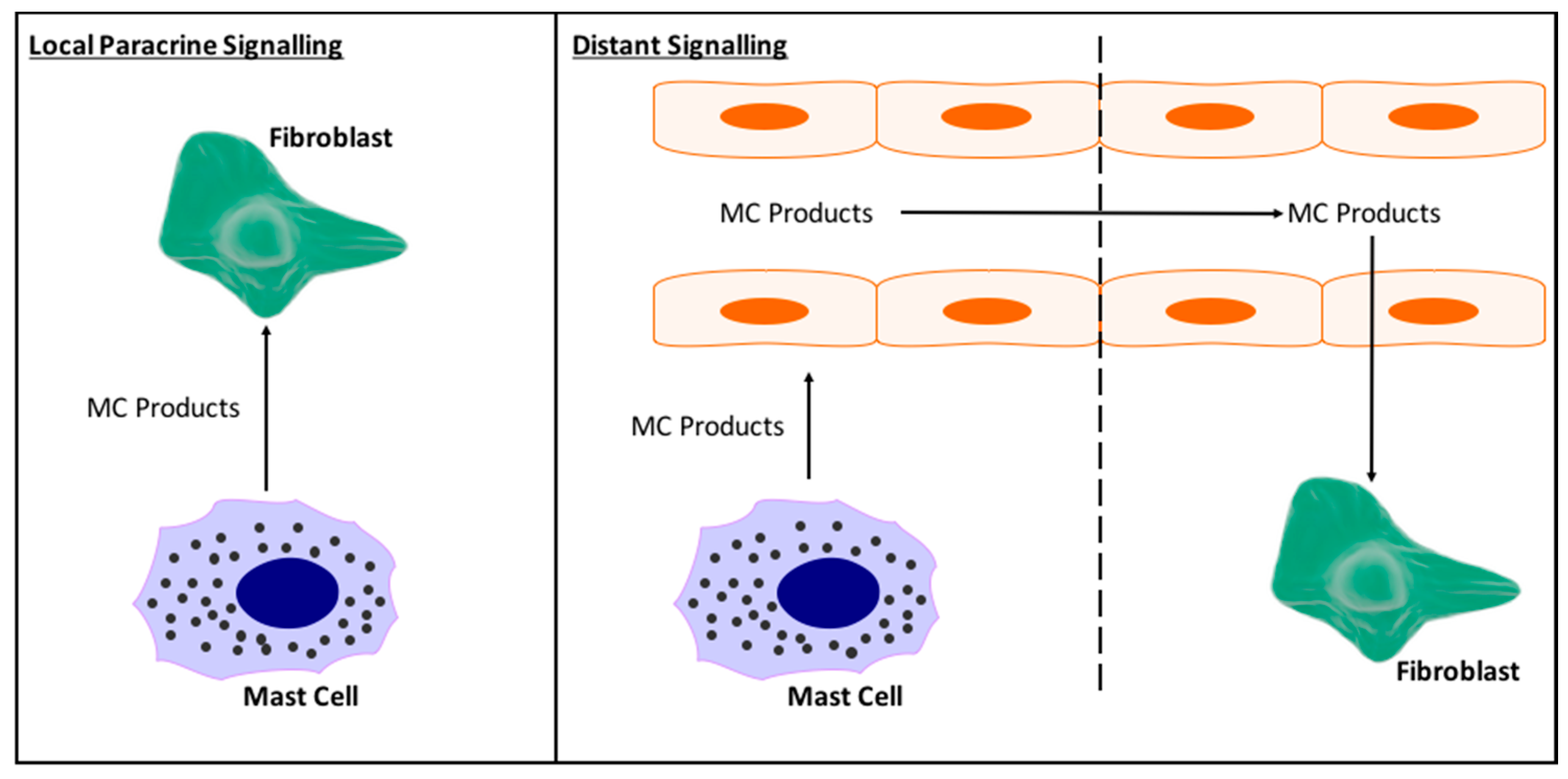
Also undetermined are the specifics of MC interactions with cardiac fibroblasts. In vitro it has been shown that MCs can act directly on cardiac fibroblasts to induce myofibroblast conversion, proliferation, and excess collagen synthesis [36,72], however, do these direct interactions actually occur in vivo, and to what extent? While cardiac MC number does vary between species, overall there are very few MCs in the heart. Clearly, although few in number they have a significant impact, however, it does raise the question of how many cardiac fibroblasts and MCs can actually interact in a paracrine manner in vivo. This we do not know. We do not even know whether released MC products circulate in the heart, giving them the opportunity to reach fibroblasts from remote parts of the heart, or whether they just act locally. Figure 5 depicts local versus distant interactions between MCs and cardiac fibroblasts.
Also likely important in MC modulation of cardiac fibrosis are interactions with other inflammatory cells (Figure 2), especially if direct MC-fibroblast interactions are minimal. Conditioned media from bone marrow-derived MCs activated with the calcium ionophore A23187 (500 ng/mL) dramatically up-regulated VCAM-1, ICAM-1, P-selectin, and E-selectin in mouse heart endothelial cells, suggesting a role for MCs in inflammatory cell recruitment [97]. In support of this, we had observed reduced numbers of macrophages in the SHR LV following treatment with nedocromil compared to untreated SHR [23]. This qualitative assessment suggests a role for MCs in the recruitment of macrophages to the fibrotic heart, however, quantitative assessment of this effect is still lacking. Should MCs regulate macrophage recruitment, then it should be determined as to whether this is a direct effect on macrophages, or whether other cells are involved (i.e., MC activation of endothelial cells). We also reported that MC stabilization reduced IFN-γ and IL-4 production, demonstrating the contribution of MCs to the overall cytokine pool, whether directly or indirectly.
Very little work has investigated MC-cardiomyocyte interactions. In our own studies, we did not observe any significant effect on cardiac hypertrophy, either by MC stabilization with nedocromil or inhibition of tryptase with FSLLRY [23,36]. Chymase appears to be able to induce cardiomyocyte death by entering these cells and inducing translocation of nuclear receptor subfamily 4A1 (NR4A1) from the nucleus to the cytoplasm. This was identified in ischemia reperfusion injury; the significance of this to cardiac fibrosis has not been explored. Media from cultured MCs can invoke death in cultured neonatal cardiomyocytes, with a neutralizing antibody against chymase opposing this effect [98]. Cardiomyocyte death is a stimulus for cardiac fibrosis. Thus, MC interactions with cardiomyocytes could be a stimulus for fibrosis, however, this is yet to be established.
5.3. What Is the Cardiac Mast Cell Phenotype and Are There Gender Differences?
A large amount of the work contributing to our understanding of the role of MCs in adverse cardiac remodeling has come from the laboratory of Joseph Janicki. Recently, the Janicki lab produced evidence indicating that differences in MC phenotype may underlie cardioprotection in pre-menopausal females. Intact and ovariectomised (ovx) female rats were exposed to pressure overload induced by transaortic constriction. LV MC density did not increase in intact females, but did increase in oxv rats [99]. Further, whilst there was a small increase in LV chymase levels in intact rats, the increase was greater in ovx animals. This was also the case for TGF-β1, presumably related to the increase in chymase. When oxv female rats were treated with estrogen, MC density, LV chymase, and TGF-β1 were reduced, as was fibrosis. This suggests that estrogen in pre-menopausal females provides a level of protection from cardiac fibrosis by reducing the ability of MCs to either: (1) respond to activation stimuli and therefore reduce levels of MC proteases that contribute to fibrosis in males; or (2) reduce production of mediators responsible for promoting fibrosis. Essentially, phenotypic differences exist between male and female MCs and this may underlie pre-menopausal cardioprotection in females (Figure 2). This is an extremely interesting concept that needs to be further explored.
These potential phenotypic differences between male and female MCs leads to another gap in our cardiac MC understanding; very little is known about the cardiac MC phenotype in general. This is an area that has been badly neglected. We know that cardiac MCs are tryptase+/chymase+ (Figure 3), as well as containing histamine and TNF-α [84,94,100]. Patella et al. [94] have demonstrated that isolated human cardiac MCs produce leukotrienes in response to activation by C5a. Beyond these few mediators, essentially nothing is known about cardiac MC phenotype. Whether cardiac MC sub-populations exist, as is the case for macrophages and T cells, is currently unknown, but should become a focus of investigation.
6. Conclusions
There is strong experimental evidence that MCs contribute to cardiac fibrosis, at least in part through the release of MC-specific proteases. However, the specifics of how MCs promote fibrosis is not clear, including the role of non-protease MC products, the stimuli that activate cardiac MCs, how cardiac MCs and fibroblasts interact in vivo, and specifics of cardiac MC phenotype (e.g., sub-populations and gender differences). These questions must be answered if targeting MCs is to eventually become a therapeutic approach in humans.
Acknowledgments
This work was supported by the National Heart Lung and Blood Institute at the National Institutes of Health [HL093215, HL132908 to Scott P. Levick]; Greater Milwaukee Foundation—Elsa Schoeneich Medical Research Fund to Scott P. Levick; the George and Mary Thomson Fellowship to Scott P. Levick; and an MCW Presidential Postdoctoral Fellow Award to Alexander Widiapradja.
Conflicts of Interest
The authors declare no conflict of interest. The funding sponsors had no role in the writing of this manuscript.
Abbreviations
| MC | Mast cell |
| MI | Myocardial Infarction |
| RV | Right Ventricle |
| LV | Left Ventricle |
| SHR | Spontaneously Hypertensive Rat |
| ECM | Extracellular Matrix |
| Sld | Steel Dickie |
| ACE | Angiotensin Converting Enzyme |
| PAR-2 | Protease Activated Receptor 2 |
| H1R | Histamine Receptor 1 |
| H2R | Histamine Receptor 2 |
| H3R | Histamine Receptor 3 |
| H4R | Histamine Receptor 4 |
| RAS | Renin Angiotensin System |
| HMC-1 | Human Mast Cell Line 1 |
| MMP | Matrix Metalloproteinase |
| TNFRI | Tumor Necrosis Factor Receptor I |
| TNFRII | Tumor Necrosis Factor Receptor II |
| C5a | Complement Factor 5a |
| Ovx | Ovariectomised |
References
- Levick, S.P.; Melendez, G.C.; Plante, E.; McLarty, J.L.; Brower, G.L.; Janicki, J.S. Cardiac mast cells: The centrepiece in adverse myocardial remodelling. Cardiovasc. Res. 2011, 89, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Engels, W.; Reiters, P.H.; Daemen, M.J.; Smits, J.F.; van der Vusse, G.J. Transmural changes in mast cell density in rat heart after infarct induction in vivo. J. Pathol. 1995, 177, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G.; Perrard, J.L.; Mendoza, L.H.; Burns, A.R.; Lindsey, M.L.; Ballantyne, C.M.; Michael, L.H.; Smith, C.W.; Entman, M.L. Stem cell factor induction is associated with mast cell accumulation after canine myocardial ischemia and reperfusion. Circulation 1998, 98, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, P.; Ren, G.; Nagar, H.; Kraemer, D.; Mendoza, L.; Michael, L.H.; Caughey, G.H.; Entman, M.L.; Frangogiannis, N.G. Mast cell tryptase may modulate endothelial cell phenotype in healing myocardial infarcts. J. Pathol. 2005, 205, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Panizo, A.; Mindán, F.J.; Galindo, M.F.; Cenarruzabeitia, E.; Hernández, M.; Díez, J. Are mast cells involved in hypertensive heart disease? J. Hypertens. 1995, 13, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Olivetti, G.; Lagrasta, C.; Ricci, R.; Sonnenblick, E.H.; Capasso, J.M.; Anversa, P. Long-term pressure-induced cardiac hypertrophy: Capillary and mast cell proliferation. AJP-Heart Circ. Phys. 1989, 257, H1766–H1772. [Google Scholar] [CrossRef] [PubMed]
- Shiota, N.; Rysä, J.; Kovanen, P.T.; Ruskoaho, H.; Kokkonen, J.O.; Lindstedt, K.A. A role for cardiac mast cells in the pathogenesis of hypertensive heart disease. J. Hypertens. 2003, 21, 1823–1825. [Google Scholar] [CrossRef]
- Li, Q.Y.; Raza-Ahmad, A.; MacAulay, M.A.; Lalonde, L.D.; Rowden, G.; Trethewey, E.; Dean, S. The relationship of mast cells and their secreted products to the volume of fibrosis in posttransplant hearts. Transplantation 1992, 53, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Estensen, R.D. Eosinophilic myocarditis: A role for mast cells? Arch. Pathol. Lab. Med. 1984, 108, 358–359. [Google Scholar] [PubMed]
- Brower, G.L.; Chancey, A.L.; Thanigaraj, S.; Matsubara, B.B.; Janicki, J.S. Cause and effect relationship between myocardial mast cell number and matrix metalloproteinase activity. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H518–H525. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.A.; Wei, C.C.; Brower, G.L.; Rynders, P.E.; Hankes, G.H.; Dillon, A.R.; Lucchesi, P.A.; Janicki, J.S.; Dell’Italia, L.J. Cardiac mast cell- and chymase-mediated matrix metalloproteinase activity and left ventricular remodeling in mitral regurgitation in the dog. J. Mol. Cell. Cardiol. 2003, 35, 311–319. [Google Scholar] [CrossRef]
- Batlle, M.; Roig, E.; Perez-Villa, F.; Lario, S.; Cejudo-Martin, P.; Garcia-Pras, E.; Ortiz, J.; Roque, M.; Orus, J.; Rigol, M.; et al. Increased expression of the renin-angiotensin system and mast cell density but not of angiotensin-converting enzyme II in late stages of human heart failure. J. Heart Lung Transplant. 2006, 25, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Fernex, M.; Sternby, N.H. Mast cells and coronary heart disease. Relationship between number of mast cells in the myocardium, severity of coronary atherosclerosis and myocardial infarction in an autopsy series of 672 cases. Acta Pathol. Microbiol. Scand. 1964, 62, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Turlington, B.S.; Edwards, W.D. Quantitation of mast cells in 100 normal and 92 diseased human hearts. Implications for interpretation of endomyocardial biopsy specimens. Am. J. Cardiovasc. Pathol. 1988, 2, 151–157. [Google Scholar] [PubMed]
- Pires, J.G.; Milanez, M.C.; Pereira, F.E. Histamine levels in tissues of Trypanosoma cruzi-infected mice. Agents Actions Suppl. 1992, 36, 96–98. [Google Scholar] [PubMed]
- Postan, M.; Correa, R.; Ferrans, V.J.; Tarleton, R.L. In vitro culture of cardiac mast cells from mice experimentally infected with Trypanosoma cruzi. Int. Arch. Allergy Immunol. 1994, 105, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Fairweather, D.; Frisancho-Kiss, S.; Gatewood, S.; Njoku, D.; Steele, R.; Barrett, M.; Rose, N.R. Mast cells and innate cytokines are associated with susceptibility to autoimmune heart disease following coxsackievirus B3 infection. Autoimmunity 2004, 37, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Kitaura-Inenaga, K.; Hara, M.; Higuchi, K.; Yamamoto, K.; Yamaki, A.; Ono, K.; Nakano, A.; Kinoshita, M.; Sasayama, S.; Matsumori, A. Gene expression of cardiac mast cell chymase and tryptase in a murine model of heart failure caused by viral myocarditis. Circ. J. 2003, 67, 881–884. [Google Scholar] [CrossRef] [PubMed]
- Palaniyandi, S.S.; Watanabe, K.; Ma, M.; Tachikawa, H.; Kodama, M.; Aizawa, Y. Involvement of mast cells in the development of fibrosis in rats with postmyocarditis dilated cardiomyopathy. Biol. Pharm. Bull. 2005, 28, 2128–2132. [Google Scholar] [CrossRef]
- Helske, S.; Syväranta, S.; Kupari, M.; Lappalainen, J.; Laine, M.; Lommi, J.; Turto, H.; Mayranpaa, M.; Werkkala, K.; Kovanen, P.T.; et al. Possible role for mast cell-derived cathepsin G in the adverse remodelling of stenotic aortic valves. Eur. Heart J. 2006, 27, 1495–1504. [Google Scholar] [CrossRef] [PubMed]
- Luitel, H.; Sydykov, A.; Schymura, Y.; Mamazhakypov, A.; Janssen, W.; Pradhan, K.; Wietelmann, A.; Kosanovic, D.; Dahal, B.K.; Weissmann, N.; et al. Pressure overload leads to an increased accumulation and activity of mast cells in the right ventricle. Physiol. Rep. 2017, 5, e13146. [Google Scholar] [CrossRef] [PubMed]
- Mina, Y.; Rinkevich-Shop, S.; Konen, E.; Goitein, O.; Kushnir, T.; Epstein, F.H.; Feinberg, M.S.; Leor, J.; Landa-Rouben, N. Mast cell inhibition attenuates myocardial damage, adverse remodeling, and dysfunction during fulminant myocarditis in the rat. J. Cardiovasc. Pharmacol. Ther. 2013, 18, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Levick, S.P.; McLarty, J.L.; Murray, D.B.; Freeman, R.M.; Carver, W.E.; Brower, G.L. Cardiac mast cells mediate left ventricular fibrosis in the hypertensive rat heart. Hypertension 2009, 53, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Palaniyandi, S.S.; Watanabe, K.; Ma, M.; Tachikawa, H.; Kodama, M.; Aizawa, Y. Inhibition of mast cells by interleukin-10 gene transfer contributes to protection against acute myocarditis in rats. Eur. J. Immunol. 2004, 34, 3508–3515. [Google Scholar] [CrossRef] [PubMed]
- Kanellakis, P.; Ditiatkovski, M.; Kostolias, G.; Bobik, A. A pro-fibrotic role for interleukin-4 in cardiac pressure overload. Cardiovasc. Res. 2012, 95, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.H.; Akazawa, H.; Tamagawa, M.; Ito, K.; Yasuda, N.; Kudo, Y.; Yamamoto, R.; Ozasa, Y.; Fujimoto, M.; Wang, P.; et al. Cardiac mast cells cause atrial fibrillation through PDGF-A-mediated fibrosis in pressure-overloaded mouse hearts. J. Clin. Investig. 2010, 120, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.G.; Jin, Q.; Fan, M.; Cong, X.L.; Han, S.F.; Gao, H.; Shan, Y. Myocardial remodeling in diabetic cardiomyopathy associated with cardiac mast cell activation. PLoS ONE 2013, 8, e60827. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jubair, S.; Levick, S.P.; Janicki, J.S. The autocrine role of tryptase in pressure overload-induced mast cell activation, chymase release and cardiac fibrosis. IJC Metab. Endocr. 2016, 10, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.; Ono, K.; Hwang, M.W.; Iwasaki, A.; Okada, M.; Nakatani, K.; Sasayama, S.; Matsumori, A. Evidence for a role of mast cells in the evolution to congestive heart failure. J. Exp. Med. 2002, 195, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chancey, A.L.; Tzeng, H.P.; Zhou, Z.; Lavine, K.J.; Gao, F.; Sivasubramanian, N.; Barger, P.M.; Mann, D.L. The development of myocardial fibrosis in transgenic mice with targeted overexpression of tumor necrosis factor requires mast cell-fibroblast interactions. Circulation 2011, 124, 2106–2116. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.; Kennedy, R.H.; Devi, S.; Wang, J.; Joseph, L.; Hauer-Jensen, M. Protective role of mast cells in homocysteine-induced cardiac remodeling. AJP-Heart Circ. Phys. 2005, 288, H2541–H2545. [Google Scholar] [CrossRef] [PubMed]
- Boerma, M.; Wang, J.; Wondergem, J.; Joseph, J.; Qiu, X.; Kennedy, R.H.; Hauer-Jensen, M. Influence of mast cells on structural and functional manifestations of radiation-induced heart disease. Cancer Res. 2005, 65, 3100–3107. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Wada, A.; Tsutamoto, T.; Ohnishi, M.; Isono, T.; Kinoshita, M. Chymase inhibition prevents cardiac fibrosis and improves diastolic dysfunction in the progression of heart failure. Circulation 2003, 107, 2555–2558. [Google Scholar] [CrossRef] [PubMed]
- Kanemitsu, H.; Takai, S.; Tsuneyoshi, H.; Nishina, T.; Yoshikawa, K.; Miyazaki, M.; Ikeda, T.; Komeda, M. Chymase inhibition prevents cardiac fibrosis and dysfunction after myocardial infarction in rats. Hypertens. Res. 2006, 29, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, C.; Hayashi, T.; Kitada, K.; Yamashita, C.; Miyamura, M.; Mori, T.; Ukimura, A.; Ohkita, M.; Jin, D.; Takai, S.; et al. Chymase plays an important role in left ventricular remodeling induced by intermittent hypoxia in mice. Hypertension 2009, 54, 164–171. [Google Scholar] [CrossRef] [PubMed]
- McLarty, J.L.; Melendez, G.C.; Brower, G.L.; Janicki, J.S.; Levick, S.P. Tryptase/Protease-activated receptor 2 interactions induce selective mitogen-activated protein kinase signaling and collagen synthesis by cardiac fibroblasts. Hypertension 2011, 58, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Shen, L.; Li, X.; Luo, T.; Wei, X.; Zhang, J.; Cao, S.; Huang, X.; Fukushima, Y.; Bin, J.; et al. Disruption of histamine H2 receptor slows heart failure progression through reducing myocardial apoptosis and fibrosis. Clin. Sci. 2014, 127, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Geissler, E.N.; McFarland, E.C.; Russell, E.S. Analysis of pleiotropism at the dominant white-spotting (W) locus of the house mouse: A description of ten new W alleles. Genetics 1981, 97, 337–361. [Google Scholar] [PubMed]
- Kitamura, Y.; Go, S.; Hatanaka, K. Decrease of mast cells in W/Wv mice and their increase by bone marrow transplantation. Blood 1978, 52, 447–452. [Google Scholar] [PubMed]
- Lyon, M.F.; Glenister, P.H. A new allele sash (Wsh) at the W-locus and a spontaneous recessive lethal in mice. Genet. Res. 1982, 39, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Niwa, Y.; Kasugai, T.; Ohno, K.; Morimoto, M.; Yamazaki, M.; Dohmae, K.; Nishimune, Y.; Kondo, K.; Kitamura, Y. Anemia and mast cell depletion in mutant rats that are homozygous at “white spotting (Ws)” locus. Blood 1991, 78, 1936–1941. [Google Scholar] [PubMed]
- Kitamura, Y. Heterogeneity of mast cells and phenotypic change between subpopulations. Annu. Rev. Immunol. 1989, 7, 59–76. [Google Scholar] [CrossRef] [PubMed]
- Nocka, K.; Tan, J.C.; Chiu, E.; Chu, T.Y.; Ray, P.; Traktman, P.; Besmer, P. Molecular bases of dominant negative and loss of function mutations at the murine c-kit/white spotting locus: W37, Wv, W41 and W. EMBO J. 1990, 9, 1805–1813. [Google Scholar] [PubMed]
- Reith, A.D.; Rottapel, R.; Giddens, E.; Brady, C.; Forrester, L.; Bernstein, A. W mutant mice with mild or severe developmental defects contain distinct point mutations in the kinase domain of the c-kit receptor. Genes Dev. 1990, 4, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Huizinga, J.D.; Thuneberg, L.; Kluppel, M.; Malysz, J.; Mikkelsen, H.B.; Bernstein, A. W/kit gene required for interstitial cells of Cajal and for intestinal pacemaker activity. Nature 1995, 373, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Waki, N.; Asai, H.; Kitamura, Y. Different repopulation profile between erythroid and nonerythroid progenitor cells in genetically anemic W/Wv mice after bone marrow transplantation. Blood 1989, 74, 1552–1556. [Google Scholar] [PubMed]
- Puddington, L.; Olson, S.; Lefrancois, L. Interactions between stem cell factor and c-Kit are required for intestinal immune system homeostasis. Immunity 1994, 1, 733–739. [Google Scholar] [CrossRef]
- Tsai, M.; Tam, S.Y.; Wedemeyer, J.; Galli, S.J. Mast cells derived from embryonic stem cells: A model system for studying the effects of genetic manipulations on mast cell development, phenotype, and function in vitro and in vivo. Int. J. Hematol. 2002, 75, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Galli, S.J.; Arizono, N.; Murakami, T.; Dvorak, A.M.; Fox, J.G. Development of large numbers of mast cells at sites of idiopathic chronic dermatitis in genetically mast cell-deficient WBB6F1-W/Wv mice. Blood 1987, 69, 1661–1666. [Google Scholar] [PubMed]
- Galli, S.J.; Zsebo, K.M.; Geissler, E.N. The kit ligand, stem cell factor. Adv. Immunol. 1994, 55, 1–96. [Google Scholar] [PubMed]
- Shimada, M.; Kitamura, Y.; Yokoyama, M.; Miyano, Y.; Maeyama, K.; Yamatodani, A.; Takahashi, Y.; Tatsuta, M. Spontaneous stomach ulcer in genetically mast-cell depleted W/Wv mice. Nature 1980, 283, 662–664. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, M.; Tatsuta, M.; Baba, M.; Kitamura, Y. Bile reflux: A possible cause of stomach ulcer in nontreated mutant mice of W/WV genotype. Gastroenterology 1982, 82 Pt 1, 857–863. [Google Scholar] [PubMed]
- Kitamura, Y.; Yokoyama, M.; Matsuda, H.; Shimada, M. Coincidental development of forestomach papilloma and prepyloric ulcer in nontreated mutant mice of W/Wv and SI/SId genotypes. Cancer Res. 1980, 40, 3392–3397. [Google Scholar] [PubMed]
- Nagle, D.L.; Kozak, C.A.; Mano, H.; Chapman, V.M.; Bucan, M. Physical mapping of the Tec and Gabrb1 loci reveals that the Wsh mutation on mouse chromosome 5 is associated with an inversion. Hum. Mol. Genet. 1995, 4, 2073–2079. [Google Scholar] [CrossRef] [PubMed]
- Mallen-St Clair, J.; Pham, C.T.; Villalta, S.A.; Caughey, G.H.; Wolters, P.J. Mast cell dipeptidyl peptidase I mediates survival from sepsis. J. Clin. Investig. 2004, 113, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Wolters, P.J.; Mallen-St Clair, J.; Lewis, C.C.; Villalta, S.A.; Baluk, P.; Erle, D.J.; Caughey, G.H. Tissue-selective mast cell reconstitution and differential lung gene expression in mast cell-deficient Kit(W-sh)/Kit(W-sh) sash mice. Clin. Exp. Allergy 2005, 35, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Tono, T.; Tsujimura, T.; Koshimizu, U.; Kasugai, T.; Adachi, S.; Isozaki, K.; Nishikawa, S.; Morimoto, M.; Nishimune, Y.; Nomura, S.; et al. c-kit Gene was not transcribed in cultured mast cells of mast cell-deficient Wsh/Wsh mice that have a normal number of erythrocytes and a normal c-kit coding region. Blood 1992, 80, 1448–1453. [Google Scholar] [PubMed]
- Grimbaldeston, M.A.; Chen, C.C.; Piliponsky, A.M.; Tsai, M.; Tam, S.Y.; Galli, S.J. Mast cell-deficient W-sash c-kit mutant kitW-sh/W-sh mice as a model for investigating mast cell biology in vivo. Am. J. Pathol. 2005, 167, 835–848. [Google Scholar] [CrossRef]
- Brannan, C.I.; Lyman, S.D.; Williams, D.E.; Eisenman, J.; Anderson, D.M.; Cosman, D.; Bedell, M.A.; Jenkins, N.A.; Copeland, N.G. Steel-Dickie mutation encodes a c-kit ligand lacking transmembrane and cytoplasmic domains. Proc. Natl. Acad. Sci. USA 1991, 88, 4671–4674. [Google Scholar] [CrossRef] [PubMed]
- Copeland, N.G.; Gilbert, D.J.; Cho, B.C.; Donovan, P.J.; Jenkins, N.A.; Cosman, D.; Anderson, D.; Lyman, S.D.; Williams, D.E. Mast cell growth factor maps near the steel locus on mouse chromosome 10 and is deleted in a number of steel alleles. Cell 1990, 63, 175–183. [Google Scholar] [CrossRef]
- Huang, E.; Nocka, K.; Beier, D.R.; Chu, T.Y.; Buck, J.; Lahm, H.W.; Wellner, D.; Leder, P.; Besmer, P. The hematopoietic growth factor KL is encoded by the Sl locus and is the ligand of the c-kit receptor, the gene product of the W locus. Cell 1990, 63, 225–233. [Google Scholar] [CrossRef]
- Zsebo, K.M.; Williams, D.A.; Geissler, E.N.; Broudy, V.C.; Martin, F.H.; Atkins, H.L.; Hsu, R.Y.; Birkett, N.C.; Okino, K.H.; Murdock, D.C.; et al. Stem cell factor is encoded at the Sl locus of the mouse and is the ligand for the c-kit tyrosine kinase receptor. Cell 1990, 63, 213–224. [Google Scholar] [CrossRef]
- Bernstein, S. Steel Dickie. Mouse News Lett. 1960, 23, 33–34. [Google Scholar]
- Shiota, N.; Jin, D.; Takai, S.; Kawamura, T.; Koyama, M.; Nakamura, N.; Miyazaki, M. Chymase is activated in the hamster heart following ventricular fibrosis during the chronic stage of hypertension. FEBS Lett. 1997, 406, 301–304. [Google Scholar] [CrossRef]
- Helske, S.; Lindstedt, K.A.; Laine, M.; Mäyränpää, M.; Werkkala, K.; Lommi, J.; Turto, H.; Kupari, M.; Kovanen, P.T. Induction of local angiotensin II-producing systems in stenotic aortic valves. J. Am. Coll. Cardiol. 2004, 44, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Tanaka, R.; Fukuyama, T.; Aoki, R.; Orito, K.; Yamane, Y. Cardiac remodeling and angiotensin II-forming enzyme activity of the left ventricle in hamsters with chronic pressure overload induced by ascending aortic stenosis. J. Vet. Med. Sci. 2006, 68, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Varagic, J.; Westwood, B.M.; Chappell, M.C.; Ferrario, C.M. Uptake and metabolism of the novel peptide angiotensin-(1-12) by neonatal cardiac myocytes. PLoS ONE 2011, 6, e15759. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Simmons, T.; Varagic, J.; Moniwa, N.; Chappell, M.C.; Ferrario, C.M. Chymase-dependent generation of angiotensin II from angiotensin-(1-12) in human atrial tissue. PLoS ONE 2011, 6, e28501. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Wei, C.C.; Tallaj, J.; Dell’Italia, L.J.; Moniwa, N.; Varagic, J.; Ferrario, C.M. Chymase mediates angiotensin-(1-12) metabolism in normal human hearts. J. Am. Soc. Hypertens. 2013, 7, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Balcells, E.; Meng, Q.C.; Johnson, W.H., Jr.; Oparil, S.; Dell’Italia, L.J. Angiotensin II formation from ACE and chymase in human and animal hearts: Methods and species considerations. Am. J. Physiol. 1997, 273 Pt 2, H1769–H1774. [Google Scholar] [CrossRef]
- Akasu, M.; Urata, H.; Kinoshita, A.; Sasaguri, M.; Ideishi, M.; Arakawa, K. Differences in tissue angiotensin II-forming pathways by species and organs in vitro. Hypertension 1998, 32, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Y.; Zhao, L.Y.; Zheng, Q.S.; Su, J.L.; Guan, H.; Shang, F.J.; Niu, X.L.; He, Y.P.; Lu, X.L. Chymase induces profibrotic response via transforming growth factor-β1/Smad activation in rat cardiac fibroblasts. Mol. Cell. Biochem. 2008, 310, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Gristwood, R.W.; Lincoln, J.C.; Owen, D.A.; Smith, I.R. Histamine release from human right atrium. Br. J. Pharmacol. 1981, 74, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, H.; Minamino, T.; Ogai, A.; Kim, J.; Asakura, M.; Komamura, K.; Sanada, S.; Fujita, M.; Hirata, A.; Wakeno, M.; et al. Blockade of histamine H2 receptors protects the heart against ischemia and reperfusion injury in dogs. J. Mol. Cell. Cardiol. 2006, 40, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Jesmin, S.; Takahashi, Y.; Hatta, E.; Kobayashi, M.; Matsuyama, K.; Kawakami, N.; Sakuma, I.; Gando, S.; Fukui, H.; et al. Histamine H1 and H2 receptor gene and protein levels are differentially expressed in the hearts of rodents and humans. J. Pharmacol. Exp. Ther. 2004, 309, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Powers, M.J.; Peterson, B.A.; Hardwick, J.C. Regulation of parasympathetic neurons by mast cells and histamine in the guinea pig heart. Auton. Neurosci. 2001, 87, 37–45. [Google Scholar] [CrossRef]
- Hardwick, J.C.; Kotarski, A.F.; Powers, M.J. Ionic mechanisms of histamine-induced responses in guinea pig intracardiac neurons. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R241–R250. [Google Scholar] [CrossRef] [PubMed]
- Fuder, H.; Ries, P.; Schwarz, P. Histamine and serotonin released from the rat perfused heart by compound 48/80 or by allergen challenge influence noradrenaline or acetylcholine exocytotic release. Fundam. Clin. Pharmacol. 1994, 8, 477–490. [Google Scholar] [CrossRef] [PubMed]
- Leary, P.J.; Tedford, R.J.; Bluemke, D.A.; Bristow, M.R.; Heckbert, S.R.; Kawut, S.M.; Krieger, E.V.; Lima, J.A.; Masri, C.S.; Ralph, D.D.; et al. Histamine H2 receptor antagonists, left ventricular morphology, and heart failure risk: The MESA study. J. Am. Coll. Cardiol. 2016, 67, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Dehlin, H.M.; Manteufel, E.J.; Monroe, A.L.; Reimer, M.H., Jr.; Levick, S.P. Substance P acting via the neurokinin-1 receptor regulates adverse myocardial remodeling in a rat model of hypertension. Int. J. Cardiol. 2013, 168, 4643–4651. [Google Scholar] [CrossRef] [PubMed]
- Silver, R.B.; Reid, A.C.; Mackins, C.J.; Askwith, T.; Schaefer, U.; Herzlinger, D.; Levi, R. Mast cells: A unique source of renin. Proc. Natl. Acad. Sci. USA 2004, 101, 13607–13612. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.; Ono, K.; Wada, H.; Sasayama, S.; Matsumori, A. Preformed angiotensin II is present in human mast cells. Cardiovasc. Drugs Ther. 2004, 18, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Mackins, C.J.; Kano, S.; Seyedi, N.; Schafer, U.; Reid, A.C.; Machida, T.; Silver, R.B.; Levi, R. Cardiac mast cell-derived renin promotes local angiotensin formation, norepinephrine release, and arrhythmias in ischemia/reperfusion. J. Clin. Investig. 2006, 116, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Levick, S.P.; Gardner, J.D.; Holland, M.; Hauer-Jensen, M.; Janicki, J.S.; Brower, G.L. Protection from adverse myocardial remodeling secondary to chronic volume overload in mast cell deficient rats. J. Mol. Cell. Cardiol. 2008, 45, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G.; Lindsey, M.L.; Michael, L.H.; Youker, K.A.; Bressler, R.B.; Mendoza, L.H.; Spengler, R.N.; Smith, C.W.; Entman, M.L. Resident cardiac mast cells degranulate and release preformed TNF-{α}, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation 1998, 98, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Gilles, S.; Zahler, S.; Welsch, U.; Sommerhoff, C.P.; Becker, B.F. Release of TNF-α during myocardial reperfusion depends on oxidative stress and is prevented by mast cell stabilizers. Cardiovasc. Res. 2003, 60, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.R.; Galli, S.J. Promotion of mouse fibroblast collagen gene expression by mast cells stimulated via the Fc epsilon RI. Role for mast cell-derived transforming growth factor β and tumor necrosis factor alpha. J. Exp. Med. 1994, 180, 2027–2037. [Google Scholar] [CrossRef] [PubMed]
- Fang, K.C.; Wolters, P.J.; Steinhoff, M.; Bidgol, A.; Blount, J.L.; Caughey, G.H. Mast cell expression of gelatinases A and B is regulated by kit ligand and TGF-β. J. Immunol. 1999, 162, 5528–5535. [Google Scholar] [PubMed]
- Iyer, R.P.; Patterson, N.L.; Fields, G.B.; Lindsey, M.L. The history of matrix metalloproteinases: Milestones, myths, and misperceptions. Am. J. Physiol. Heart Circ. Phys. 2012, 303, H919–H930. [Google Scholar] [CrossRef] [PubMed]
- Chancey, A.L.; Brower, G.L.; Janicki, J.S. Cardiac mast cell-mediated activation of gelatinase and alteration of ventricular diastolic function. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H2152–H2158. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, A.; Mustin, D.; Forman, M.F.; Brower, G.L.; Janicki, J.S.; Carver, W. Effects of mast cells on the behavior of isolated heart fibroblasts: Modulation of collagen remodeling and gene expression. J. Cell. Physiol. 2002, 191, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Oyamada, S.; Bianchi, C.; Takai, S.; Chu, L.M.; Sellke, F.W. Chymase inhibition reduces infarction and matrix metalloproteinase-9 activation and attenuates inflammation and fibrosis after acute myocardial ischemia/reperfusion. J. Pharmacol. Exp. Ther. 2011, 339, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Patella, V.; Marino, I.; Lamparter, B.; Arbustini, E.; Adt, M.; Marone, G. Human heart mast cells. Isolation, purification, ultrastructure, and immunologic characterization. J. Immunol. 1995, 154, 2855–2865. [Google Scholar] [PubMed]
- Patella, V.; de Crescenzo, G.; Ciccarelli, A.; Marinò, I.; Adt, M.; Marone, G. Human heart mast cells: A definitive case of mast cell heterogeneity. Int. Arch. Allergy Immunol. 1995, 106, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Fureder, W.; Agis, H.; Willheim, M.; Bankl, H.C.; Maier, U.; Kishi, K.; Muller, M.R.; Czerwenka, K.; Radaszkiewicz, T.; Butterfield, J.H.; et al. Differential expression of complement receptors on human basophils and mast cells. Evidence for mast cell heterogeneity and CD88/C5aR expression on skin mast cells. J. Immunol. 1995, 155, 3152–3160. [Google Scholar] [PubMed]
- Ito, B.R.; Engler, R.L.; del Balzo, U. Role of cardiac mast cells in complement C5a-induced myocardial ischemia. Am. J. Physiol. 1993, 264 Pt 2, H1346–H1354. [Google Scholar] [CrossRef]
- Zhang, J.; Alcaide, P.; Liu, L.; Sun, J.; He, A.; Luscinskas, F.W.; Shi, G.P. Regulation of endothelial cell adhesion molecule expression by mast cells, macrophages, and neutrophils. PLoS ONE 2011, 6, e14525. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.; Matsumori, A.; Ono, K.; Kido, H.; Hwang, M.W.; Miyamoto, T.; Iwasaki, A.; Okada, M.; Nakatani, K.; Sasayama, S. Mast cells cause apoptosis of cardiomyocytes and proliferation of other intramyocardial cells in vitro. Circulation 1999, 100, 1443–1449. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jubair, S.; Janicki, J.S. Estrogen inhibits mast cell chymase release to prevent pressure overload-induced adverse cardiac remodeling. Hypertension 2015, 65, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Marone, G.; Triggiani, M.; Cirillo, R.; Giacummo, A.; Hammarstrom, S.; Condorelli, M. IgE-mediated activation of human heart in vitro. Agents Actions 1986, 18, 194–196. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
MC stabilization with nedocromil, or MC deficiency prevents cardiac fibrosis. (A) Quantification and representative picrosirius red-stained images (20× magnification) for left ventricle (LV) collagen volume fraction for Wistar Kyoto rats (WKY), spontaneously hypertensive rats (SHR), and SHR treated with the MC stabilizer, nedocromil (Ned, 30 mg/kg/day), * = p < 0.05 vs WKY, † = p < 0.05 vs SHR; (B) representative images of picrosirius red stained LV collagen in control mice (LM/c-kit+/−), TNF-α overexpressing mice (MHCsTNF/c-kit+/−), and TNF-α overexpressing mice crossed with MC-deficient mice (MHCsTNF/c-kit−/−); (C) quantification of collagen volume fraction in control LM//c-kit+/− mice, MHCsTNF/c-kit+/− mice, and MHCsTNF/c-kit−/− mice; and (D) LV pressure-volume relationship for control LM//c-kit+/− mice, MHCsTNF/c-kit+/− mice, and MHCsTNF/c-kit−/− mice. * = p < 0.05 vs LM/c-kit+/−, † = p < 0.05 vs MHCsTNF/c-kit+/−. (Copied with permission from Levick et al., Hypertension, 2009;53:1041–1047 (A); and Zhang et al., Circulation, 2011;124:2106–2116 (B–D)).
Figure 1.
MC stabilization with nedocromil, or MC deficiency prevents cardiac fibrosis. (A) Quantification and representative picrosirius red-stained images (20× magnification) for left ventricle (LV) collagen volume fraction for Wistar Kyoto rats (WKY), spontaneously hypertensive rats (SHR), and SHR treated with the MC stabilizer, nedocromil (Ned, 30 mg/kg/day), * = p < 0.05 vs WKY, † = p < 0.05 vs SHR; (B) representative images of picrosirius red stained LV collagen in control mice (LM/c-kit+/−), TNF-α overexpressing mice (MHCsTNF/c-kit+/−), and TNF-α overexpressing mice crossed with MC-deficient mice (MHCsTNF/c-kit−/−); (C) quantification of collagen volume fraction in control LM//c-kit+/− mice, MHCsTNF/c-kit+/− mice, and MHCsTNF/c-kit−/− mice; and (D) LV pressure-volume relationship for control LM//c-kit+/− mice, MHCsTNF/c-kit+/− mice, and MHCsTNF/c-kit−/− mice. * = p < 0.05 vs LM/c-kit+/−, † = p < 0.05 vs MHCsTNF/c-kit+/−. (Copied with permission from Levick et al., Hypertension, 2009;53:1041–1047 (A); and Zhang et al., Circulation, 2011;124:2106–2116 (B–D)).

Figure 2.
Schematic depicting potential MC activation stimuli and interactions with other cell types that lead to cardiac fibrosis. Candidates for cardiac MC activation include IgE, TNF-α, and C5a. These then cause the release of MC mediators including the proteases tryptase and chymase, TNF-α, histamine, and TGF-β1. These mediators can then have direct effects on cardiac fibroblasts, but may also contribute to an inflammatory response that then activates cardiac fibroblasts via numerous cytokines. IL-10 and estrogen likely oppose cardiac MC activation/degranulation.
Figure 2.
Schematic depicting potential MC activation stimuli and interactions with other cell types that lead to cardiac fibrosis. Candidates for cardiac MC activation include IgE, TNF-α, and C5a. These then cause the release of MC mediators including the proteases tryptase and chymase, TNF-α, histamine, and TGF-β1. These mediators can then have direct effects on cardiac fibroblasts, but may also contribute to an inflammatory response that then activates cardiac fibroblasts via numerous cytokines. IL-10 and estrogen likely oppose cardiac MC activation/degranulation.

Figure 3.
Immunolabelling of chymase (A, green) and tryptase (B, red) in cardiac mast cells isolated from rats (400× magnification; Copied with permission from Morgan et al., Inflamm Res, 2008;57:1–6).
Figure 3.
Immunolabelling of chymase (A, green) and tryptase (B, red) in cardiac mast cells isolated from rats (400× magnification; Copied with permission from Morgan et al., Inflamm Res, 2008;57:1–6).

Figure 4.
Picrosirius red-stained images of perivascular fibrosis in normal, paced (vehicle), and paced plus the chymase inhibitor SUNC8257 (Chy-I) dog hearts (100× magnification; Copied with permission from Matsumoto et al., Circulation, 2003;107:2555–2558).
Figure 4.
Picrosirius red-stained images of perivascular fibrosis in normal, paced (vehicle), and paced plus the chymase inhibitor SUNC8257 (Chy-I) dog hearts (100× magnification; Copied with permission from Matsumoto et al., Circulation, 2003;107:2555–2558).

Figure 5.
Schematic depicting the possible interactions between MCs and fibroblasts in the heart. (Left) MCs may act in a paracrine manner to signal only to fibroblasts in their local area. This would limit direct MC-fibroblast interactions as a mechanism by which MCs cause fibrosis; (Right) Alternatively, MC products may be taken up in the general coronary circulation allowing their products to be distributed to fibroblasts throughout the heart. This would allow for greater MC-fibroblast interactions.
Figure 5.
Schematic depicting the possible interactions between MCs and fibroblasts in the heart. (Left) MCs may act in a paracrine manner to signal only to fibroblasts in their local area. This would limit direct MC-fibroblast interactions as a mechanism by which MCs cause fibrosis; (Right) Alternatively, MC products may be taken up in the general coronary circulation allowing their products to be distributed to fibroblasts throughout the heart. This would allow for greater MC-fibroblast interactions.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Summary of in vivo studies associating mast cells (MCs) with cardiac fibrosis.
| Species | Model/Pathology | Outcome | Heart Chamber | References |
|---|---|---|---|---|
| Human | Fibrosis | ↑ MC | LV | [13,14] |
| Rat | Pulmonary hypertension | ↑ MC | RV | [6] |
| Human | Transplantation | MC number correlated with fibrosis | LV | [8] |
| Mouse | Myocarditis | ↑ histamine, ↑ MC correlated with fibrosis | LV | [15,19] |
| Rat | Hypertension | ↑ MC correlated with fibrosis | LV | [5] |
| Mouse | Pulmonary hypertension | ↑ MC | RV | [21] |
LV = Left ventricle, RV = Right ventricle.
Table 2.
In vivo studies establishing cause and effect between MCs and cardiac fibrosis.
| Species | Intervention | Pathology | Outcome | Heart Chamber | Reference |
|---|---|---|---|---|---|
| MC Stabilizers | |||||
| Rat | Cromolyn | Myocarditis | ↓ fibrosis | LV | [19,22] |
| Rat | Nedocromil | Hypertension | ↓ fibrosis | LV | [23] |
| Mouse | Cromolyn | Transaortic constriction | ↓ fibrosis | LV | [25] |
| Mouse | Cromolyn | Transaortic constriction | ↓ fibrosis | Atria | [26] |
| Mouse | Nedocromil | STZ-induced diabetes | ↓ fibrosis | LV | [27] |
| Rat | Nedocromil | Transaortic constriction | ↓ fibrosis | LV | [28] |
| MC-deficient Rodents | |||||
| Mouse | KitW/Wv | Abdominal aortic banding | ↓ perivascular fibrosis | LV | [29] |
| Mouse | KitW/Wv | Transaortic constriction | ↓ fibrosis | Atria | [26] |
| Mouse | KitW/W-sh | TNF-α overexpression | ↓ fibrosis, ↓ diastolic dysfunction | LV | [30] |
| Rat | Ws/Ws | Hyperhomocysteinemia | ↑ fibrosis | LV | [31] |
| Rat | Ws/Ws | Radiation | ↑ fibrosis | LV | [32] |
| Targeting Proteases | |||||
| Canine | Chymase inhibitor (SUNC8257) | Pacing-induced heart failure | ↓ collagen I and III mRNA, ↓ fibrosis | LV | [33] |
| Rat | Chymase inhibitor (NK3201) | Myocardial infarction | ↓ collagen I and III mRNA, ↓ fibrosis, ↓ E/A | LV | [34] |
| Mouse | Chymase inhibitor (NK3201) | Intermittent hypoxia | ↓ perivascular fibrosis | LV | [35] |
| Rat | PAR-2 antagonist (FSLLRY, tryptase) | Hypertension | ↓ fibrosis | LV | [36] |
| Mouse | H2R−/− | Transaortic constriction | ↓ fibrosis, ↑ systolic function | LV | [37] |
LV = Left ventricle, RV = Right ventricle, PAR-2 = Protease activated receptor-24. MC products.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Levick, S.P.; Widiapradja, A. Mast Cells: Key Contributors to Cardiac Fibrosis. Int. J. Mol. Sci. 2018, 19, 231. https://doi.org/10.3390/ijms19010231
AMA Style
Levick SP, Widiapradja A. Mast Cells: Key Contributors to Cardiac Fibrosis. International Journal of Molecular Sciences. 2018; 19(1):231. https://doi.org/10.3390/ijms19010231
Chicago/Turabian StyleLevick, Scott P., and Alexander Widiapradja. 2018. "Mast Cells: Key Contributors to Cardiac Fibrosis" International Journal of Molecular Sciences 19, no. 1: 231. https://doi.org/10.3390/ijms19010231
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.