Glycoprotein 90K Promotes E-Cadherin Degradation in a Cell Density-Dependent Manner via Dissociation of E-Cadherin–p120-Catenin Complex

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
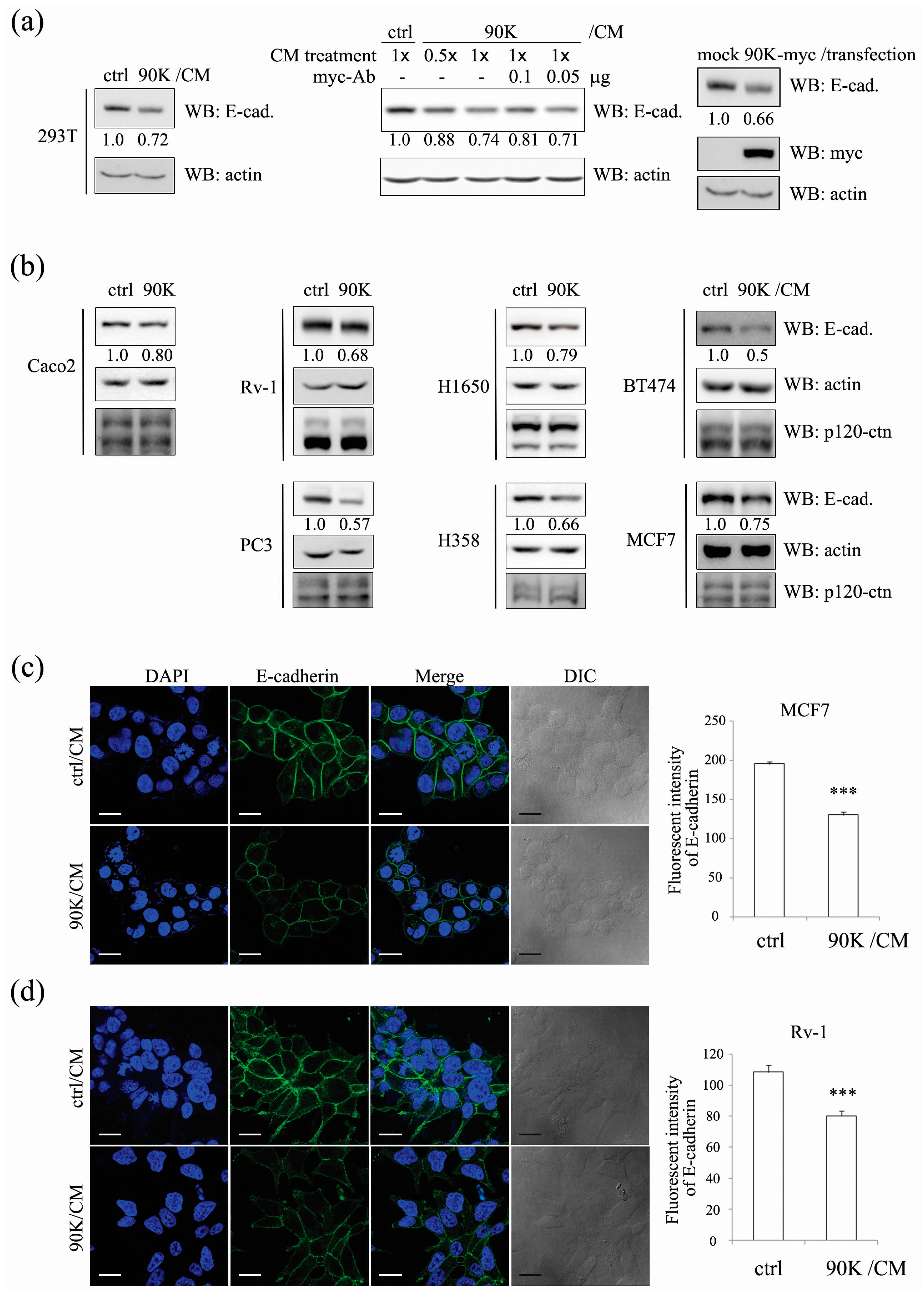
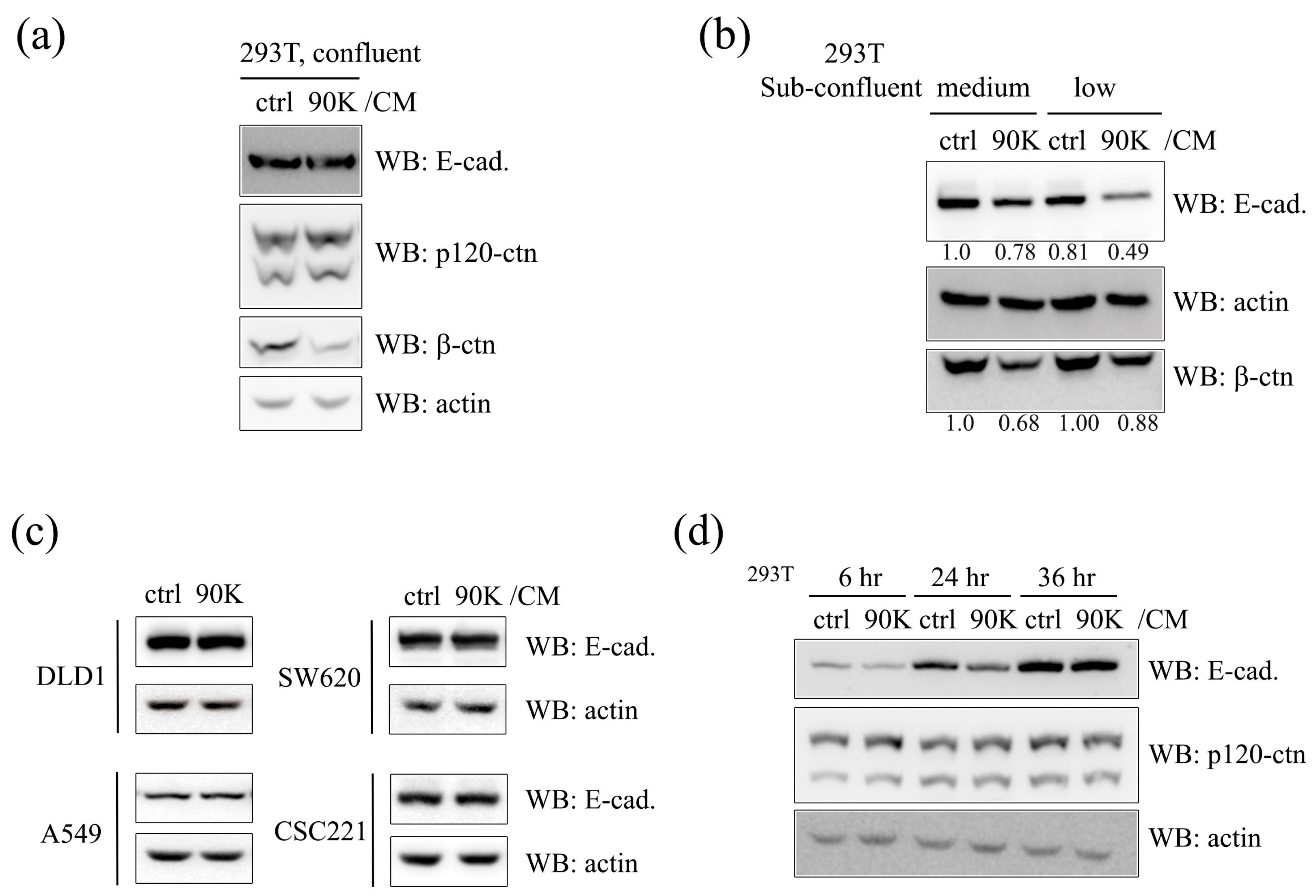
2.1. 90K Negatively Regulates E-Cadherin Levels in a Cell-Population-Dependent Manner
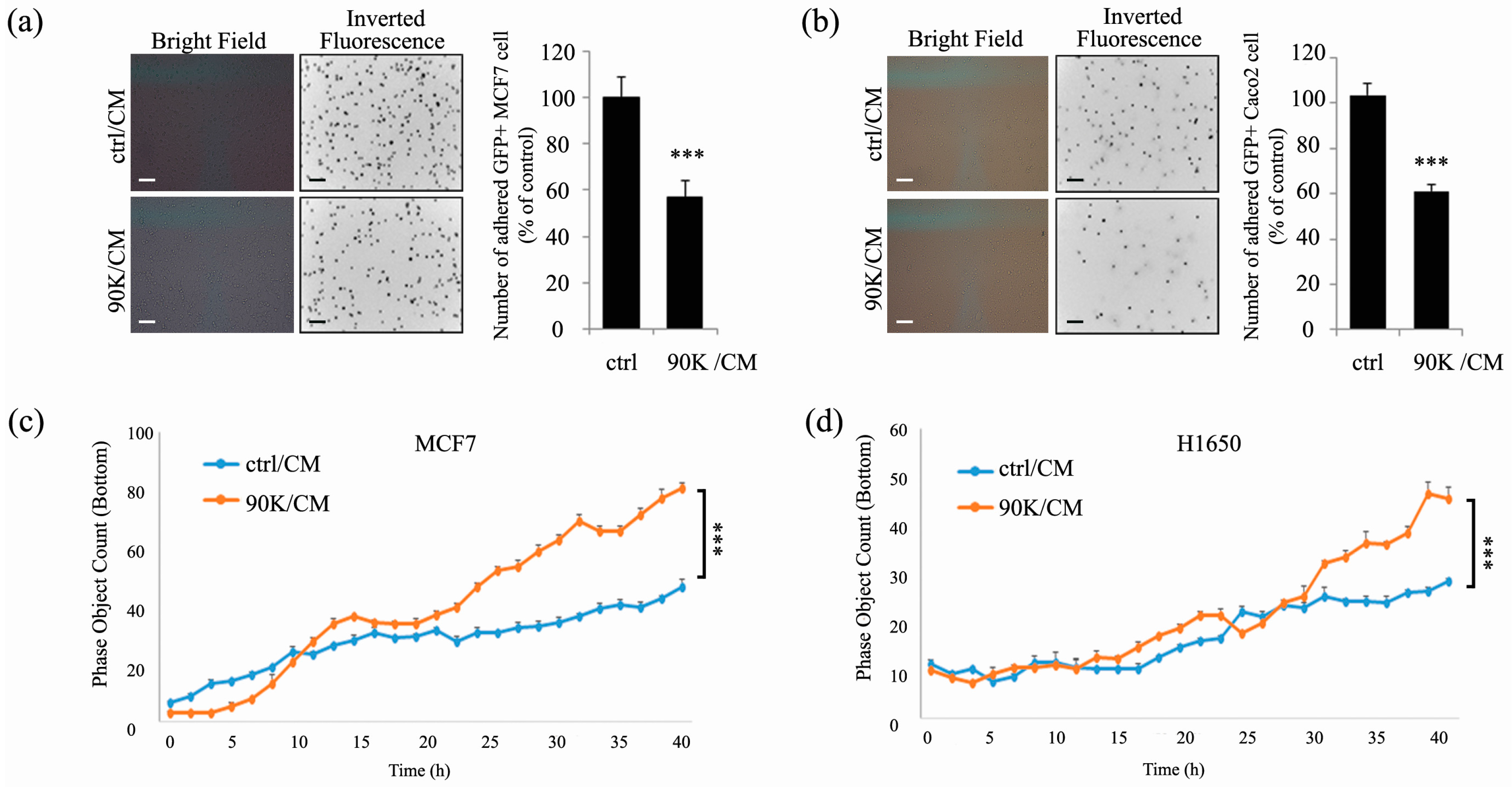
2.2. 90K Decreases Adhesion and Increases Invasion of Subconfluent Cancer Cells
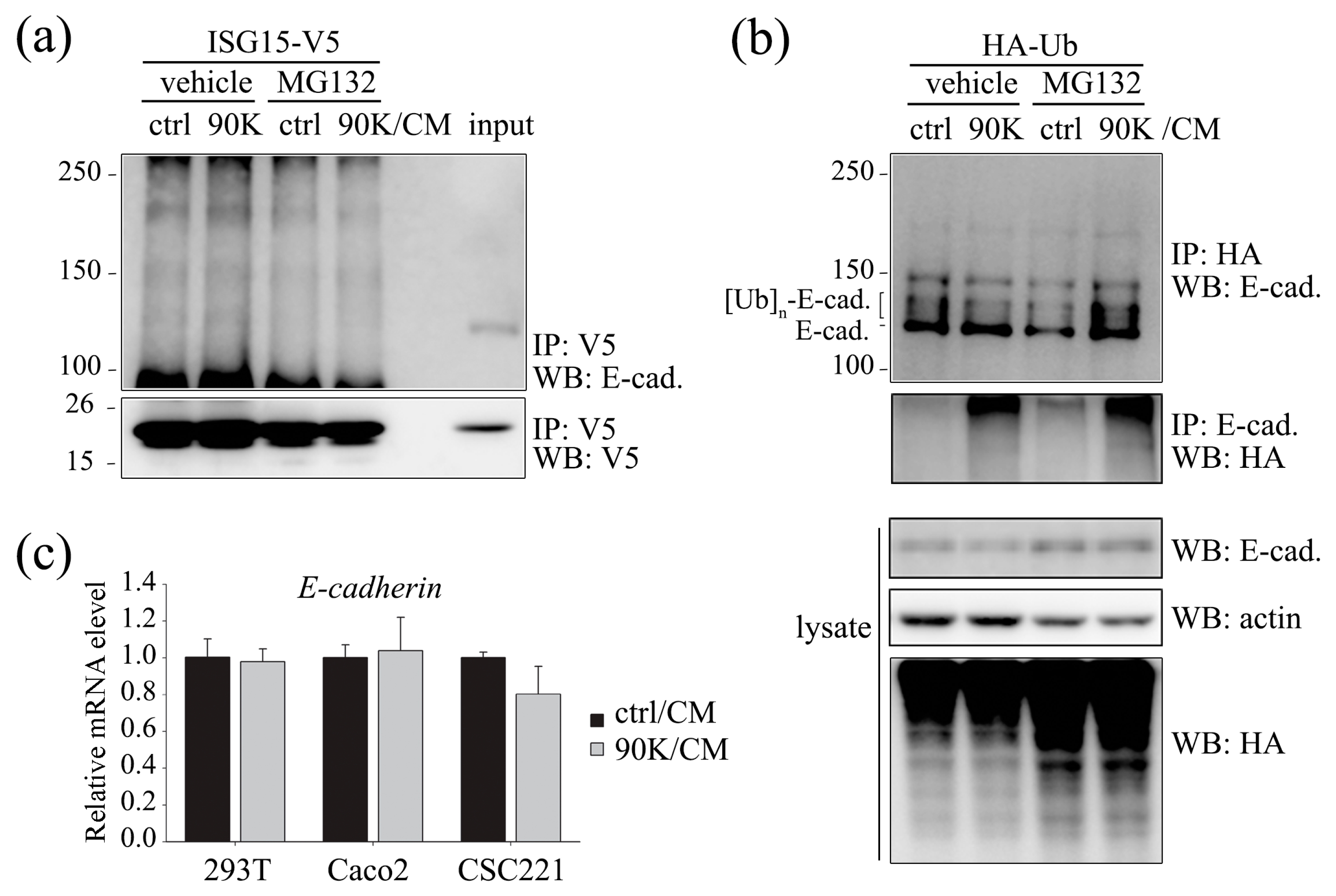
2.3. 90K Modulates E-Cadherin Levels via Ubiquitination-Mediated Proteasomal Degradation but Not via ISGylation
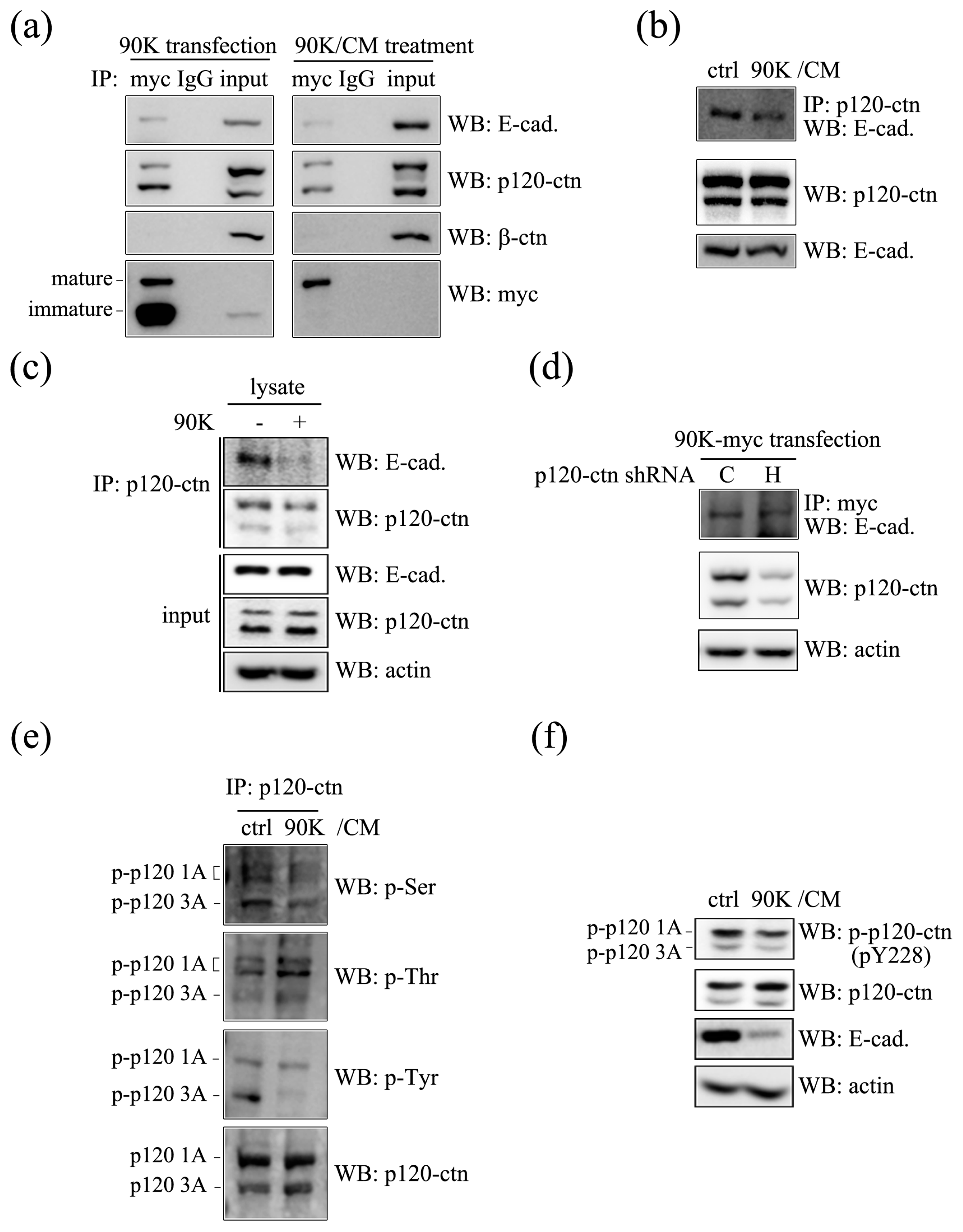
2.4. 90K Forms a Complex with E-Cadherin and Decreases the Association between p120-Catenin and E-Cadherin through Affecting Phosphorylation Status of p120-Catenin
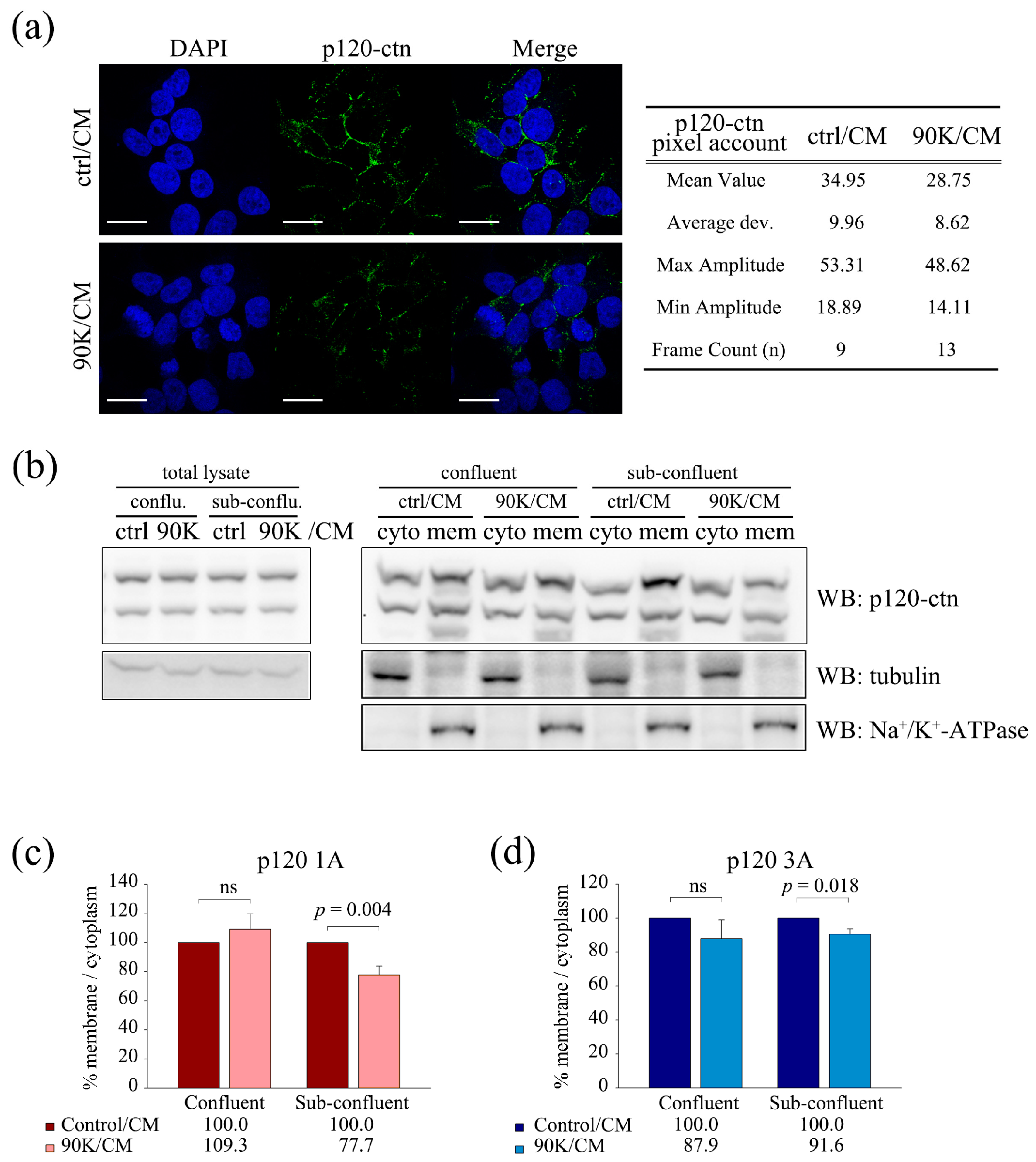
2.5. 90K Decreases Membrane Localization of p120-Catenin
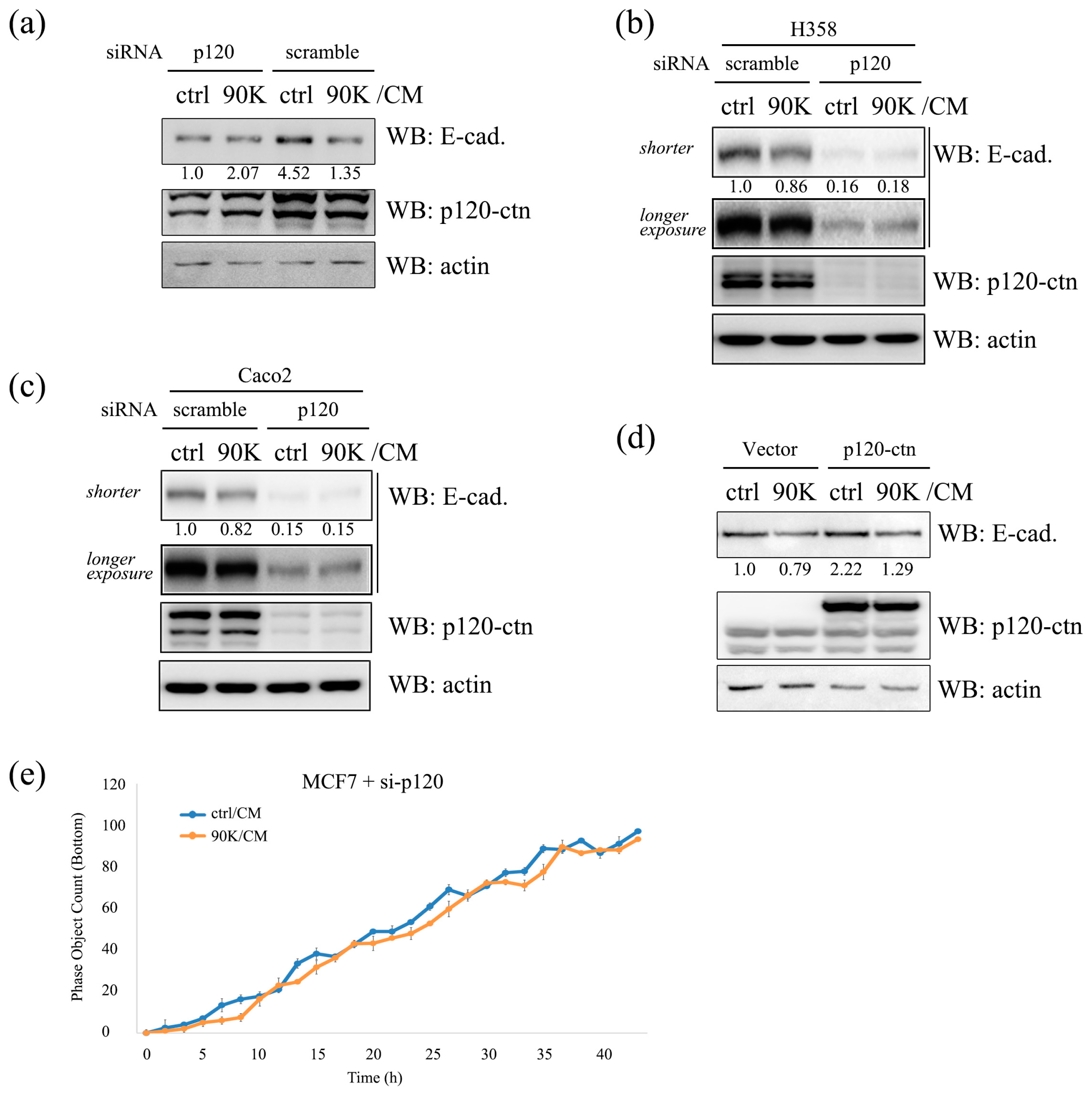
2.6. p120-Catenin Plays a Pivotal Role in 90K-Induced Downregulation of E-Cadherin and Promotion of Cancer Cell Invasion
3. Discussion
4. Materials and Methods
4.1. Plasmids and Antibodies
4.2. Cell Culture, Transfection, and Reagents
4.3. 90K Conditioned Media Preparation
4.4. Immunoblotting and Immunoprecipitation
4.5. Immunostaining and Image Acquisition
4.6. Cell Fractionation
4.7. Cell Adhesion Assay
4.8. Live-Cell Analysis of Cell Invasion
4.9. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Natoli, C.; Ortona, L.; Tamburrini, E.; Tinari, N.; Di Stefano, P.; D’Egidio, M.; Ghinelli, F.; Sighinolfi, L.; D’Ostilio, N.; Piazza, M.; et al. Elevated serum levels of a 90,000 daltons tumor-associated antigen in cancer and in infection by human immunodeficiency virus (HIV). Anticancer Res. 1994, 14, 1457–1460. [Google Scholar] [PubMed]
- Artini, M.; Natoli, C.; Tinari, N.; Costanzo, A.; Marinelli, R.; Balsano, C.; Porcari, P.; Angelucci, D.; D’Egidio, M.; Levrero, M.; et al. Elevated serum levels of 90K/MAC-2 BP predict unresponsiveness to α-interferon therapy in chronic HCV hepatitis patients. J. Hepatol. 1996, 25, 212–217. [Google Scholar] [CrossRef]
- Hepojoki, J.; Strandin, T.; Hetzel, U.; Sironen, T.; Klingstrom, J.; Sane, J.; Makela, S.; Mustonen, J.; Meri, S.; Lundkvist, A.; et al. Acute hantavirus infection induces galectin-3-binding protein. J. Gen. Virol. 2014, 95, 2356–2364. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.T.; Liu, Y.H.; Chen, Y.H.; Lin, C.Y.; Huang, C.H.; Yen, M.C.; Kuo, P.L. Serum Galectin-9 and Galectin-3-Binding Protein in Acute Dengue Virus Infection. Int. J. Mol. Sci. 2016, 17, 832. [Google Scholar] [CrossRef] [PubMed]
- Iacobelli, S.; Arno, E.; D’Orazio, A.; Coletti, G. Detection of antigens recognized by a novel monoclonal antibody in tissue and serum from patients with breast cancer. Cancer Res. 1986, 46, 3005–3010. [Google Scholar] [PubMed]
- Grassadonia, A.; Tinari, N.; Iurisci, I.; Piccolo, E.; Cumashi, A.; Innominato, P.; D’Egidio, M.; Natoli, C.; Piantelli, M.; Iacobelli, S. 90K (Mac-2 BP) and galectins in tumor progression and metastasis. Glycoconj. J. 2004, 19, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Ulmer, T.A.; Keeler, V.; Loh, L.; Chibbar, R.; Torlakovic, E.; Andre, S.; Gabius, H.J.; Laferte, S. Tumor-associated antigen 90K/Mac-2-binding protein: Possible role in colon cancer. J. Cell. Biochem. 2006, 98, 1351–1366. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.L.; Chen, H.H.; Pen, C.T.; Yeh, W.L.; Huang, E.Y.; Hsiao, C.C.; Yang, K.D. Circulating Galectin-1 and 90K/Mac-2BP Correlated with the Tumor Stages of Patients with Colorectal Cancer. BioMed Res. Int. 2015, 2015, 306964. [Google Scholar] [CrossRef] [PubMed]
- Bair, E.L.; Nagle, R.B.; Ulmer, T.A.; Laferte, S.; Bowden, G.T. 90K/Mac-2 binding protein is expressed in prostate cancer and induces promatrilysin expression. Prostate 2006, 66, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.P.; Choi, S.C.; Kim, J.H.; Song, E.Y.; Kim, J.W.; Yoon, D.Y.; Yeom, Y.I.; Lim, J.S.; Paik, S.G.; Lee, H.G. Up-regulation of Mac-2 binding protein by hTERT in gastric cancer. Int. J. Cancer 2007, 120, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, A.; Tinari, N.; Buttitta, F.; Chella, A.; Angeletti, C.A.; Sacco, R.; Mucilli, F.; Ullrich, A.; Iacobelli, S. Expression of 90K (Mac-2 BP) correlates with distant metastasis and predicts survival in stage I non-small cell lung cancer patients. Cancer Res. 2002, 62, 2535–2539. [Google Scholar] [PubMed]
- Tinari, N.; Lattanzio, R.; Querzoli, P.; Natoli, C.; Grassadonia, A.; Alberti, S.; Hubalek, M.; Reimer, D.; Nenci, I.; Bruzzi, P.; et al. High expression of 90K (Mac-2 BP) is associated with poor survival in node-negative breast cancer patients not receiving adjuvant systemic therapies. Int. J. Cancer 2009, 124, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Endo, H.; Muramatsu, T.; Furuta, M.; Uzawa, N.; Pimkhaokham, A.; Amagasa, T.; Inazawa, J.; Kozaki, K. Potential of tumor-suppressive miR-596 targeting LGALS3BP as a therapeutic agent in oral cancer. Carcinogenesis 2013, 34, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, R.; Yamasaki, K.; Abiru, S.; Komori, A.; Nagaoka, S.; Saeki, A.; Hashimoto, S.; Bekki, S.; Kugiyama, Y.; Kuno, A.; et al. Serum wisteria floribunda agglutinin-positive mac-2 binding protein values predict the development of hepatocellular carcinoma among patients with chronic hepatitis c after sustained virological response. PLoS ONE 2015, 10, e0129053. [Google Scholar] [CrossRef] [PubMed]
- Waragai, Y.; Suzuki, R.; Takagi, T.; Sugimoto, M.; Asama, H.; Watanabe, K.; Kikuchi, H.; Hikichi, T.; Masamune, A.; Kang, Y.; et al. Clinical significance of serum Wisteria floribunda agglutinin-positive Mac-2 binding protein in pancreatic ductal adenocarcinoma. Pancreatology 2016, 16, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Iacobelli, S.; Sismondi, P.; Giai, M.; D’Egidio, M.; Tinari, N.; Amatetti, C.; Di Stefano, P.; Natoli, C. Prognostic value of a novel circulating serum 90K antigen in breast cancer. Br. J. Cancer 1994, 69, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Escrevente, C.; Grammel, N.; Kandzia, S.; Zeiser, J.; Tranfield, E.M.; Conradt, H.S.; Costa, J. Sialoglycoproteins and N-glycans from secreted exosomes of ovarian carcinoma cells. PLoS ONE 2013, 8, e78631. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, E.; Tinari, N.; D’Addario, D.; Rossi, C.; Iacobelli, V.; La Sorda, R.; Lattanzio, R.; D’Egidio, M.; Di Risio, A.; Piantelli, M.; et al. Prognostic relevance of LGALS3BP in human colorectal carcinoma. J. Transl. Med. 2015, 13, 248. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Bae, J.A.; Seo, Y.W.; Kho, D.H.; Sun, E.G.; Lee, S.E.; Cho, S.H.; Joo, Y.E.; Ahn, K.Y.; Chung, I.J.; et al. Glycoprotein 90K, downregulated in advanced colorectal cancer tissues, interacts with CD9/CD82 and suppresses the Wnt/β-catenin signal via ISGylation of β-catenin. Gut 2010, 59, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Yoon, S.; Kim, H.; Kim, K.K. 90K Glycoprotein Promotes Degradation of Mutant β-Catenin Lacking the ISGylation or Phosphorylation Sites in the N-terminus. Neoplasia 2016, 18, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Pikkarainen, T.; Nurmi, T.; Sasaki, T.; Bergmann, U.; Vainio, S. Role of the extracellular matrix-located Mac-2 binding protein as an interactor of the Wnt proteins. Biochem. Biophys. Res. Commun. 2017, 491, 953–957. [Google Scholar] [CrossRef] [PubMed]
- Stampolidis, P.; Ullrich, A.; Iacobelli, S. LGALS3BP, lectin galactoside-binding soluble 3 binding protein, promotes oncogenic cellular events impeded by antibody intervention. Oncogene 2015, 34, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Van Roy, F. Beyond E-cadherin: Roles of other cadherin superfamily members in cancer. Nat. Rev. Cancer 2014, 14, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Schmalhofer, O.; Brabletz, S.; Brabletz, T. E-cadherin, β-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009, 28, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Hlubek, F.; Spaderna, S.; Schmalhofer, O.; Hiendlmeyer, E.; Jung, A.; Kirchner, T. Invasion and metastasis in colorectal cancer: Epithelial-mesenchymal transition, mesenchymal-epithelial transition, stem cells and β-catenin. Cells Tissues Organs 2005, 179, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Van Roy, F.; Berx, G. The cell-cell adhesion molecule E-cadherin. Cell. Mol. Life Sci. 2008, 65, 3756–3788. [Google Scholar] [CrossRef] [PubMed]
- Stairs, D.B.; Bayne, L.J.; Rhoades, B.; Vega, M.E.; Waldron, T.J.; Kalabis, J.; Klein-Szanto, A.; Lee, J.S.; Katz, J.P.; Diehl, J.A.; et al. Deletion of p120-catenin results in a tumor microenvironment with inflammation and cancer that establishes it as a tumor suppressor gene. Cancer Cell 2011, 19, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-Y.; Kim, H. Decreases in invasive motility of the MCF7 and Caco2 cells by 90K were observed when cell reaches their confluency. Sunchon National University, 2017; unpublished work. [Google Scholar]
- Alema, S.; Salvatore, A.M. p120 catenin and phosphorylation: Mechanisms and traits of an unresolved issue. Biochim. Biophys. Acta 2007, 1773, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Ishiyama, N.; Lee, S.H.; Liu, S.; Li, G.Y.; Smith, M.J.; Reichardt, L.F.; Ikura, M. Dynamic and static interactions between p120 catenin and E-cadherin regulate the stability of cell-cell adhesion. Cell 2010, 141, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Roura, S.; Miravet, S.; Piedra, J.; Garcia de Herreros, A.; Dunach, M. Regulation of E-cadherin/Catenin association by tyrosine phosphorylation. J. Biol. Chem. 1999, 274, 36734–36740. [Google Scholar] [CrossRef] [PubMed]
- Piedra, J.; Miravet, S.; Castano, J.; Palmer, H.G.; Heisterkamp, N.; Garcia de Herreros, A.; Dunach, M. p120 Catenin-associated Fer and Fyn tyrosine kinases regulate β-catenin Tyr-142 phosphorylation and β-catenin-α-catenin Interaction. Mol. Cell. Biol. 2003, 23, 2287–2297. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, M.; Ohkubo, T. Tyrosine phosphorylation of p120(ctn) in v-Src transfected L cells depends on its association with E-cadherin and reduces adhesion activity. J. Cell Sci. 2001, 114, 503–512. [Google Scholar] [PubMed]
- Vandenbroucke St Amant, E.; Tauseef, M.; Vogel, S.M.; Gao, X.P.; Mehta, D.; Komarova, Y.A.; Malik, A.B. PKCα activation of p120-catenin serine 879 phospho-switch disassembles VE-cadherin junctions and disrupts vascular integrity. Circ. Res. 2012, 111, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, P.; Fernandes, M.S.; Figueiredo, J.; Caldeira, J.; Carvalho, J.; Pinheiro, H.; Leite, M.; Melo, S.; Oliveira, P.; Simoes-Correia, J.; et al. E-cadherin dysfunction in gastric cancer--cellular consequences, clinical applications and open questions. FEBS Lett. 2012, 586, 2981–2989. [Google Scholar] [CrossRef] [PubMed]
- Brooks, S.A.; Lomax-Browne, H.J.; Carter, T.M.; Kinch, C.E.; Hall, D.M. Molecular interactions in cancer cell metastasis. Acta Histochem. 2010, 112, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Ki, H.; Park, H.S.; Kim, K. Presenilin-1 D257A and D385A mutants fail to cleave Notch in their endoproteolyzed forms, but only presenilin-1 D385A mutant can restore its γ-secretase activity with the compensatory overexpression of normal C-terminal fragment. J. Biol. Chem. 2005, 280, 22462–22472. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; He, Y.; Yang, I.; Zeng, Y.; Kim, Y.; Seo, Y.W.; Murnane, M.J.; Jung, C.; Lee, J.H.; Min, J.J.; et al. delta-Catenin promotes E-cadherin processing and activates β-catenin-mediated signaling: Implications on human prostate cancer progression. Biochim. Biophys. Acta 2012, 1822, 509–521. [Google Scholar] [CrossRef] [PubMed]







© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.-Y.; Yoon, S.; Sun, E.G.; Zhou, R.; Bae, J.A.; Seo, Y.-W.; Chae, J.-I.; Paik, M.-J.; Ha, H.-H.; Kim, H.; et al. Glycoprotein 90K Promotes E-Cadherin Degradation in a Cell Density-Dependent Manner via Dissociation of E-Cadherin–p120-Catenin Complex. Int. J. Mol. Sci. 2017, 18, 2601. https://doi.org/10.3390/ijms18122601
Park S-Y, Yoon S, Sun EG, Zhou R, Bae JA, Seo Y-W, Chae J-I, Paik M-J, Ha H-H, Kim H, et al. Glycoprotein 90K Promotes E-Cadherin Degradation in a Cell Density-Dependent Manner via Dissociation of E-Cadherin–p120-Catenin Complex. International Journal of Molecular Sciences. 2017; 18(12):2601. https://doi.org/10.3390/ijms18122601
Chicago/Turabian StylePark, So-Yeon, Somy Yoon, Eun Gene Sun, Rui Zhou, Jeong A. Bae, Young-Woo Seo, Jung-Il Chae, Man-Jeong Paik, Hyung-Ho Ha, Hangun Kim, and et al. 2017. "Glycoprotein 90K Promotes E-Cadherin Degradation in a Cell Density-Dependent Manner via Dissociation of E-Cadherin–p120-Catenin Complex" International Journal of Molecular Sciences 18, no. 12: 2601. https://doi.org/10.3390/ijms18122601