BRD9 Inhibition, Alone or in Combination with Cytostatic Compounds as a Therapeutic Approach in Rhabdoid Tumors

Abstract
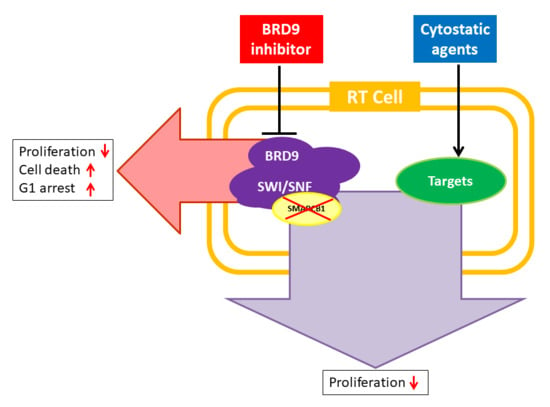
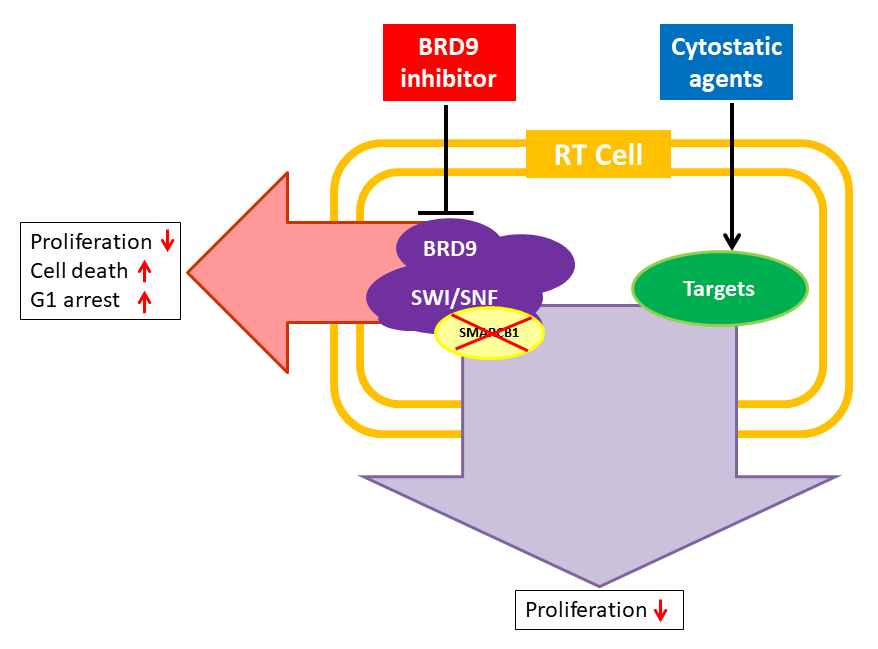
:
1. Introduction
2. Results
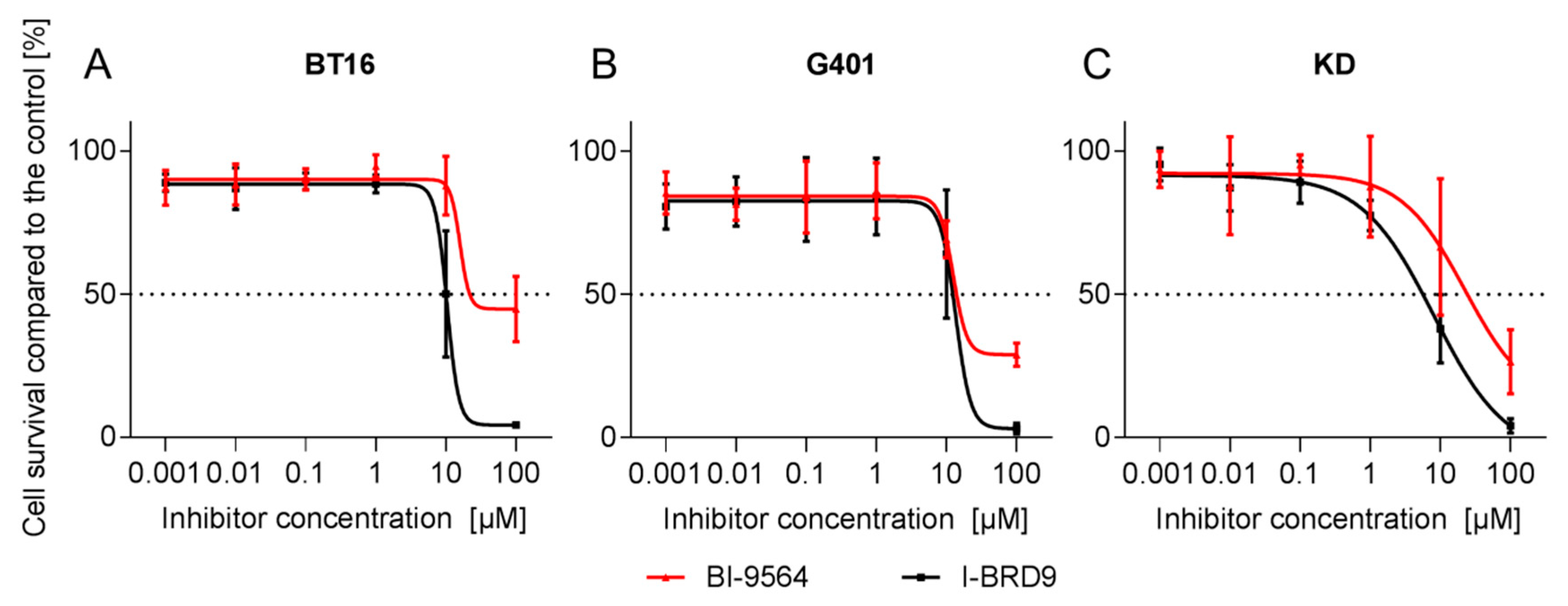
2.1. Small-Molecule BRD9 Inhibitors Decrease Rhabdoid Tumor Cell Proliferation In Vitro
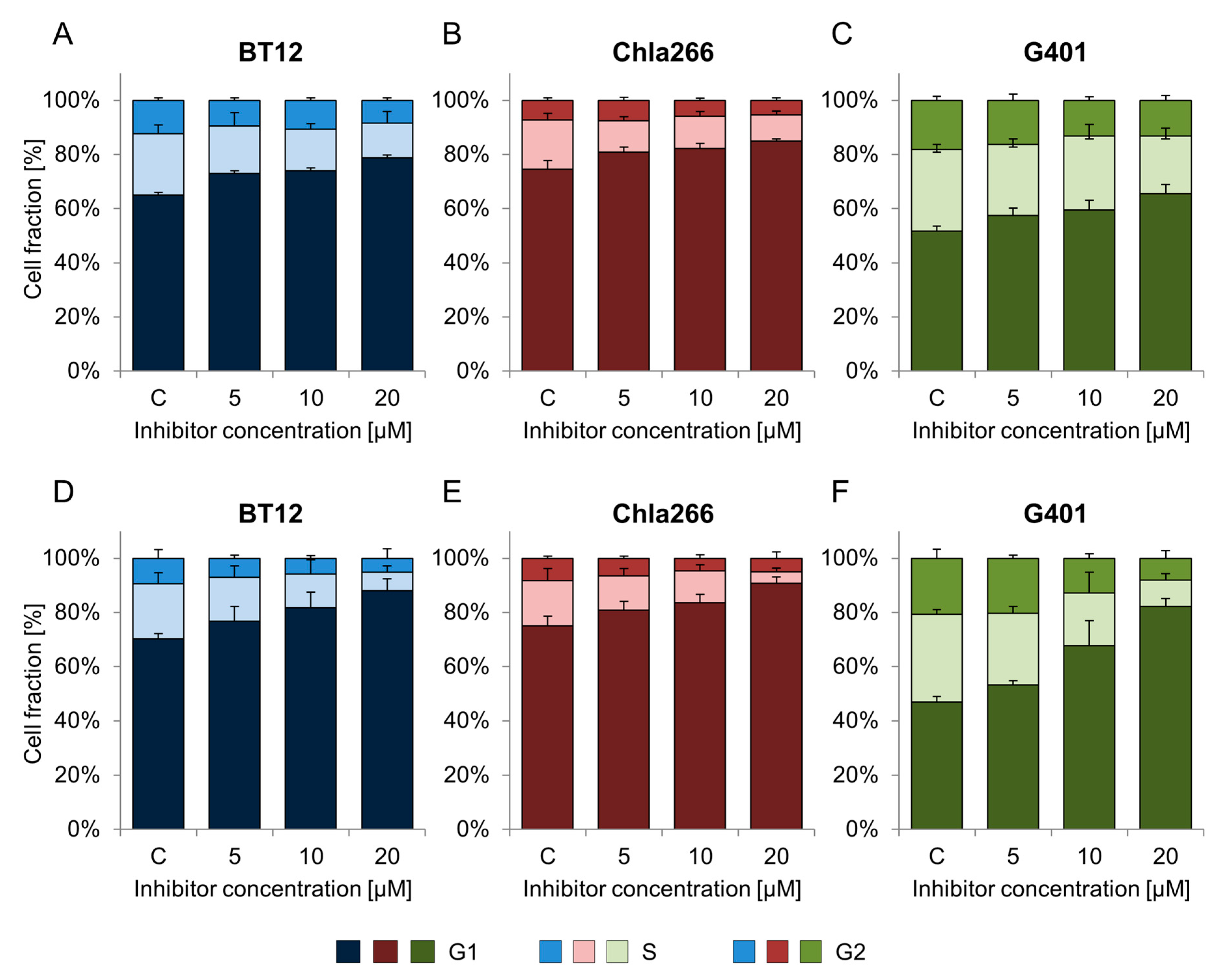
2.2. BRD9 Inhibitors Induce G1 Cell Cycle Arrest in RT Cell Lines
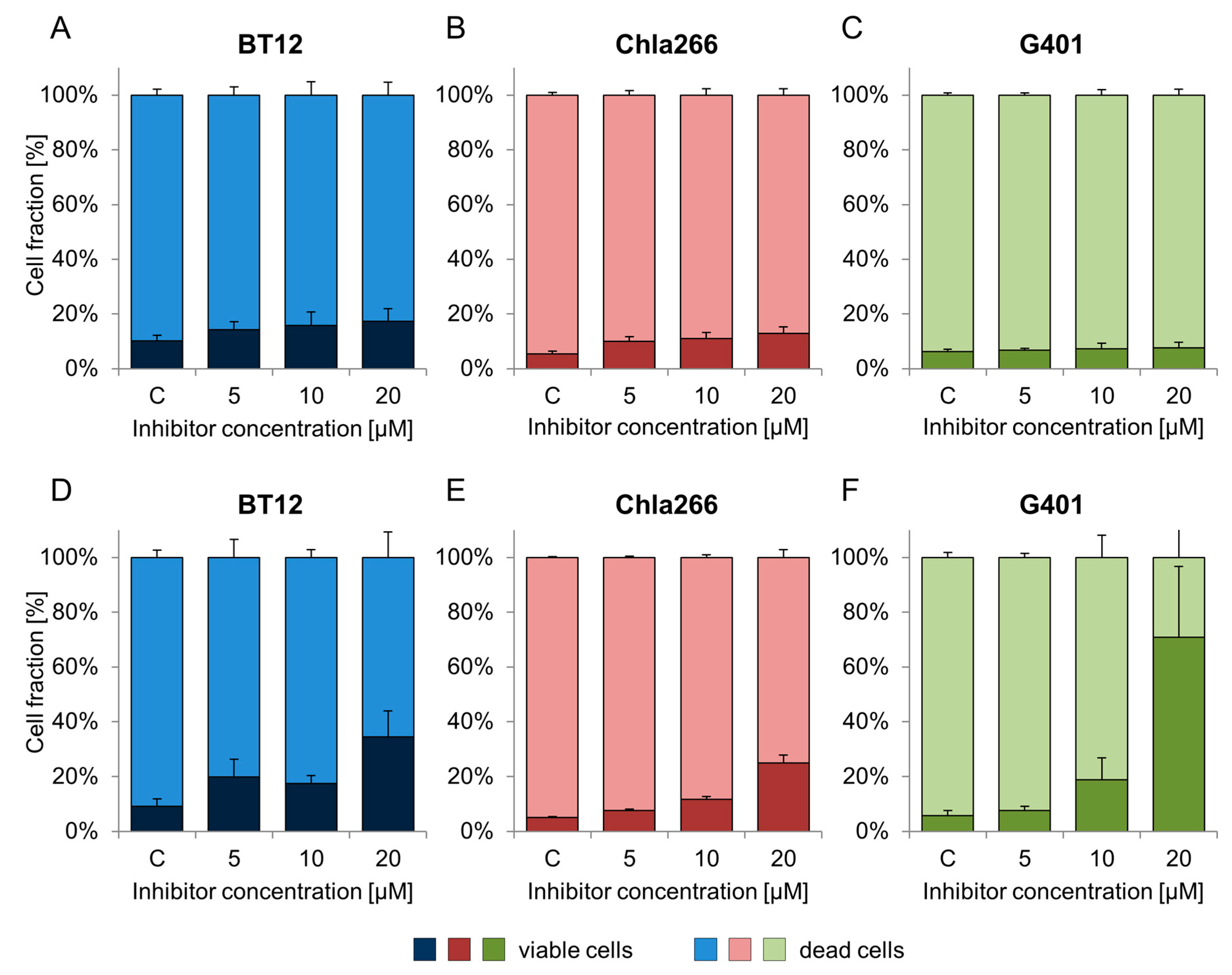
2.3. Treatment with BRD9 Inhibitors Reduces the Viability of RT Cells In Vitro
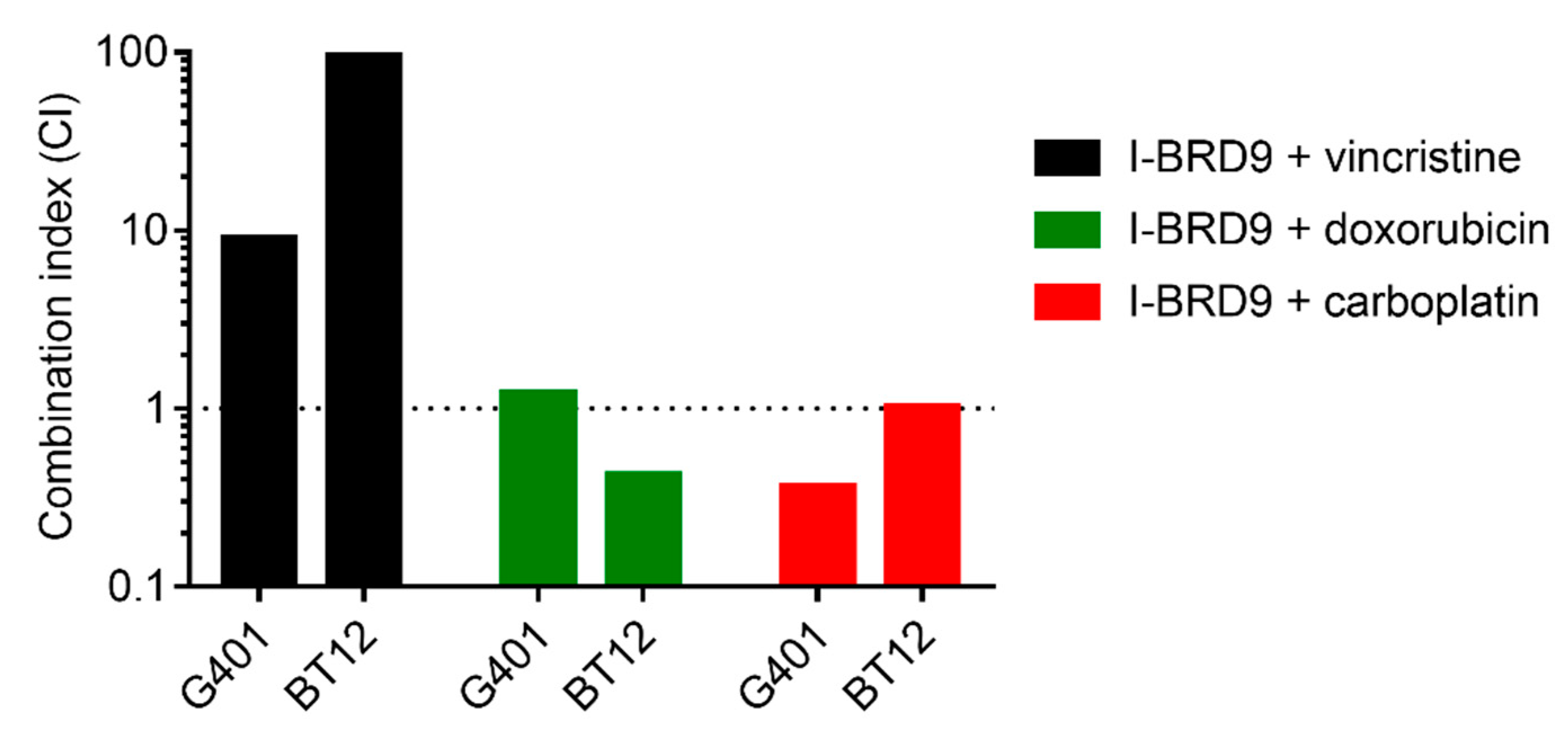
2.4. I-BRD9 Synergistically Inhibits RT Cell Growth in Combination with Carboplatin and Doxorubicin
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. BRD9 Inhibitors and Cytostatics
4.3. Cytotoxicity Assay
4.4. Apoptosis Assay and Cell Cycle Analysis
4.5. Statistical Analyses
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fossey, M.; Li, H.; Afzal, S.; Carret, A.-S.; Eisenstat, D.D.; Fleming, A.; Hukin, J.; Hawkins, C.; Jabado, N.; Johnston, D.; et al. Atypical teratoid rhabdoid tumor in the first year of life: The Canadian ATRT registry experience and review of the literature. J. Neurooncol. 2017, 132, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Schrey, D.; Carceller Lechón, F.; Malietzis, G.; Moreno, L.; Dufour, C.; Chi, S.; Lafay-Cousin, L.; von Hoff, K.; Athanasiou, T.; Marshall, L.V.; et al. Multimodal therapy in children and adolescents with newly diagnosed atypical teratoid rhabdoid tumor: Individual pooled data analysis and review of the literature. J. Neurooncol. 2016, 126, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Zaky, W.; Ji, G.D.L.; Haley, K.; Allen, J.; Atlas, M.; Bertolone, S.; Cornelius, A.; Gardner, S.; Patel, R.; Pradhan, K.; et al. Intensive Induction Chemotherapy Followed by Myeloablative Chemotherapy With Autologous Hematopoietic Progenitor Cell Rescue for Young Children Newly-Diagnosed With Central Nervous System Atypical Teratoid/Rhabdoid Tumors: The Head Start III Experience. Pediatr. Blood Cancer 2014, 61, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Versteege, I.; Sévenet, N.; Lange, J.; Rousseau-Merck, M.F.; Ambros, P.; Handgretinger, R.; Aurias, A.; Delattre, O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 1998, 394, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Biggs, P.J.; Garen, P.D.; Powers, J.M.; Garvin, A.J. Malignant rhabdoid tumor of the central nervous system. Hum. Pathol. 1987, 18, 332–337. [Google Scholar] [CrossRef]
- Johann, P.D.; Erkek, S.; Zapatka, M.; Kerl, K.; Buchhalter, I.; Hovestadt, V.; Jones, D.T.; Sturm, D.; Hermann, C.; Segura Wang, M.; et al. Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell 2016, 29, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Kerl, K.; Ries, D.; Unland, R.; Borchert, C.; Moreno, N.; Hasselblatt, M.; Jürgens, H.; Kool, M.; Görlich, D.; Eveslage, M.; et al. The histone deacetylase inhibitor SAHA acts in synergism with fenretinide and doxorubicin to control growth of rhabdoid tumor cells. BMC Cancer 2013, 13, 286. [Google Scholar] [CrossRef] [PubMed]
- Phelan, M.L.; Sif, S.; Narlikar, G.J.; Kingston, R.E. Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol. Cell 1999, 3, 247–253. [Google Scholar] [CrossRef]
- Hohmann, A.F.; Vakoc, C.R. A rationale to target the SWI/SNF complex for cancer therapy. Trends Genet. 2014, 30, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.W.M.; Orkin, S.H. The SWI/SNF complex—Chromatin and cancer. Nat. Rev. Cancer 2004, 4, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Pulice, J.L.; Kadoch, C. Composition and Function of Mammalian SWI/SNF Chromatin Remodeling Complexes in Human Disease. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Jagani, Z.; Mora-Blanco, E.L.; Sansam, C.G.; McKenna, E.S.; Wilson, B.; Chen, D.; Klekota, J.; Tamayo, P.; Nguyen, P.T.L.; Tolstorukov, M.; et al. Supplemental: Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nat. Med. 2010, 16, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Mora-Blanco, E.L.; Mishina, Y.; Tillman, E.J.; Cho, Y.-J.; Thom, C.S.; Pomeroy, S.L.; Shao, W.; Roberts, C.W.M. Activation of β-catenin/TCF targets following loss of the tumor suppressor SNF5. Oncogene 2014, 33, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.G.; Wang, X.; Shen, X.; McKenna, E.S.; Lemieux, M.E.; Cho, Y.-J.; Koellhoffer, E.C.; Pomeroy, S.L.; Orkin, S.H.; Roberts, C.W.M. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 2010, 18, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Barton, M.C. Bromodomain Histone Readers and Cancer. J. Mol. Biol. 2017, 429, 2003–2010. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.W. Small Molecule Bromodomain Inhibitors: Extending the Druggable Genome, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2012; Volume 51. [Google Scholar]
- Flynn, E.M.; Huang, O.W.; Poy, F.; Oppikofer, M.; Bellon, S.F.; Tang, Y.; Cochran, A.G. A Subset of Human Bromodomains Recognizes Butyryllysine and Crotonyllysine Histone Peptide Modifications. Structure 2015, 23, 1801–1814. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.H.; Allis, C.D. Roles of histone acetyltransferases and deacetylases in gene regulation. BioEssays 1998, 20, 615–626. [Google Scholar] [CrossRef]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, A.F.; Martin, L.J.; Minder, J.L.; Roe, J.-S.; Shi, J.; Steurer, S.; Bader, G.; McConnell, D.; Pearson, M.; Gerstberger, T.; et al. Sensitivity and engineered resistance of myeloid leukemia cells to BRD9 inhibition. Nat. Chem. Biol. 2016, 12, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-T.; Chen, S.-M.; Pan, L.-B.; Yao, J.; Ma, H.-T. Loss of function of SWI/SNF chromatin remodeling genes leads to genome instability of human lung cancer. Oncol. Rep. 2014, 33, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Xi, W.; Sansam, C.G.; Thom, C.S.; Metzger, D.; Evans, J.A.; Nguyen, P.T.L.; Roberts, C.W.M. Oncogenesis caused by loss of the SNF5 tumor suppressor is dependent on activity of BRG1, the ATPase of the SWI/SNF chromatin remodeling complex. Cancer Res. 2009, 69, 8094–8101. [Google Scholar]
- Wilson, B.G.; Roberts, C.W. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 2011, 11, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Koegl, M.; Bader, G.; Cockcroft, X.L.; Fedorov, O.; Fiegen, D.; Gerstberger, T.; Hofmann, M.H.; Hohmann, A.F.; Kessler, D.; et al. Structure-Based Design of an in Vivo Active Selective BRD9 Inhibitor. J. Med. Chem. 2016, 59, 4462–4475. [Google Scholar] [CrossRef] [PubMed]
- Theodoulou, N.H.; Bamborough, P.; Bannister, A.J.; Becher, I.; Bit, R.A.; Che, K.H.; Chung, C.; Dittmann, A.; Drewes, G.; Drewry, D.H.; et al. Discovery of I-BRD9, a Selective Cell Active Chemical Probe for Bromodomain Containing Protein 9 Inhibition. J. Med. Chem. 2016, 59, 1425–1439. [Google Scholar] [CrossRef] [PubMed]
- Bourdeaut, F.; Chi, S.N.; Frühwald, M.C. Rhabdoid tumors: Integrating biological insights with clinical success. A report from the SMARCB1 and Rhabdoid Tumor Symposium, Paris, 12–14 December 2013. Cancer Genet. 2014, 207, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Frühwald, M.C.; Biegel, J.A.; Bourdeaut, F.; Roberts, C.W.M.; Chi, S.N. Atypical teratoid/rhabdoid tumors—Current concepts, advances in biology, and potential future therapies. Neuro Oncol. 2016, 18, 764–778. [Google Scholar] [CrossRef] [PubMed]
- Sredni, S.T.; Halpern, A.L.; Hamm, C.A.; de Fátima Bonaldo, M.; Tomita, T. Histone deacetylases expression in atypical teratoid rhabdoid tumors. Child’s Nerv. Syst. 2013, 29, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Wöhrle, S.; Weiss, A.; Ito, M.; Kauffmann, A.; Murakami, M.; Jagani, Z.; Thuery, A.; Bauer-Probst, B.; Reimann, F.; Stamm, C.; et al. Fibroblast Growth Factor Receptors as Novel Therapeutic Targets in SNF5-Deleted Malignant Rhabdoid Tumors. PLoS ONE 2013, 8, e77652. [Google Scholar] [CrossRef] [PubMed]
- Geoerger, B.; Bourdeaut, F.; DuBois, S.G.; Fischer, M.; Geller, J.I.; Gottardo, N.G.; Marabelle, A.; Pearson, A.D.J.; Modak, S.; Cash, T.; et al. A Phase I Study of the CDK4/6 Inhibitor Ribociclib (LEE011) in Pediatric Patients with Malignant Rhabdoid Tumors, Neuroblastoma, and Other Solid Tumors. Clin. Cancer Res. 2017, 23, 2433–2441. [Google Scholar] [CrossRef] [PubMed]
- Kurmasheva, R.T.; Sammons, M.; Favours, E.; Wu, J.; Kurmashev, D.; Cosmopoulos, K.; Keilhack, H.; Klaus, C.R.; Houghton, P.J.; Smith, M.A. Initial testing (stage 1) of tazemetostat (EPZ-6438), a novel EZH2 inhibitor, by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 2017, 64, e26218. [Google Scholar] [CrossRef] [PubMed]
- Maris, J.M.; Morton, C.L.; Gorlick, R.; Kolb, E.A.; Lock, R.; Carol, H.; Keir, S.T.; Reynolds, C.P.; Kang, M.H.; Wu, J.; et al. Initial testing of the aurora kinase A inhibitor MLN8237 by the Pediatric Preclinical Testing Program (PPTP). Pediatr. Blood Cancer 2010, 55, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Sowa, M.E.; Ottinger, M.; Smith, J.A.; Shi, Y.; Harper, J.W.; Howley, P.M. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol. Cell. Biol. 2011, 31, 2641–2652. [Google Scholar] [CrossRef] [PubMed]
- Theodoulou, N.H.; Tomkinson, N.C.O.; Prinjha, R.K.; Humphreys, P.G. Progress in the Development of non-BET Bromodomain Chemical Probes. ChemMedChem 2016, 11, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Morel, D.; Almouzni, G.; Soria, J.-C.; Postel-Vinay, S. Targeting chromatin defects in selected solid tumors based on oncogene addiction, synthetic lethality and epigenetic antagonism. Ann. Oncol. 2016, 28, 254–269. [Google Scholar] [CrossRef] [PubMed]
- Bagnyukova, T.V.; Restifo, D.; Beeharry, N.; Gabitova, L.; Li, T.; Serebriiskii, I.G.; Golemis, E.A.; Astsaturov, I. DUSP6 regulates drug sensitivity by modulating DNA damage response. Br. J. Cancer 2013, 109, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Groom, L.A.; Sneddon, A.A.; Alessi, D.R.; Dowd, S.; Keyse, S.M. Differential regulation of the MAP, SAP and RK/p38 kinases by Pyst1, a novel cytosolic dual-specificity phosphatase. EMBO J. 1996, 15, 3621–3632. [Google Scholar] [PubMed]
- Song, H.; Wu, C.; Wei, C.; Li, D.; Hua, K.; Song, J.; Xu, H.; Chen, L.; Fang, L. Silencing of DUSP6 gene by RNAi-mediation inhibits proliferation and growth in MDA-MB-231 breast cancer cells: An in vitro study. Int. J. Clin. Exp. Med. 2015, 8, 10481–10490. [Google Scholar] [PubMed]
- Olvedy, M.; Tisserand, J.C.; Luciani, F.; Boeckx, B.; Wouters, J.; Lopez, S.; Rambow, F.; Aibar, S.; Thienpont, B.; Barra, J.; et al. Comparative oncogenomics identifies tyrosine kinase FES as a tumor suppressor in melanoma. J. Clin. Investig. 2017, 127, 2310–2325. [Google Scholar] [CrossRef] [PubMed]
- Condorelli, F.; Stec-Martyna, E.; Zaborowska, J.; Felli, L.; Gnemmi, I.; Ponassi, M.; Rosano, C. Role of the Non-Receptor Tyrosine Kinase Fes in Cancer. Curr. Med. Chem. 2011, 18, 2913–2920. [Google Scholar] [CrossRef] [PubMed]
- Craig, A.W.B. FES/FER kinase signaling in hematopoietic cells and leukemias. Front. Biosci. 2012, 17, 861–875. [Google Scholar] [CrossRef]
- Noll, J.E.; Hewett, D.R.; Williams, S.A.; Vandyke, K.; Kok, C.; To, L.B.; Zannettino, A.C.W. SAMSN1 Is a Tumor Suppressor Gene in Multiple Myeloma. Neoplasia 2014, 16, 572–585. [Google Scholar] [CrossRef] [PubMed]
- Brandt, S.; Ellwanger, K.; Beuter-Gunia, C.; Schuster, M.; Hausser, A.; Schmitz, I.; Beer-Hammer, S. SLy2 targets the nuclear SAP30/HDAC1 complex. Int. J. Biochem. Cell Biol. 2010, 42, 1472–1481. [Google Scholar] [CrossRef] [PubMed]
- Kanda, M.; Shimizu, D.; Sueoka, S.; Nomoto, S.; Oya, H.; Takami, H.; Ezaka, K.; Hashimoto, R.; Tanaka, Y.; Kobayashi, D.; et al. Prognostic relevance of SAMSN1 expression in gastric cancer. Oncol. Lett. 2016, 12, 4708–4716. [Google Scholar] [CrossRef] [PubMed]
- Sueoka, S.; Kanda, M.; Sugimoto, H.; Shimizu, D.; Nomoto, S.; Oya, H.; Takami, H.; Ezaka, K.; Hashimoto, R.; Tanaka, Y.; et al. Suppression of SAMSN1 Expression is Associated with the Malignant Phenotype of Hepatocellular Carcinoma. Ann. Surg. Oncol. 2015, 22, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Stojanova, A.; Tu, W.B.; Ponzielli, R.; Kotlyar, M.; Chan, P.K.; Boutros, P.C.; Khosravi, F.; Jurisica, I.; Raught, B.; Penn, L.Z. MYC interaction with the tumor suppressive SWI/SNF complex member INI1 regulates transcription and cellular transformation. Cell Cycle 2016, 15, 1693–1705. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.; Eastman, A. Microtubule destabilising agents: Far more than just antimitotic anticancer drugs. Br. J. Clin. Pharmacol. 2016, 83, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Cappetta, D.; Rossi, F.; Piegari, E.; Quaini, F.; Berrino, L.; Urbanek, K.; De Angelis, A. Doxorubicin targets multiple players: A new view of an old problem. Pharmacol. Res. 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Freeman, M.D.; Mazu, T.; Miles, J.S.; Darling-Reed, S.; Flores-Rozas, H. Inactivation of chromatin remodeling factors sensitizes cells to selective cytotoxic stress. Biol. Targets Ther. 2014, 8, 269–280. [Google Scholar]
- Flores-Rozas, H.; Freeman, M.; Mazu, T.; Miles, J.; Darling-Reed, S. Inactivation of chromatin remodeling factors sensitizes cells to selective cytotoxic stress. Biol. Targets Ther. 2014, 8, 269. [Google Scholar] [CrossRef] [PubMed]
- Dilruba, S.; Kalayda, G.V. Platinum-based drugs: Past, present and future. Cancer Chemother. Pharmacol. 2016, 77, 1103–1124. [Google Scholar] [CrossRef] [PubMed]
- Kothandapani, A.; Gopalakrishnan, K.; Kahali, B.; Reisman, D.; Patrick, S.M. Downregulation of SWI/SNF chromatin remodeling factor subunits modulates cisplatin cytotoxicity. Exp. Cell Res. 2012, 318, 1973–1986. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (µM) | BI-9564 | I-BRD9 | ||
|---|---|---|---|---|
| Cell Line | 72 h | 144 h | 72 h | 144 h |
| BT12 | >100 | 17.2 | 21.2 | 8.2 |
| BT16 | 15.9 | - | 11.2 | - |
| Chla266 | >100 | 33.7 | 13.3 | 24.7 |
| G401 | 12.5 | 10.8 | 13.4 | 6.1 |
| KD | 22.3 | 10.8 | 8.1 | 7.1 |
| Treatment | Cell Line | |||
|---|---|---|---|---|
| BT12 | Chla266 | G401 | ||
| BI-9564 | C | 65.0 ± 3.9 | 74.1 ± 3.2 | 51.7 ± 2.0 |
| 5 µM | 73.1 ± 4.6 | 80.0 ± 1.8 | 57.6 ± 2.6 | |
| 10 µM | 74.0 ± 3.4 | 81.2 ± 1.8 * | 59.5 ± 3.6 * | |
| 20 µM | 78.9 ± 6.3 * | 83.6 ± 0.9 * | 65.5 ± 3.3 * | |
| I-BRD9 | C | 69.0 ± 1.9 | 74.2 ± 3.6 | 46.7 ± 1.9 |
| 5 µM | 75.0 ± 5.3 | 79.5 ± 3.2 | 53.0 ± 1.6 | |
| 10 µM | 80.1 ± 5.8 | 82.7 ± 2.9 * | 67.1 ± 9.1 * | |
| 20 µM | 85.6 ± 4.3 * | 89.3 ± 2.5 * | 78.1 ± 2.7 * | |
| Treatment | Cell Line | |||
|---|---|---|---|---|
| BT12 | Chla266 | G401 | ||
| BI-9564 | C | 10.1 ± 2.2 | 5.4 ± 1.0 | 6.2 ± 0.8 |
| 5 µM | 14.2 ±3.0 | 10.0 ± 1.7 * | 6.8 ± 0.8 | |
| 10 µM | 15.9 ± 4.9 | 11.0 ± 2.3 * | 7.3 ± 2.0 | |
| 20 µM | 17.3 ± 4.7 | 12.9 ± 2.4 * | 7.6 ± 2.1 | |
| I-BRD9 | C | 9.1 ± 2.8 | 5.0 ± 0.3 | 5.7 ± 1.9 |
| 5 µM | 19.9 ± 6.6 | 7.7 ± 0.5 | 7.7 ± 1.5 | |
| 10 µM | 17.6 ± 2.8 | 11.7 ± 1.0 * | 18.8 ± 8.1 | |
| 20 µM | 34.6 ± 9.4 * | 25.0 ± 2.9 * | 70.8 ± 26.0 * | |
| Treatment | BT12 | G401 | ||||
|---|---|---|---|---|---|---|
| IC50 (µM) | CI | R2 | IC50 (µM) | CI | R2 | |
| Carboplatin + I-BRD9 | 40.9 | 1.01 | 0.83 | 9.1 | 0.37 | 0.94 |
| Doxorubicin + I-BRD9 | 0.94 | 0.42 | 0.94 | 0.096 | 1.2 | 0.84 |
| Vincristine + I-BRD9 | 16.7 | 96.1 | 0.59 | 0.008 | 9.0 | 0.88 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krämer, K.F.; Moreno, N.; Frühwald, M.C.; Kerl, K. BRD9 Inhibition, Alone or in Combination with Cytostatic Compounds as a Therapeutic Approach in Rhabdoid Tumors. Int. J. Mol. Sci. 2017, 18, 1537. https://doi.org/10.3390/ijms18071537
Krämer KF, Moreno N, Frühwald MC, Kerl K. BRD9 Inhibition, Alone or in Combination with Cytostatic Compounds as a Therapeutic Approach in Rhabdoid Tumors. International Journal of Molecular Sciences. 2017; 18(7):1537. https://doi.org/10.3390/ijms18071537
Chicago/Turabian StyleKrämer, Katja F., Natalia Moreno, Michael C. Frühwald, and Kornelius Kerl. 2017. "BRD9 Inhibition, Alone or in Combination with Cytostatic Compounds as a Therapeutic Approach in Rhabdoid Tumors" International Journal of Molecular Sciences 18, no. 7: 1537. https://doi.org/10.3390/ijms18071537