
Combining miRNA and mRNA Expression Profiles in Wilms Tumor Subtypes

 and
and
Abstract
:1. Introduction
2. Results
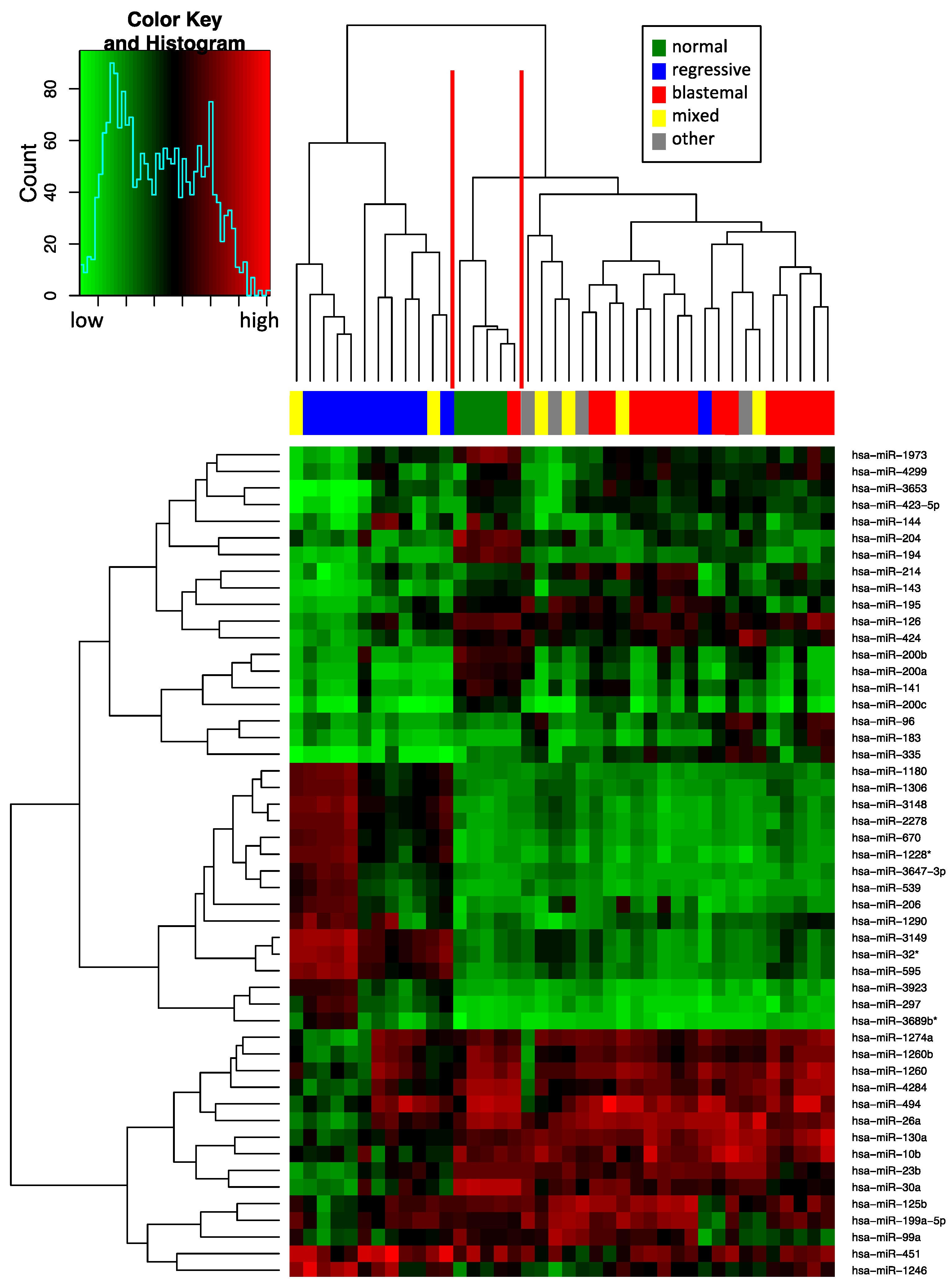
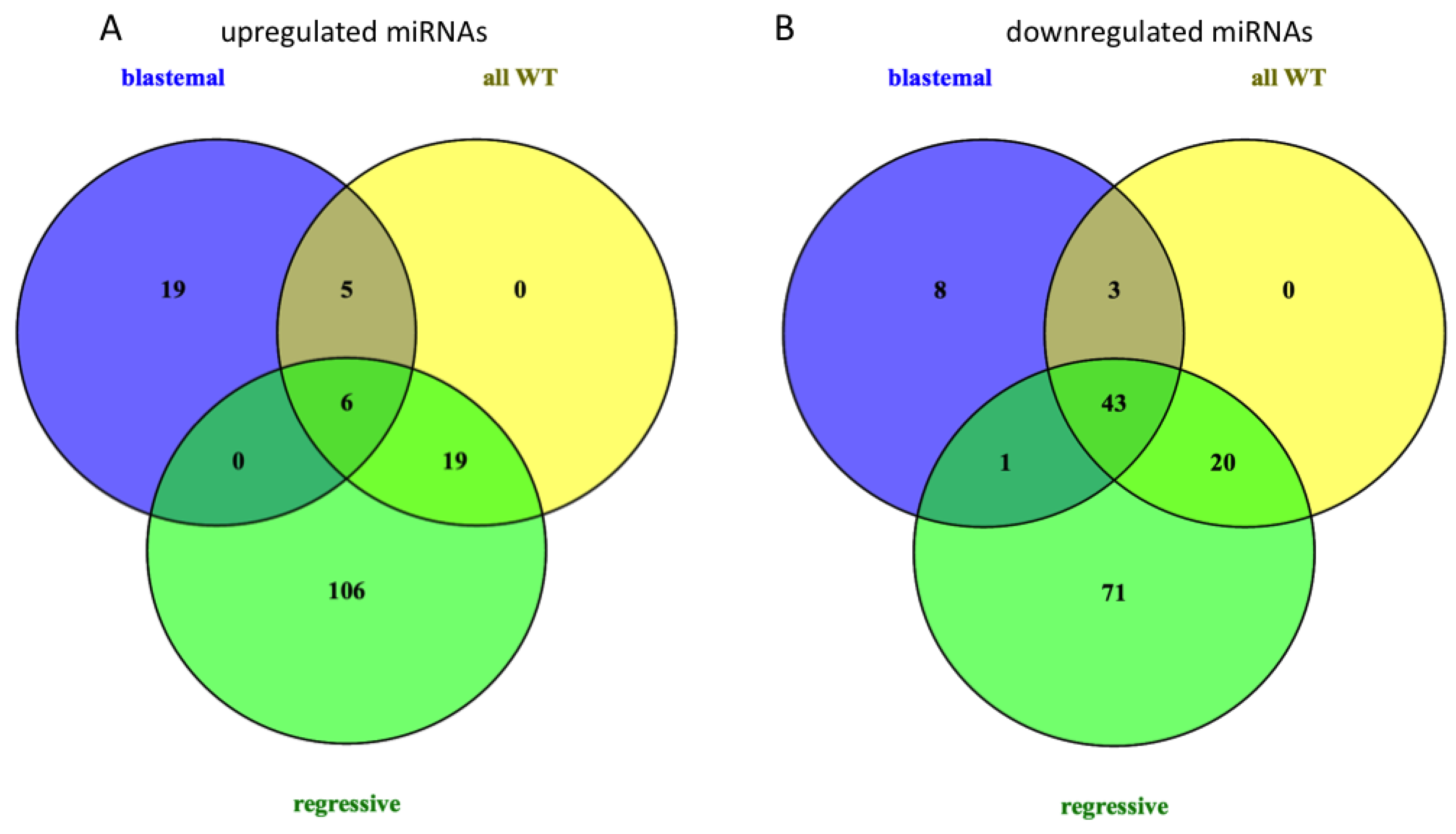
2.1. Deregulated miRNA Expression in Wilms Tumor (WT) Compared to Normal Tissue
2.2. Reverse Transcription Quantitative Realtime PCR (RT-qPCR) Validation of the miRNA Expression in WT Compared to Normal Tissue
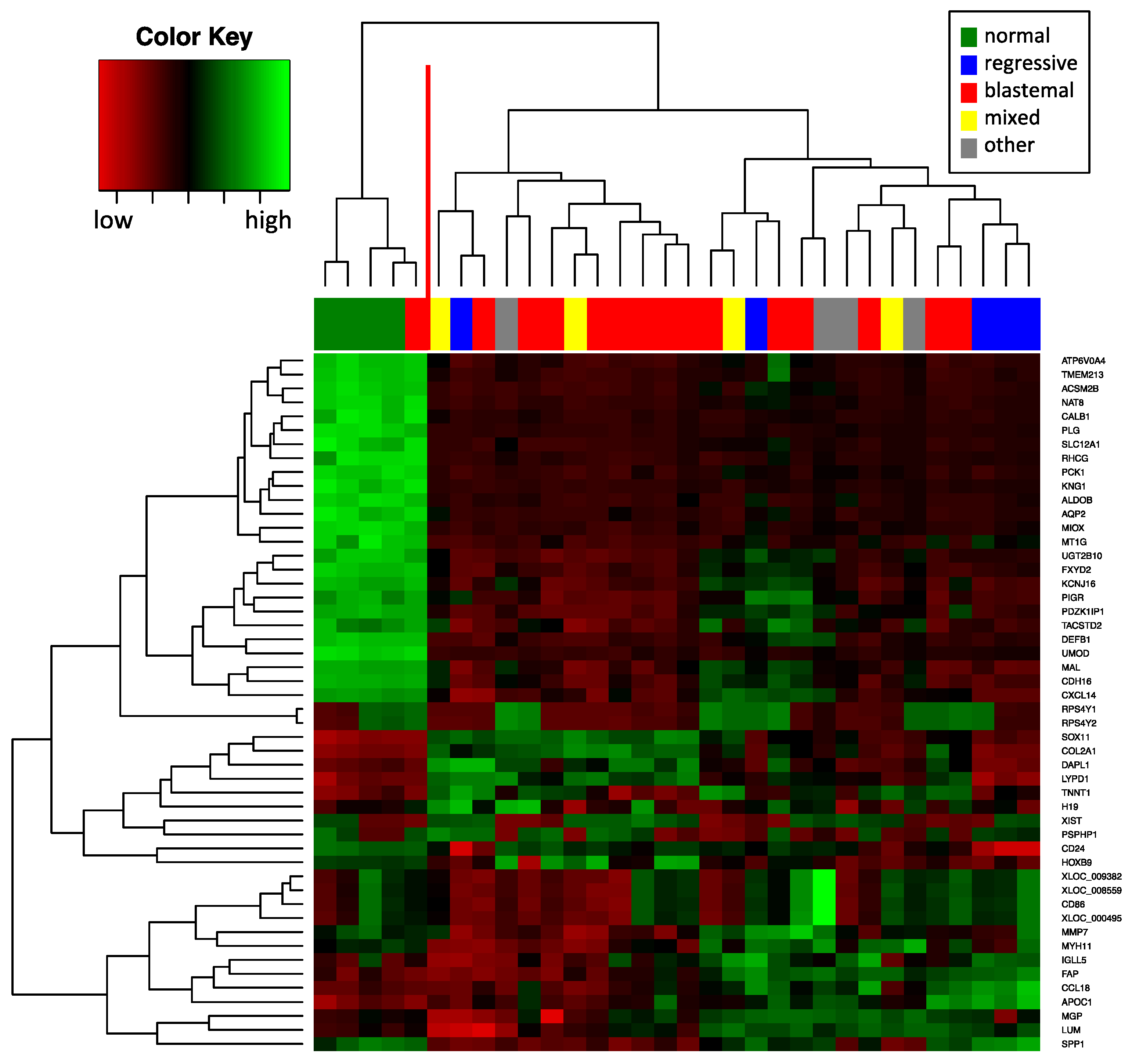
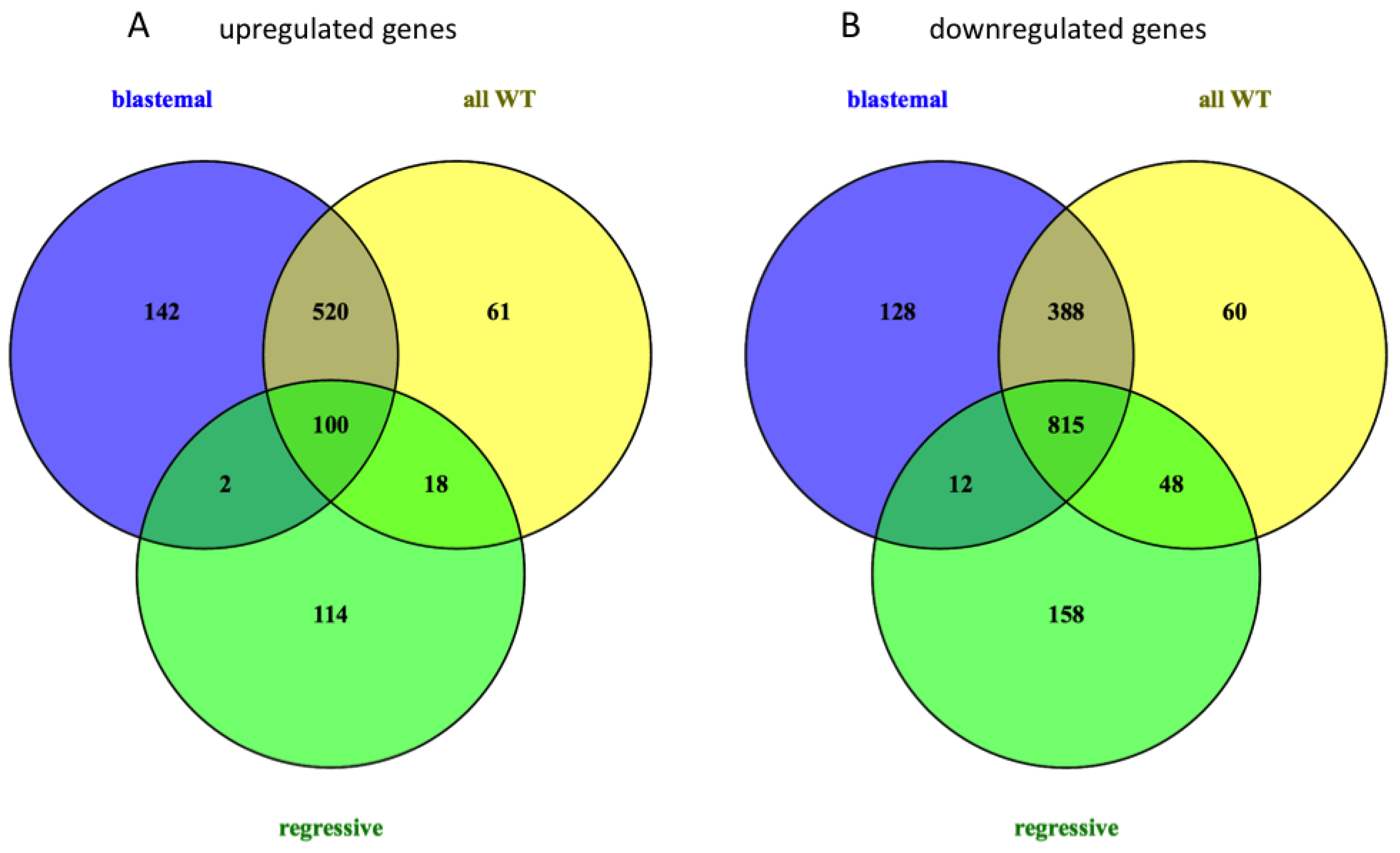
2.3. Deregulated mRNA Expression in WT Compared to Normal Tissue
2.4. Relation of mRNA and miRNA Expression
2.5. Chromosome Mapping of Deregulated miRNAs and mRNAs
3. Discussion
4. Materials and Methods
4.1. Patient Samples
4.2. RNA Isolation
4.3. miRNA Expression Profile
4.4. RT-qPCR Validation
4.5. Gene Expression Profile
4.6. miRNA-mRNA Correlation and Pathway Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| COG | Children’s Oncology Group |
| FC | fold change |
| LOH | loss of heterozygosity |
| RT-qPCR | reverse transcription quantitative PCR |
| SIOP | International Society of Paediatric Oncology |
| WT | Wilms tumor |
References
- Davidoff, A.M. Wilms tumor. Adv. Pediatr. 2012, 59, 247–267. [Google Scholar] [CrossRef] [PubMed]
- Furtwangler, R.; Nourkami, N.; Alkassar, M.; von Schweinitz, D.; Schenk, J.P.; Rube, C.; Siemer, S.; Leuschner, I.; Graf, N. Update on relapses in unilateral nephroblastoma registered in 3 consecutive SIOP/GPOH studies—A report from the GPOH-nephroblastoma study group. Klinische Padiatrie 2011, 223, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Schenk, J.P.; Graf, N.; Gunther, P.; Ley, S.; Goppl, M.; Kulozik, A.; Rohrschneider, W.K.; Troger, J. Role of MRI in the management of patients with nephroblastoma. Eur. Radiol. 2008, 18, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Schenk, J.P.; Gunther, P.; Schrader, C.; Ley, S.; Furtwangler, R.; Leuschner, I.; Edelhauser, M.; Graf, N.; Troger, J. Childhood kidney tumors—The relevance of imaging. Der Radiologe 2005, 45, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Vujanic, G.M.; Sandstedt, B.; Harms, D.; Kelsey, A.; Leuschner, I.; de Kraker, J. Revised international society of paediatric oncology (SIOP) working classification of renal tumors of childhood. Med. Pediatr. Oncol. 2002, 38, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Grundy, P.E.; Breslow, N.E.; Li, S.; Perlman, E.; Beckwith, J.B.; Ritchey, M.L.; Shamberger, R.C.; Haase, G.M.; D’Angio, G.J.; Donaldson, M.; et al. Loss of heterozygosity for chromosomes 1p and 16q is an adverse prognostic factor in favorable-histology Wilms tumor: A report from the national Wilms tumor study group. J. Clin. Oncol. 2005, 23, 7312–7321. [Google Scholar] [CrossRef] [PubMed]
- Messahel, B.; Williams, R.; Ridolfi, A.; A’Hern, R.; Warren, W.; Tinworth, L.; Hobson, R.; Al-Saadi, R.; Whyman, G.; Brundler, M.A.; et al. Allele loss at 16q defines poorer prognosis Wilms tumour irrespective of treatment approach in the UKW1–3 clinical trials: A children’s cancer and leukaemia group (CCLG) study. Eur. J. Cancer 2009, 45, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, S.; Zirn, B.; Alkassar, M.; Ambros, P.; Graf, N.; Gessler, M. Loss of 11q and 16q in Wilms tumors is associated with anaplasia, tumor recurrence, and poor prognosis. Genes Chromosomes Cancer 2007, 46, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Spreafico, F.; Gamba, B.; Mariani, L.; Collini, P.; D’Angelo, P.; Pession, A.; di Cataldo, A.; Indolfi, P.; Nantron, M.; Terenziani, M.; et al. Loss of heterozygosity analysis at different chromosome regions in Wilms tumor confirms 1p allelic loss as a marker of worse prognosis: A study from the italian association of pediatric hematology and oncology. J. Urol. 2013, 189, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Graf, N.T.H.; Pritchard-Jones, K.; Bergeron, C.; Sandstedt, B.; Leuschner, I.; Vujanic, G.; Boccon-Gibod, L.; van den Heuvel-Eibrink, M.; de Camargo, B.; Spreafico, F.; et al. Is the absolute blastema volume after preoperative chemotherapy in nephroblastoma relevant for prognosis? Pediatr. Blood Cancer 2011, 57, 741–742. [Google Scholar]
- Krutovskikh, V.A.; Herceg, Z. Oncogenic microRNAs (OncomiRs) as a new class of cancer biomarkers. BioEssays: News Rev. Mol. Cell. Dev. Biol. 2010, 32, 894–904. [Google Scholar] [CrossRef]
- Cortez, M.A.; Ivan, C.; Zhou, P.; Wu, X.; Ivan, M.; Calin, G.A. MicroRNAs in cancer: From bench to bedside. Adv. Cancer Res. 2010, 108, 113–157. [Google Scholar] [PubMed]
- Babashah, S.; Soleimani, M. The oncogenic and tumour suppressive roles of microRNAs in cancer and apoptosis. Eur. J. Cancer 2011, 47, 1127–1137. [Google Scholar] [CrossRef] [PubMed]
- Shenouda, S.K.; Alahari, S.K. MicroRNA function in cancer: Oncogene or a tumor suppressor? Cancer Metastasis Rev. 2009, 28, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Kent, O.A.; Mendell, J.T. A small piece in the cancer puzzle: MicroRNAs as tumor suppressors and oncogenes. Oncogene 2006, 25, 6188–6196. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.N.; Yan, M.D.; Lai, H.C.; Huang, R.L.; Chou, Y.C.; Lin, W.C.; Yeh, L.T.; Lin, Y.W. Downregulation of miR-29 contributes to cisplatin resistance of ovarian cancer cells. Int. J. Cancer 2014, 134, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Ma, N.; Wang, D.; Zhang, Y.; Zhou, J.; Wu, G.; Zhao, R.; Huang, H.; Wang, X.; Qiao, Y.; et al. miR-152 and miR-185 co-contribute to ovarian cancer cells cisplatin sensitivity by targeting DNMT1 directly: A novel epigenetic therapy independent of decitabine. Oncogene 2014, 33, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Hofsli, E.; Sjursen, W.; Prestvik, W.S.; Johansen, J.; Rye, M.; Trano, G.; Wasmuth, H.H.; Hatlevoll, I.; Thommesen, L. Identification of serum microRNA profiles in colon cancer. Br. J. Cancer 2013, 108, 1712–1719. [Google Scholar] [CrossRef] [PubMed]
- Kaduthanam, S.; Gade, S.; Meister, M.; Brase, J.C.; Johannes, M.; Dienemann, H.; Warth, A.; Schnabel, P.A.; Herth, F.J.; Sultmann, H.; et al. Serum miR-142–3p is associated with early relapse in operable lung adenocarcinoma patients. Lung Cancer 2013, 80, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.; Leidinger, P.; Gislefoss, R.; Haugen, A.; Langseth, H.; Staehler, P.; Lenhof, H.P.; Meese, E. Stable serum miRNA profiles as potential tool for non-invasive lung cancer diagnosis. RNA Biol. 2011, 8, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wang, M.; Lin, G.; Sun, S.; Li, X.; Qi, J.; Li, J. Serum microRNA-155 as a potential biomarker to track disease in breast cancer. PLoS ONE 2012, 7, e47003. [Google Scholar] [CrossRef] [PubMed]
- Zhao, A.; Li, G.; Peoc’h, M.; Genin, C.; Gigante, M. Serum miR-210 as a novel biomarker for molecular diagnosis of clear cell renal cell carcinoma. Exp. Mol. Pathol. 2013, 94, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.K.; Sabbaghian, N.; Xu, B.; Addidou-Kalucki, S.; Bernard, C.; Zou, D.; Reeve, A.E.; Eccles, M.R.; Cole, C.; Choong, C.S.; et al. Biallelic DICER1 mutations occur in Wilms tumours. J. Pathol. 2013, 230, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Lee, H.; Ghahremani, S.; Kempert, P.; Ischander, M.; Teitell, M.A.; Nelson, S.F.; Martinez-Agosto, J.A. Expanding the phenotype of mutations in DICER1: Mosaic missense mutations in the RNase IIIb domain of DICER1 cause GLOW syndrome. J. Med. Genet. 2014, 51, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Torrezan, G.T.; Ferreira, E.N.; Nakahata, A.M.; Barros, B.D.; Castro, M.T.; Correa, B.R.; Krepischi, A.C.; Olivieri, E.H.; Cunha, I.W.; Tabori, U.; et al. Recurrent somatic mutation in DROSHA induces microRNA profile changes in Wilms tumour. Nat. Commun. 2014, 5, 4039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegert, J.; Ishaque, N.; Vardapour, R.; Georg, C.; Gu, Z.; Bieg, M.; Ziegler, B.; Bausenwein, S.; Nourkami, N.; Ludwig, N.; et al. Mutations in the SIX1/2 pathway and the DROSHA/DGCR8 miRNA microprocessor complex underlie high-risk blastemal type Wilms tumors. Cancer Cell 2015, 27, 298–311. [Google Scholar] [CrossRef] [PubMed]
- Walz, A.L.; Ooms, A.; Gadd, S.; Gerhard, D.S.; Smith, M.A.; Guidry Auvil, J.M.; Meerzaman, D.; Chen, Q.R.; Hsu, C.H.; Yan, C.; et al. Recurrent DGCR8, DROSHA, and SIX homeodomain mutations in favorable histology Wilms tumors. Cancer Cell 2015, 27, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, J.; Backes, C.; Nourkami-Tutdibi, N.; Leidinger, P.; Deutscher, S.; Beier, M.; Gessler, M.; Graf, N.; Lenhof, H.P.; Keller, A.; et al. Treatment-independent miRNA signature in blood of Wilms tumor patients. BMC Genom. 2012, 13, 379. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.A.; Bryan, K.; Williams, R.; Popov, S.; Vujanic, G.; Coulomb, A.; Boccon-Gibod, L.; Graf, N.; Pritchard-Jones, K.; O’Sullivan, M. MiRNA profiles as a predictor of chemoresponsiveness in Wilms’ tumor blastema. PLoS ONE 2013, 8, e53417. [Google Scholar] [CrossRef] [PubMed]
- Backes, C.; Keller, A.; Kuentzer, J.; Kneissl, B.; Comtesse, N.; Elnakady, Y.A.; Muller, R.; Meese, E.; Lenhof, H.P. Genetrail—Advanced gene set enrichment analysis. Nucleic Acids Res. 2007, 35, W186–W192. [Google Scholar] [CrossRef] [PubMed]
- Rakheja, D.; Chen, K.S.; Liu, Y.; Shukla, A.A.; Schmid, V.; Chang, T.C.; Khokhar, S.; Wickiser, J.E.; Karandikar, N.J.; Malter, J.S.; et al. Somatic mutations in DROSHA and DICER1 impair microRNA biogenesis through distinct mechanisms in Wilms tumours. Nat. Commun. 2014, 2, 4802. [Google Scholar] [CrossRef] [PubMed]
- Spreafico, F.; Ciceri, S.; Gamba, B.; Torri, F.; Terenziani, M.; Collini, P.; Macciardi, F.; Radice, P.; Perotti, D. Chromosomal anomalies at 1q, 3, 16q, and mutations of SIX1 and DROSHA genes underlie Wilms tumor recurrences. Oncotarget 2016. [Google Scholar] [CrossRef]
- Senanayake, U.; Das, S.; Vesely, P.; Alzoughbi, W.; Frohlich, L.F.; Chowdhury, P.; Leuschner, I.; Hoefler, G.; Guertl, B. miR-192, miR-194, miR-215, miR-200c and miR-141 are downregulated and their common target ACVR2b is strongly expressed in renal childhood neoplasms. Carcinogenesis 2012, 33, 1014–1021. [Google Scholar] [CrossRef] [PubMed]
- Kort, E.J.; Farber, L.; Tretiakova, M.; Petillo, D.; Furge, K.A.; Yang, X.J.; Cornelius, A.; Teh, B.T. The E2F3-oncomir-1 axis is activated in Wilms’ tumor. Cancer Res. 2008, 68, 4034–4038. [Google Scholar] [CrossRef] [PubMed]
- Veronese, A.; Lupini, L.; Consiglio, J.; Visone, R.; Ferracin, M.; Fornari, F.; Zanesi, N.; Alder, H.; D’Elia, G.; Gramantieri, L.; et al. Oncogenic role of miR-483-3p at the IGF2/483 locus. Cancer Res. 2010, 70, 3140–3149. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nature Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, H.; Enokida, H.; Itesako, T.; Tatarano, S.; Kinoshita, T.; Fuse, M.; Kojima, S.; Nakagawa, M.; Seki, N. Epithelial-mesenchymal transition-related microRNA-200s regulate molecular targets and pathways in renal cell carcinoma. J. Hum. Genet. 2013, 58, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Wang, Z.; Fillmore, R.; Xi, Y. MiR-200, a new star miRNA in human cancer. Cancer Lett. 2014, 344, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.; Hoang-Vu, C.; Dralle, H.; Huttelmaier, S. Downregulation of microRNAs directs the EMT and invasive potential of anaplastic thyroid carcinomas. Oncogene 2010, 29, 4237–4244. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, I.S.; Chang, K.C.; Tsai, Y.T.; Ke, J.Y.; Lu, P.J.; Lee, K.H.; Yeh, S.D.; Hong, T.M.; Chen, Y.L. MicroRNA-320 suppresses the stem cell-like characteristics of prostate cancer cells by downregulating the Wnt/β-catenin signaling pathway. Carcinogenesis 2013, 34, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Funamizu, N.; Lacy, C.R.; Parpart, S.T.; Takai, A.; Hiyoshi, Y.; Yanaga, K. MicroRNA-301b promotes cell invasiveness through targeting TP63 in pancreatic carcinoma cells. Int. J. Oncol. 2014, 44, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; An, Y.; Lv, N.; Chen, J.; Tu, M.; Sun, J.; Wu, P.; Wei, J.; Jiang, K.; Miao, Y. MiRNA-181b increases the sensitivity of pancreatic ductal adenocarcinoma cells to gemcitabine in vitro and in nude mice by targeting BCL-2. Oncol. Rep. 2013, 29, 1769–1776. [Google Scholar] [PubMed]
- Comincini, S.; Allavena, G.; Palumbo, S.; Morini, M.; Durando, F.; Angeletti, F.; Pirtoli, L.; Miracco, C. MicroRNA-17 regulates the expression of ATG7 and modulates the autophagy process, improving the sensitivity to temozolomide and low-dose ionizing radiation treatments in human glioblastoma cells. Cancer Biol. Ther. 2013, 14, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Lucotti, S.; Rainaldi, G.; Evangelista, M.; Rizzo, M. Fludarabine treatment favors the retention of miR-485–3p by prostate cancer cells: Implications for survival. Mol. Cancer 2013, 12, 52. [Google Scholar] [CrossRef] [PubMed]
- Streppel, M.M.; Pai, S.; Campbell, N.R.; Hu, C.; Yabuuchi, S.; Canto, M.I.; Wang, J.S.; Montgomery, E.A.; Maitra, A. MicroRNA 223 is upregulated in the multistep progression of barrett’s esophagus and modulates sensitivity to chemotherapy by targeting PARP1. Clin. Cancer Res. 2013, 19, 4067–4078. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sai, K.; Chen, F.R.; Chen, Z.P. MiR-181b modulates glioma cell sensitivity to temozolomide by targeting MEK1. Cancer Chemother. Pharmacol. 2013, 72, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Shan, X.; Wang, T.; Shu, Y.; Liu, P. MiR-181b modulates multidrug resistance by targeting BCL2 in human cancer cell lines. Int. J. Cancer 2010, 127, 2520–2529. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Zheng, Z.M.; Li, X.N.; Li, Z.F.; Wang, Y.; Geng, Y.F.; Bai, L.; Zhang, X.B. MiR-223 modulates multidrug resistance via downregulation of ABCB1 in hepatocellular carcinoma cells. Exp. Biol. Med. 2013. [Google Scholar] [CrossRef] [PubMed]
- Hawthorn, L.; Cowell, J.K. Analysis of Wilms tumors using SNP mapping array-based comparative genomic hybridization. PLoS ONE 2011, 6, e18941. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Sahu, D.K.; Goel, M.; Kant, R.; Gupta, D.K. Retrospective analysis of FFPE based Wilms’ tumor samples through copy number and somatic mutation related molecular inversion probe based array. Gene 2015, 565, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Feichtinger, R.G.; Neureiter, D.; Royer-Pokora, B.; Mayr, J.A.; Zimmermann, F.A.; Jones, N.; Koegler, C.; Ratschek, M.; Sperl, W.; Kofler, B. Heterogeneity of mitochondrial energy metabolism in classical triphasic Wilms’ tumor. Front. Biosci. 2011, 3, 187–193. [Google Scholar] [CrossRef]
- Chilukamarri, L.; Hancock, A.L.; Malik, S.; Zabkiewicz, J.; Baker, J.A.; Greenhough, A.; Dallosso, A.R.; Huang, T.H.; Royer-Pokora, B.; Brown, K.W.; et al. Hypomethylation and aberrant expression of the glioma pathogenesis-related 1 gene in Wilms tumors. Neoplasia 2007, 9, 970–978. [Google Scholar] [CrossRef] [PubMed]
- Koller, K.; Pichler, M.; Koch, K.; Zandl, M.; Stiegelbauer, V.; Leuschner, I.; Hoefler, G.; Guertl, B. Nephroblastomas show low expression of micror-204 and high expression of its target, the oncogenic transcription factor MEIS1. Pediatr. Dev. Pathol. 2014, 17, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Garofalo, M.; Martelli, M.P.; Briesewitz, R.; Wang, L.; Fernandez-Cymering, C.; Volinia, S.; Liu, C.G.; Schnittger, S.; Haferlach, T.; et al. Distinctive microRNA signature of acute myeloid leukemia bearing cytoplasmic mutated nucleophosmin. Proc. Natl. Acad. Sci. USA 2008, 105, 3945–3950. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, H.Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar]
- Ludwig, N.; Kim, Y.J.; Mueller, S.C.; Backes, C.; Werner, T.V.; Galata, V.; Sartorius, E.; Bohle, R.M.; Keller, A.; Meese, E. Posttranscriptional deregulation of signaling pathways in meningioma subtypes by differential expression of miRNAs. Neuro-Oncology 2015, 17, 1250–1260. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, E.C.; Backes, C.; Knorck, A.; Ludwig, N.; Leidinger, P.; Hoxha, C.; Schwar, G.; Grossmann, T.; Muller, S.C.; Hart, M.; et al. Deep characterization of blood cell miRNomes by NGs. Cell. Mol. Life Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.A.; Leidinger, P.; Backes, C.; Keller, A.; Karpinski, P.; Sasiadek, M.M.; Blin, N.; Meese, E. A set of specific miRNAs is connected with murine and human gastric cancer. Genes Chromosomes Cancer 2013, 52, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Abu-Halima, M.; Backes, C.; Leidinger, P.; Keller, A.; Lubbad, A.M.; Hammadeh, M.; Meese, E. MicroRNA expression profiles in human testicular tissues of infertile men with different histopathologic patterns. Fertil. Steril. 2014, 101, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Dweep, H.; Sticht, C.; Pandey, P.; Gretz, N. Mirwalk-database: Prediction of possible mirna binding sites by “walking” the genes of three genomes. J. Biomed. Inf. 2011, 44, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.D.; Lin, F.M.; Wu, W.Y.; Liang, C.; Huang, W.C.; Chan, W.L.; Tsai, W.T.; Chen, G.Z.; Lee, C.J.; Chiu, C.M.; et al. Mirtarbase: A database curates experimentally validated microrna-target interactions. Nucleic Acids Res. 2011, 39, D163–D169. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 15 Most Downregulated miRNAs in WT Subtypes Compared to Normal Kidneys | |||||
| Blastemal | Regressive | All WT | |||
| miRNA | FC 1 | miRNA | FC 1 | miRNA | FC 1 |
| miR-194 | 48.95 | miR-30a | 143.28 | miR-200a | 46.47 |
| miR-204 | 43.09 | miR-194 | 64.67 | miR-194 | 46.22 |
| miR-200a | 33.71 | miR-200a | 38.3 | miR-204 | 36.81 |
| miR-192 | 33.2 | miR-204 | 36.5 | miR-200b | 35.38 |
| miR-429 | 29.99 | miR-30a * | 34.58 | miR-141 | 31.93 |
| miR-215 | 25.52 | miR-200b | 31.58 | miR-192 | 28.55 |
| miR-200c | 24.62 | miR-192 | 26.64 | miR-429 | 28.46 |
| miR-200b | 24.39 | miR-1973 | 26.05 | miR-215 | 24.02 |
| miR-30a * | 18.33 | miR-141 | 24.1 | miR-1973 | 22.35 |
| miR-514 | 15.77 | miR-4284 | 23.57 | miR-200c | 21.46 |
| miR-141 | 14.47 | miR-200c | 21.44 | miR-30a * | 18.81 |
| miR-1973 | 12.75 | miR-429 | 20.79 | miR-30a | 16.95 |
| miR-30a | 11.46 | miR-215 | 19.34 | miR-514 | 9.56 |
| miR-138 | 7.57 | miR-30b | 18.61 | miR-30c-2 * | 7.06 |
| miR-30c-2 * | 6.16 | miR-26a | 14.51 | miR-30c | 7.01 |
| 15 Most Upregulated miRNAs in WT Subtypes Compared to Normal Kidneys | |||||
| Blastemal | Regressive | All WT | |||
| miRNA | FC 1 | miRNA | FC 1 | miRNA | FC 1 |
| miR-199b-5p | 7.87 | miR-3149 | 25.05 | miR-483-3p | 8.11 |
| miR-130b | 6.16 | miR-595 | 23.95 | miR-34a | 5.47 |
| miR-335 | 5.50 | miR-1290 | 23.61 | miR-1207-5p | 5.18 |
| miR-483-3p | 4.61 | miR-32 * | 20.63 | miR-199b-5p | 4.58 |
| miR-183 | 4.48 | miR-1225-5p | 20.55 | miR-1249 | 3.81 |
| miR-301b | 4.00 | miR-3148 | 19.09 | miR-130b | 3.80 |
| miR-301a | 3.88 | miR-1228 * | 15.87 | miR-1202 | 3.66 |
| miR-34b * | 3.79 | miR-670 | 15.06 | miR-320c | 3.59 |
| miR-34a | 3.70 | miR-4270 | 13.80 | miR-1225-5p | 3.50 |
| miR-18a | 3.58 | miR-2278 | 13.23 | miR-320d | 3.34 |
| miR-199a-3p | 3.23 | miR-1207-5p | 13.21 | miR-4270 | 3.25 |
| miR-342-3p | 3.08 | miR-1306 | 13.07 | miR-4281 | 3.01 |
| miR-181c * | 2.97 | miR-4281 | 12.19 | miR-320e | 2.96 |
| miR-1207-5p | 2.95 | miR-1246 | 11.66 | miR-296-5p | 2.67 |
| miR-342-5p | 2.82 | miR-574-5p | 10.91 | miR-335 | 2.63 |
| miRNA | Fold Change | Direction of Regulation | Significance (p-Value) | Comparison | |||
|---|---|---|---|---|---|---|---|
| Array | RT-qPCR | Array | RT-qPCR | Array | RT-qPCR | ||
| miR-181b | 1.21 | 3.52 | up | up | ns 1 | 3.40 × 10−4 | blastema vs. normal |
| 3.20 | 6.28 | up | up | 0.021 | 0.006 | regressive vs. normal | |
| 2.41 | 5.88 | up | up | ns | 7.45 × 10−8 | all WT vs. normal | |
| miR-223 | 1.74 | 1.99 | up | up | ns | 0.273 | blastema vs. normal |
| 6.32 | 60.34 | up | up | 0.027 | 0.002 | regressive vs. normal | |
| 2.10 | 8.97 | up | up | ns | 0.008 | all WT vs. normal | |
| miR-320a | 2.69 | 2.08 | up | up | 0.013 | 0.002 | blastema vs. normal |
| 1.82 | 3.64 | down | up | ns | 0.026 | regressive vs. normal | |
| 1.11 | 2.85 | up | up | ns | 4.11 × 10−5 | all WT vs. normal | |
| miR-485-3p | 1.02 | 2.20 | up | up | ns | 0.065 | blastema vs. normal |
| 3.97 | 11.11 | up | up | 0.001 | 0.023 | regressive vs. normal | |
| 2.02 | 3.62 | up | up | 0.021 | 0.007 | all WT vs. normal | |
| Group of Pathways | Individual Pathways |
|---|---|
| Metabolism | Tryptophan metabolism |
| Starch and sucrose metabolism | |
| Retinol metabolism | |
| Phenylalanine metabolism | |
| Pentose and glucuronate interconversions | |
| Metabolism of xenobiotics by cytochrome P450 | |
| Metabolic pathways | |
| Histidine metabolism | |
| Glycine, serine and threonine metabolism | |
| Fatty acid metabolism | |
| Fatty acid degradation | |
| Drug metabolism—other enzymes | |
| Drug metabolism—cytochrome P450 | |
| Carbon metabolism | |
| Butanoate metabolism | |
| Ascorbate and aldarate metabolism | |
| Arginine and proline metabolism | |
| Arachidonic acid metabolism | |
| Alanine, aspartate and glutamate metabolism | |
| Signal transduction and signaling molecules | Calcium signaling pathway |
| Cell adhesion molecules (CAMs) | |
| Extracellular matrix (ECM)-receptor interaction | |
| Neuroactive ligand-receptor interaction | |
| PI3K-Akt signaling pathway | |
| Cellular processes | Phagosome |
| Cell cycle | |
| Focal adhesion | |
| Organismal systems (immune, endocrine, digestive, excretory, sensory) | Renin-angiotensin system |
| Proximal tubule bicarbonate reclamation | |
| Protein digestion and absorption | |
| PPAR signaling pathway | |
| Olfactory transduction | |
| Mineral absorption | |
| Gastric acid secretion | |
| Endocrine and other factor-regulated calcium reabsorption | |
| Complement and coagulation cascades | |
| Bile secretion | |
| Aldosterone-regulated sodium reabsorption | |
| Human Diseases | Chemical carcinogenesis |
| Amoebiasis |
| Chromosome (chr) | miRNA | ||||||||
| Blastemal | Regressive | All WT | |||||||
| Expected | Observed | p | Expected | Observed | p | Expected | Observed | p | |
| Chr 1 | 3.97 | 11 | 0.01 | – | – | – | 5.09 | 12 | 0.02 |
| Chr X | – | – | – | 12.43 | 31 | <0.001 | 5.76 | 18 | <0.001 |
| Chromosome (chr) | Genes | ||||||||
| Blastemal | Regressive | All WT | |||||||
| Expected | Observed | p | Expected | Observed | p | Expected | Observed | p | |
| Chr 4 | 40.55 | 61 | 0.01 | – | – | – | 40.71 | 64 | 0.002 |
| Mitochondrial genome | 0.71 | 11 | <0.001 | 0.55 | 9 | <0.001 | 0.71 | 9 | <0.001 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ludwig, N.; Werner, T.V.; Backes, C.; Trampert, P.; Gessler, M.; Keller, A.; Lenhof, H.-P.; Graf, N.; Meese, E. Combining miRNA and mRNA Expression Profiles in Wilms Tumor Subtypes. Int. J. Mol. Sci. 2016, 17, 475. https://doi.org/10.3390/ijms17040475
Ludwig N, Werner TV, Backes C, Trampert P, Gessler M, Keller A, Lenhof H-P, Graf N, Meese E. Combining miRNA and mRNA Expression Profiles in Wilms Tumor Subtypes. International Journal of Molecular Sciences. 2016; 17(4):475. https://doi.org/10.3390/ijms17040475
Chicago/Turabian StyleLudwig, Nicole, Tamara V. Werner, Christina Backes, Patrick Trampert, Manfred Gessler, Andreas Keller, Hans-Peter Lenhof, Norbert Graf, and Eckart Meese. 2016. "Combining miRNA and mRNA Expression Profiles in Wilms Tumor Subtypes" International Journal of Molecular Sciences 17, no. 4: 475. https://doi.org/10.3390/ijms17040475