
Prediction of Toxin Genes from Chinese Yellow Catfish Based on Transcriptomic and Proteomic Sequencing

 ,
,
Abstract
:
1. Introduction
2. Results and Discussion
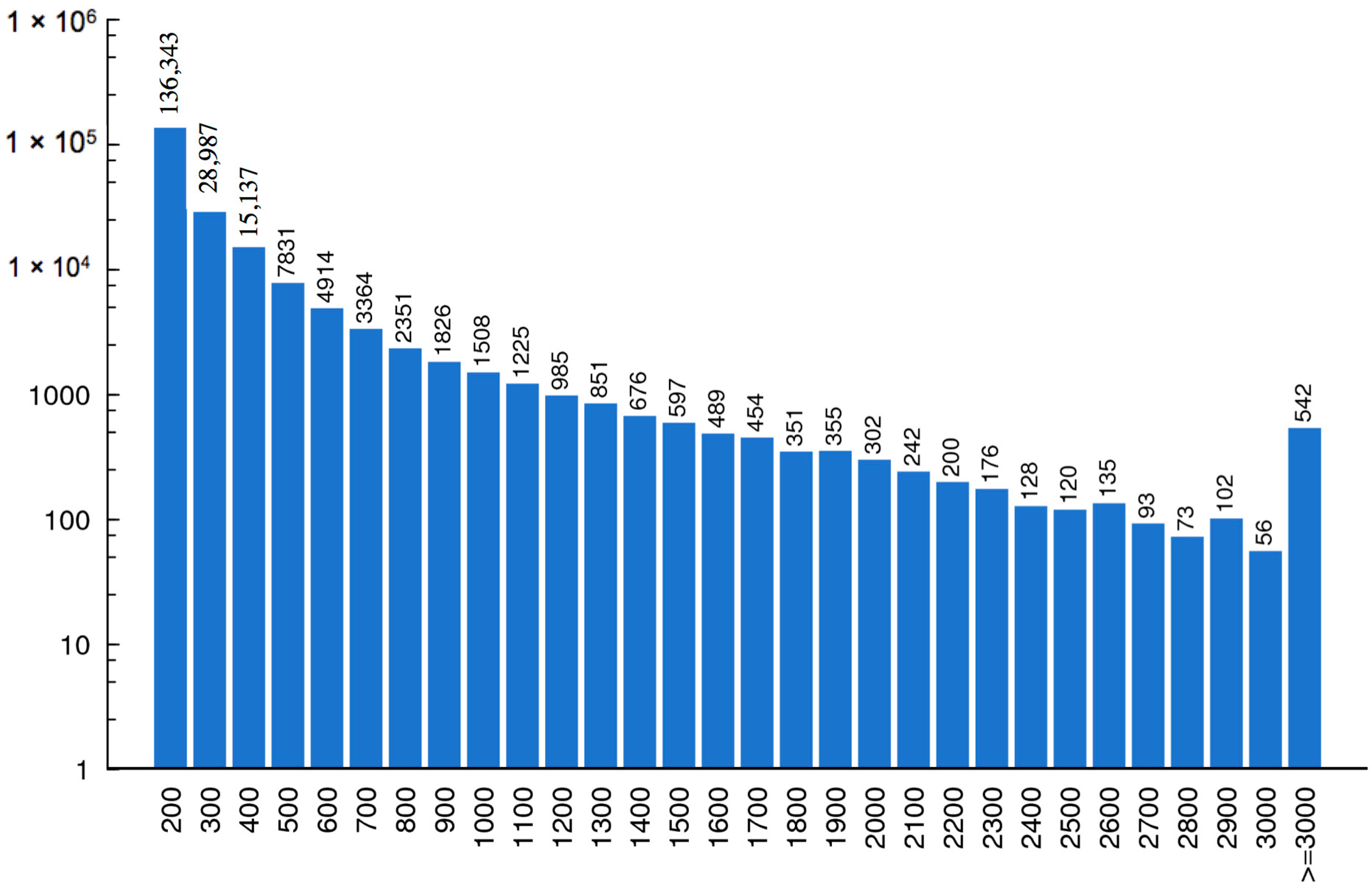
2.1. LC-MS/MS Data Analysis
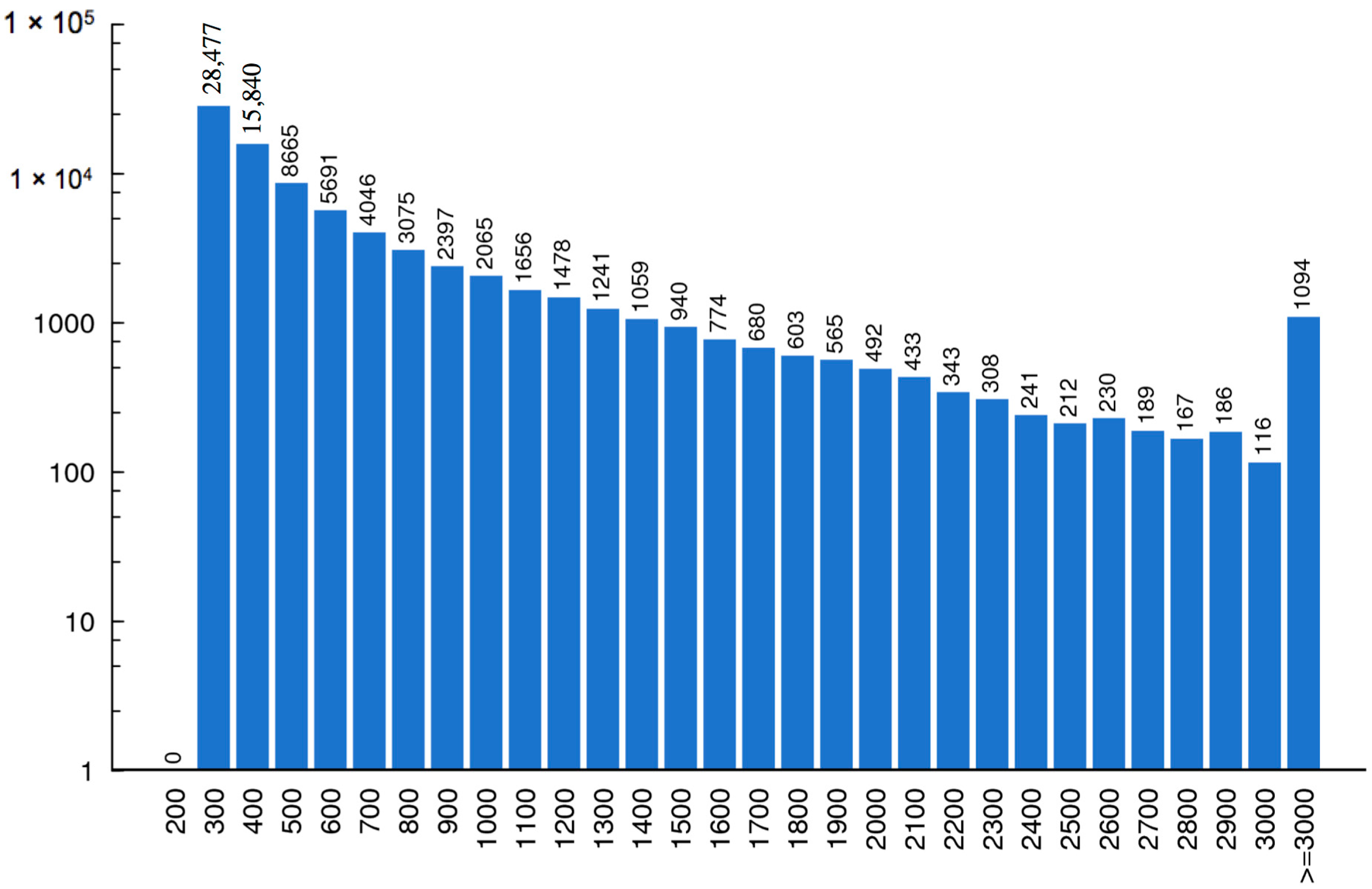
2.2. Assembly and ORF Prediction
2.3. Construction of Toxin Database
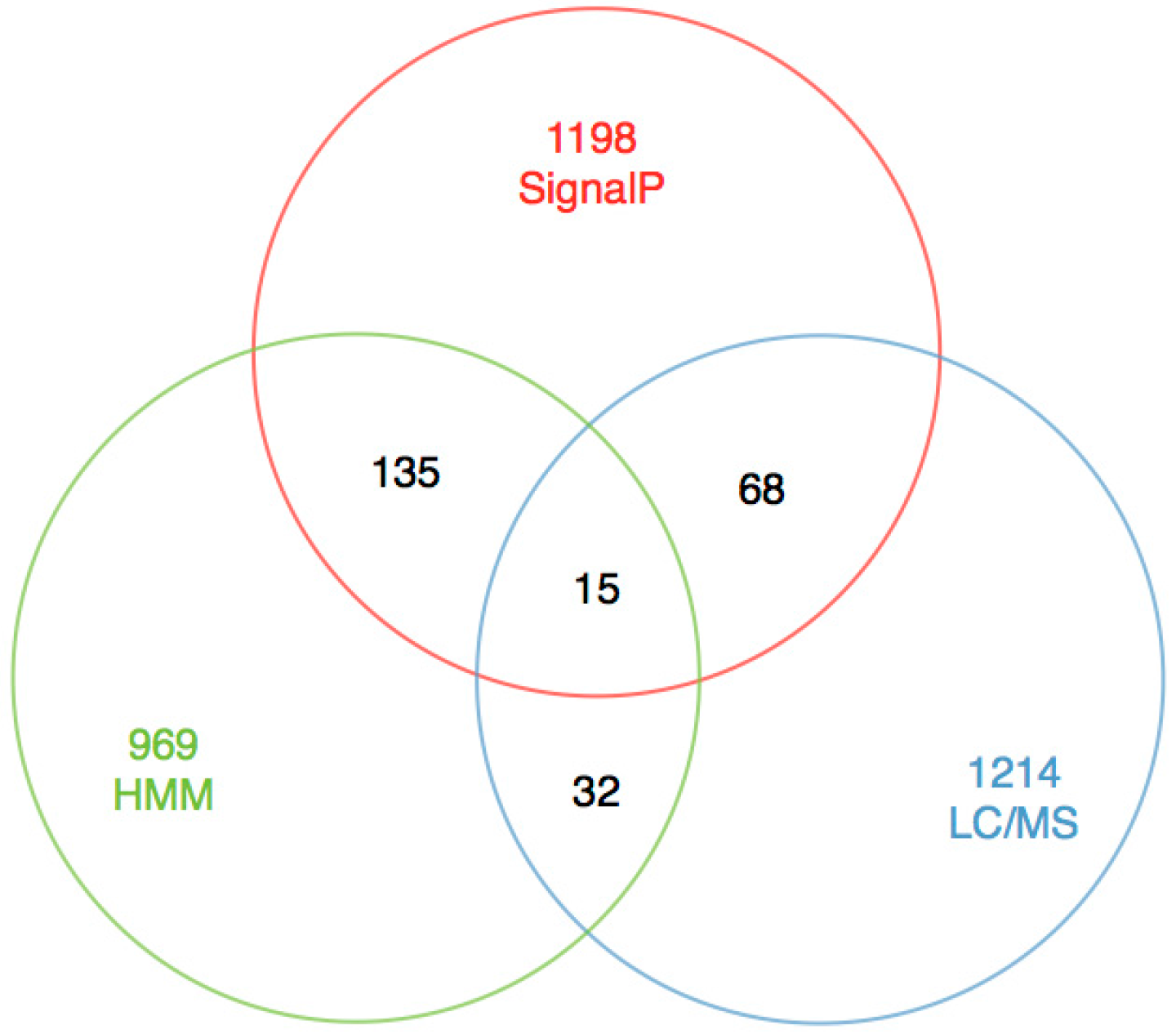
2.4. Excavation of Toxin Genes
2.5. Toxin Classification
2.5.1. Veficolin
2.5.2. Ink Toxins
2.5.3. Adamalysin
2.5.4. Zn-α2-Glycoprotein
2.5.5. Cysteine-Rich Secretory Proteins Toxin (CRISP)
2.6. Verification
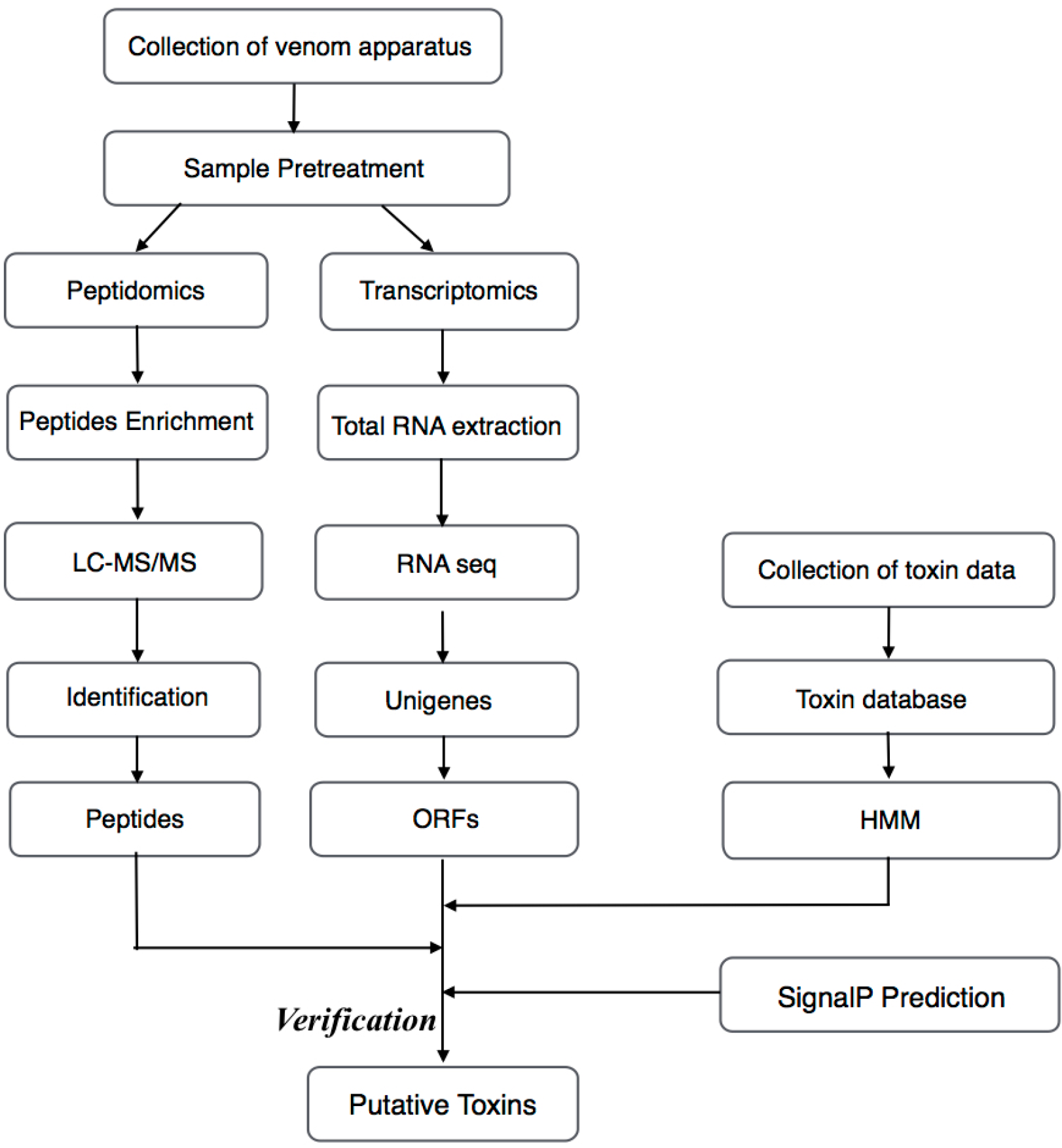
3. Materials and Methods
3.1. Specimens
3.2. MS
3.2.1. Venom Sample Preparation
3.2.2. Nano LC-MS/MS Analysis
3.3. RNA-Seq
3.4. RT-PCR
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fry, B.G.; Roelants, K.; Winter, K.; Hodgson, W.C.; Griesman, L.; Kwok, H.F.; Scanlon, D.; Karas, J.; Shaw, C.; Wong, L.; et al. Novel venom proteins produced by differential domain-expression strategies in beaded lizards and gila monsters (genus heloderma). Mol. Biol. Evol. 2010, 27, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.L.; Wheeler, W.C. Venom evolution widespread in fish: A phylogenetic road map for the bioprospecting of piscine venoms. J. Hered. 2006, 97, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.T.; Khan, A.M.; Brusic, V. Bioinformatics for venom and toxin sciences. Brief. Bioinform. 2003, 4, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Church, J.E.; Hodgson, W.C. The pharmacological activity of fish venoms. Toxicon 2002, 40, 1083–1093. [Google Scholar] [CrossRef]
- Bringans, S.; Eriksen, S.; Kendrick, T.; Gopalakrishnakone, P.; Livk, A.; Lock, R.; Lipscombe, R. Proteomic analysis of the venom of Heterometrus longimanus (Asian black scorpion). Proteomics 2008, 8, 1081–1096. [Google Scholar] [CrossRef] [PubMed]
- Menschaert, G.; Vandekerckhove, T.T.; Baggerman, G.; Schoofs, L.; Luyten, W.; van Criekinge, W. Peptidomics coming of age: A review of contributions from a bioinformatics angle. J. Proteome Res. 2010, 9, 2051–2061. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Leung, H.C.; Yiu, S.-M.; Chin, F.Y. Meta-IDBA: A de novo assembler for metagenomic data. Bioinformatics 2011, 27, i94–i101. [Google Scholar] [CrossRef] [PubMed]
- Prashanth, J.R.; Lewis, R.J.; Dutertre, S. Towards an integrated venomics approach for accelerated conopeptide discovery. Toxicon 2012, 60, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Elias, J.E.; Gygi, S.P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 2007, 4, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Brosch, M.; Yu, L.; Hubbard, T.; Choudhary, J. Accurate and sensitive peptide identification with mascot percolator. J. Proteome Res. 2009, 8, 3176–3181. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Trinity: Reconstructing a full-length transcriptome without a genome from rna-seq data. Nat. Biotechnol. 2013, 29, 644. [Google Scholar] [CrossRef] [PubMed]
- Klassen, J.L.; Currie, C.R. Orfcor: Identifying and accommodating orf prediction inconsistencies for phylogenetic analysis. PLoS ONE 2013, 8, e58387. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. Uniprot: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar]
- Jungo, F.; Bougueleret, L.; Xenarios, I.; Poux, S. The uniprotkb/Swiss-prot tox-prot program: A central hub of integrated venom protein data. Toxicon 2012, 60, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Kaas, Q.; Yu, R.; Jin, A.-H.; Dutertre, S.; Craik, D.J. Conoserver: Updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- He, Q.-Y.; He, Q.-Z.; Deng, X.-C.; Yao, L.; Meng, E.; Liu, Z.-H.; Liang, S.-P. ATDB: A uni-database platform for animal toxins. Nucleic Acids Res. 2008, 36, D293–D297. [Google Scholar] [CrossRef] [PubMed]
- Borry, P.; Fryns, J.P.; Schotsmans, P.; Dierickx, K. Attitudes towards carrier testing in minors: A systematic review. Genet. Couns. 2005, 16, 341–352. [Google Scholar] [PubMed]
- Herzig, V.; Wood, D.L.; Newell, F.; Chaumeil, P.-A.; Kaas, Q.; Binford, G.J.; Nicholson, G.M.; Gorse, D.; King, G.F. Arachnoserver 2.0, an updated online resource for spider toxin sequences and structures. Nucleic Acids Res. 2010, 39, D653–D657. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J. Blat—The blast-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped blast and psi-blast: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Eddy, S.R. Hmmer web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed]
- Fry, B.G. From genome to “venome”: Molecular origin and evolution of the snake venom proteome inferred from phylogenetic analysis of toxin sequences and related body proteins. Genome Res. 2005, 15, 403–420. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Schmidt, B.; Maskell, D.L. Msaprobs: Multiple sequence alignment based on pair hidden markov models and partition function posterior probabilities. Bioinformatics 2010, 26, 1958–1964. [Google Scholar] [CrossRef] [PubMed]
- Foppa, I.M.; Cheng, P.Y.; Reynolds, S.B.; Shay, D.K.; Carias, C.; Bresee, J.S.; Kim, I.K.; Gambhir, M.; Fry, A.M. Deaths averted by influenza vaccination in the U.S. during the seasons 2005/06 through 2013/14. Vaccine 2015, 33, 3003–3009. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.A.; Wang, Z. Next-generation transcriptome assembly. Nat. Rev. Genet. 2011, 12, 671–682. [Google Scholar] [CrossRef] [PubMed]
- OmPraba, G.; Chapeaurouge, A.; Doley, R.; Devi, K.R.; Padmanaban, P.; Venkatraman, C.; Velmurugan, D.; Lin, Q.; Kini, R.M. Identification of a novel family of snake venom proteins veficolins from Cerberus rynchops using a venom gland transcriptomics and proteomics approach. J. Proteome Res. 2010, 9, 1882–1893. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, H.; Muramoto, K.; Yamazaki, M. Aplysianin-A, an antibacterial and antineoplastic glycoprotein in the albumen gland of a sea hare, aplysia kurodai. Experientia 1986, 42, 1065–1067. [Google Scholar] [CrossRef] [PubMed]
- Kisugi, J.; Ohye, H.; Kamiya, H.; Yamazaki, M. Biopolymers from marine invertebrates. X. Mode of action of an antibacterial glycoprotein, aplysianin E, from eggs of a sea hare, Aplysia kurodai. Chem. Pharm. Bull. 1989, 37, 3050–3053. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Morita, T. Structure and function of snake venom cysteine-rich secretory proteins. Toxicon 2004, 44, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Butzke, D.; Machuy, N.; Thiede, B.; Hurwitz, R.; Goedert, S.; Rudel, T. Hydrogen peroxide produced by aplysia ink toxin kills tumor cells independent of apoptosis via peroxiredoxin I sensitive pathways. Cell Death Differ. 2004, 11, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Butzke, D.; Hurwitz, R.; Thiede, B.; Goedert, S.; Rudel, T. Cloning and biochemical characterization of apit, a new l-amino acid oxidase from aplysia punctata. Toxicon 2005, 46, 479. [Google Scholar] [CrossRef] [PubMed]
- Kurecki, T.; Laskowski, M.S.; Kress, L. Purification and some properties of two proteinases from Crotalus adamanteus venom that inactivate human α 1-proteinase inhibitor. J. Biol. Chem. 1978, 253, 8340–8345. [Google Scholar] [PubMed]
- Bode, W.; Gomis-Rüth, F.-X.; Stöckler, W. Astacins, serralysins, snake venom and matrix metalloproteinases exhibit identical zinc-binding environments (hexxhxxgxxh and Met-turn) and topologies and should be grouped into a common family, the “metzincins”. FEBS Lett. 1993, 331, 134–140. [Google Scholar] [CrossRef]
- Stöcker, W.; Grams, F.; Reinemer, P.; Bode, W.; Baumann, U.; Gomis-Rüth, F.X.; Mckay, D.B. The metzincins—Topological and sequential relations between the astacins, adamalysins, serralysins, and matrixins (collagenases) define a super family of zinc-peptidases. Protein Sci. 1995, 4, 823–840. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.I.; Waheed, A.; Yadav, S.; Singh, T.P.; Ahmad, F. Zinc α2-glycoprotein: A multidisciplinary protein. Mol. Cancer Res. 2008, 6, 892–906. [Google Scholar] [CrossRef] [PubMed]
- Albertus, D.L.; Seder, C.W.; Chen, G.; Wang, X.; Hartojo, W.; Lin, L.; Silvers, A.; Thomas, D.G.; Giordano, T.J.; Chang, A.C. AZGP1 autoantibody predicts survival and histone deacetylase inhibitors increase expression in lung adenocarcinoma. J. Thorac. Oncol. 2008, 3, 1236–1244. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Okazaki, R.; Shibata, M.; Hasegawa, Y.; Satoh, K.; Tajima, T.; Takeuchi, Y.; Fujita, T.; Nakahara, K.; Yamashita, T. Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. J. Clin. Endocrinol. Metab. 2002, 87, 4957–4960. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Tolić, N.; Liu, T.; Zhao, R.; Petritis, B.O.; Gritsenko, M.A.; Camp, D.G.; Moore, R.J.; Purvine, S.O.; Esteva, F.J. Blood peptidome-degradome profile of breast cancer. PLoS ONE 2010, 5, e13133. [Google Scholar] [CrossRef] [PubMed]
- Zelanis, A.; Tashima, A.K.; Pinto, A.F.; Paes Leme, A.F.; Stuginski, D.R.; Furtado, M.F.; Sherman, N.E.; Ho, P.L.; Fox, J.W.; Serrano, S.M. Bothrops Jararaca venom proteome rearrangement upon neonate to adult transition. Proteomics 2011, 11, 4218–4228. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Cao, R.; Jiang, L.; Liang, S. A combined de novo protein sequencing and cDNA library approach to the venomic analysis of Chinese spider Araneus ventricosus. J. Proteom. 2013, 78, 416–427. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group of Species | Taxonomy Name | Numbers of Sequences |
|---|---|---|
| Snakes | Serpents | 1406 |
| Scorpions | Scorpions | 1510 |
| Spiders | Araneae | 1047 |
| Cone snails | Conus | 3860 |
| Sea anemones | Actiniaria | 308 |
| Insects | Hexapoda | 162 |
| Fish | Teleostei | 31 |
| Mammals | Mammalias | 106 |
| Lizards | Heloderma | 241 |
| Jellyfish | Cubomedusae/Scyphozoa | 175 |
| Sea stars | Asteroidea | 8 |
| Hydra | Hydroida | 14 |
| Worms | Cerebratulus | 5 |
| Forg ,Toad | Amphibia | 64 |
| Sea-urchin | Echinoidea | 2 |
| Sea hare | Aplysia | 22 |
| Annelida | 11 | |
| Scolopendra | Myriapoda | 9 |
| All | Metazoa | 8863 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, B.; Li, X.; Lin, Z.; Ruan, Z.; Wang, M.; Liu, J.; Tong, T.; Li, J.; Huang, Y.; Wen, B.; et al. Prediction of Toxin Genes from Chinese Yellow Catfish Based on Transcriptomic and Proteomic Sequencing. Int. J. Mol. Sci. 2016, 17, 556. https://doi.org/10.3390/ijms17040556
Xie B, Li X, Lin Z, Ruan Z, Wang M, Liu J, Tong T, Li J, Huang Y, Wen B, et al. Prediction of Toxin Genes from Chinese Yellow Catfish Based on Transcriptomic and Proteomic Sequencing. International Journal of Molecular Sciences. 2016; 17(4):556. https://doi.org/10.3390/ijms17040556
Chicago/Turabian StyleXie, Bing, Xiaofeng Li, Zhilong Lin, Zhiqiang Ruan, Min Wang, Jie Liu, Ting Tong, Jia Li, Yu Huang, Bo Wen, and et al. 2016. "Prediction of Toxin Genes from Chinese Yellow Catfish Based on Transcriptomic and Proteomic Sequencing" International Journal of Molecular Sciences 17, no. 4: 556. https://doi.org/10.3390/ijms17040556