
VLDL from Metabolic Syndrome Individuals Enhanced Lipid Accumulation in Atria with Association of Susceptibility to Atrial Fibrillation

 ,
,
Abstract
:
1. Introduction
2. Results
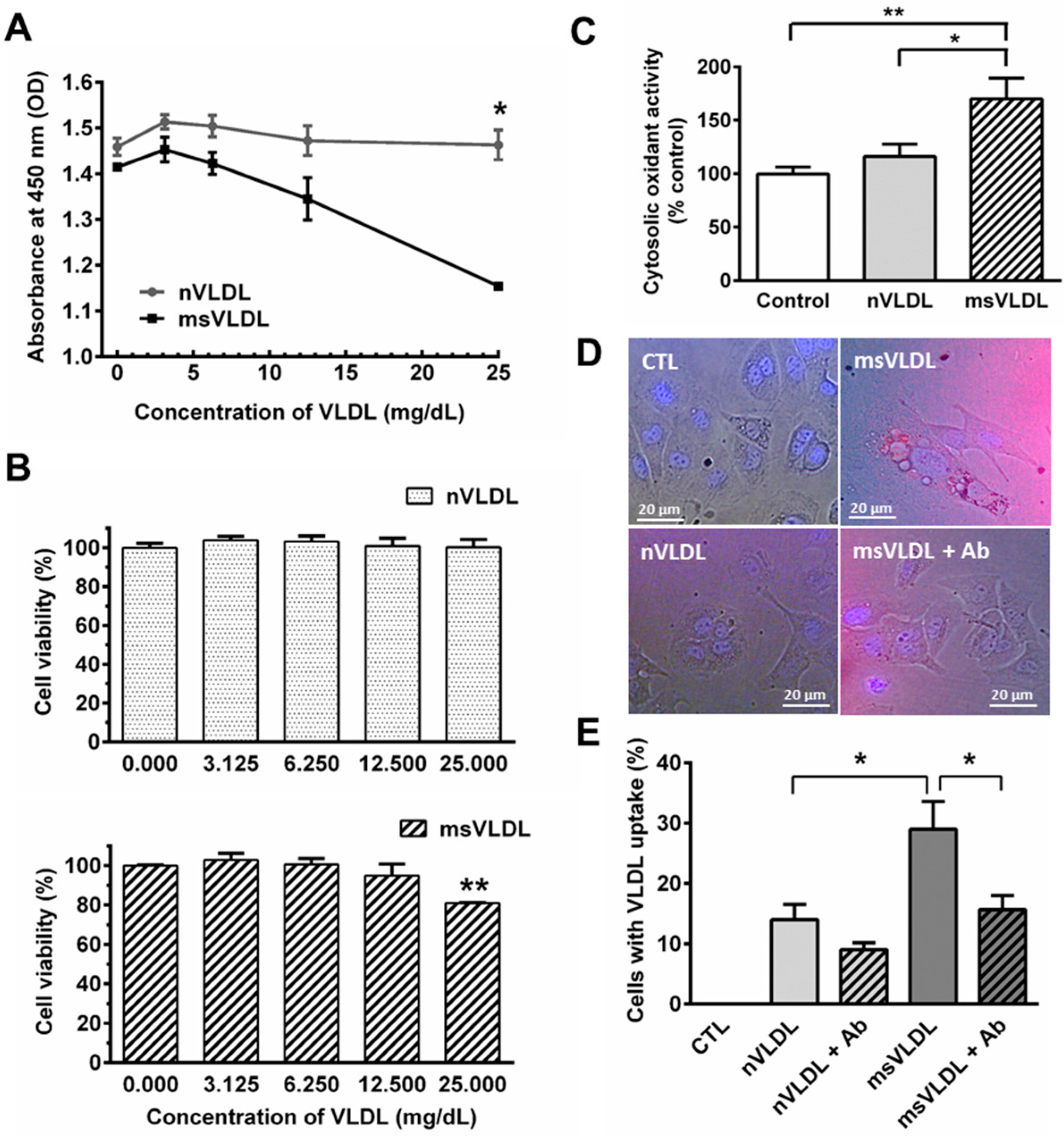
2.1. Very Low Density Lipoproteins (VLDL) in MetS (MetS-VLDL) Increased Oxidative Stress and Cytotoxicity to HL-1 Atrial Myocytes
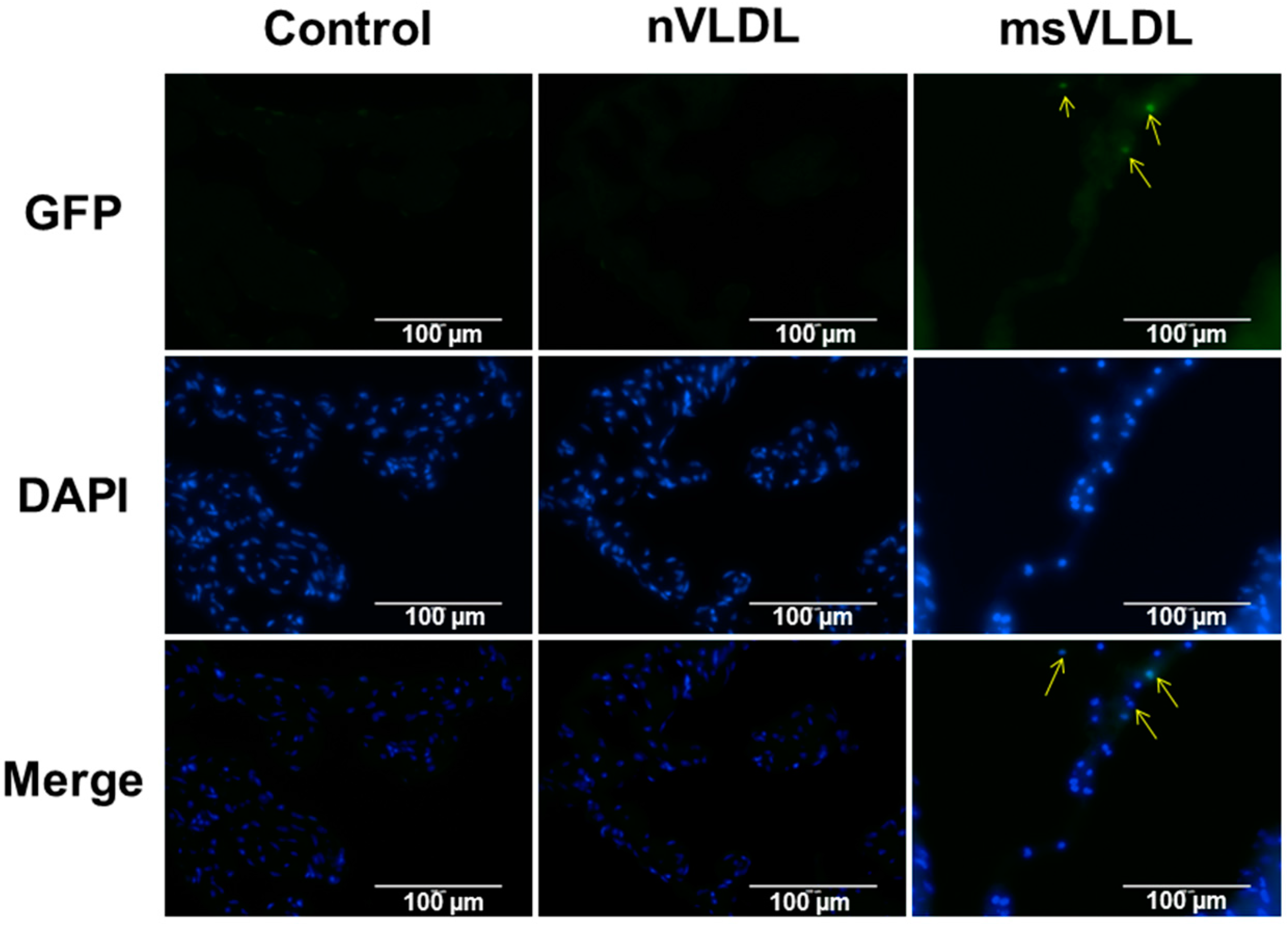
2.2. Uptake of VLDLs by VLDL Receptors of Atrial Myocytes

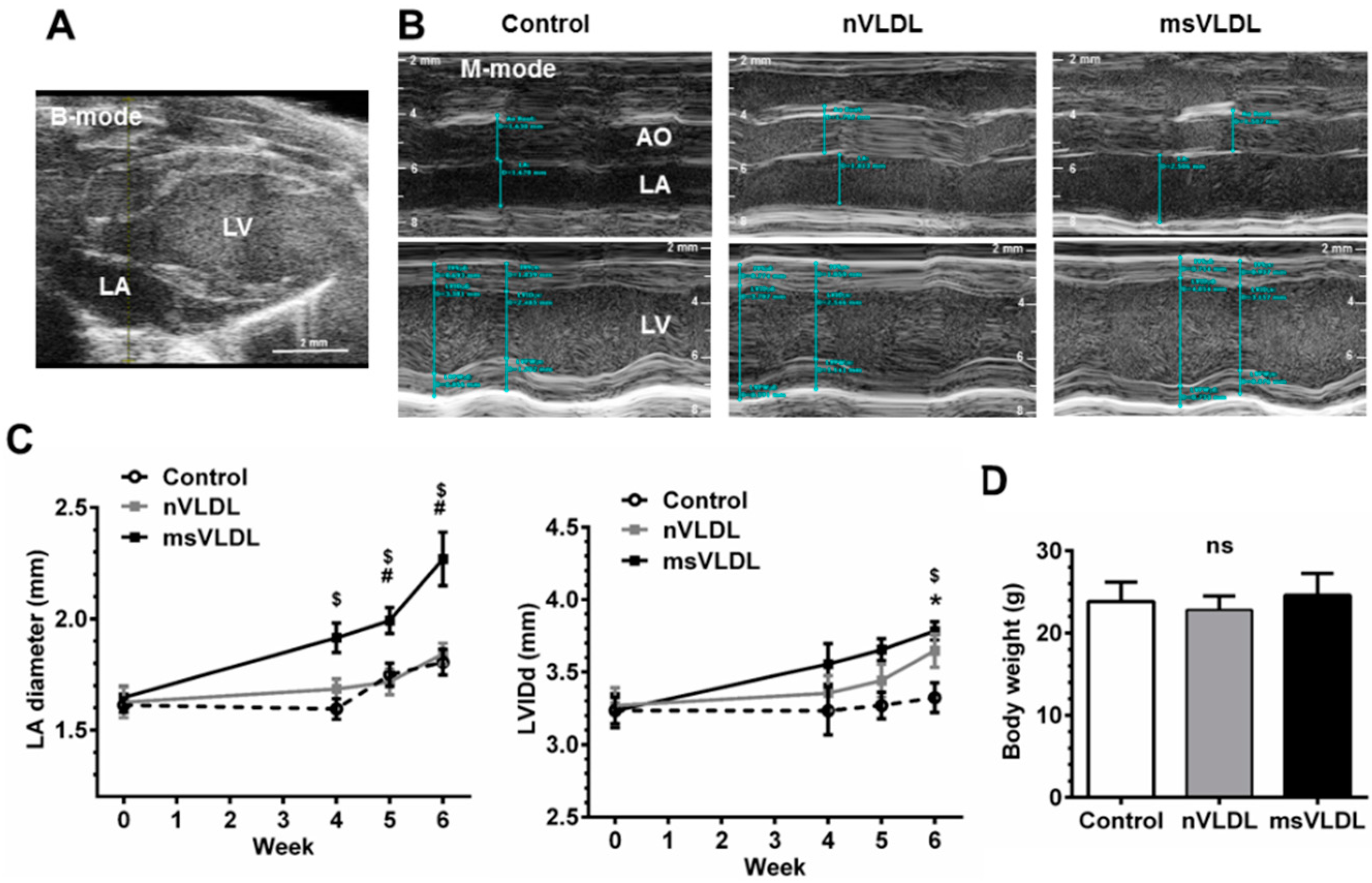
2.3. Both Normal-VLDL and MetS-VLDL Increased Left Ventricular (LV) Mass but Only MetS-VLDL Caused Left Atrial Dilation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Control (n = 10) | nVLDL (n = 7) | msVLDL (n = 10) | p Value |
|---|---|---|---|---|
| BW (g) | 23.8 ± 2.4 | 22.8 ± 1.7 | 24.6 ± 2.7 | 0.1699 |
| HR (bpm) | 230 ± 20 | 237 ± 59 | 259 ± 38 | 0.5720 |
| Measurement (mm) | ||||
| Ao Root | 1.71 ± 0.12 | 1.68 ± 0.07 | 1.69 ± 0.13 | 0.9274 |
| LA | 1.64 ± 0.23 | 1.84 ± 0.13 | 2.18 ± 0.24 $,# | <0.0001 |
| IVSd | 0.91 ± 0.13 | 0.76 ± 0.09 * | 0.78 ± 0.06 $ | 0.0054 |
| LVIDd | 3.13 ± 0.30 | 3.78 ± 0.19 * | 3.78 ± 0.20 $ | <0.0001 |
| LVPWd | 0.87 ± 0.10 | 0.85 ± 0.16 | 0.77 ± 0.11 | 0.2141 |
| LVIDs | 2.11 ± 0.33 | 2.59 ± 0.23 * | 2.77 ± 0.36 $ | 0.0003 |
| Calculation | ||||
| EF (%) | 62.5 ± 7.7 | 59.8 ± 8.1 | 52.9 ± 9.6 $ | 0.0529 |
| FS (%) | 32.9 ± 5.4 | 31.5 ± 5.4 | 27.0 ± 6.5 | 0.0853 |
| LV Mass (mg) | 92.3 ± 13.4 | 107.6 ± 16.2 | 102.7 ± 9.0 | 0.0553 |
| LVEDV (µL) | 39.3 ± 8.8 | 61.3 ± 7.2 * | 61.6 ± 7.9 $ | <0.0001 |
| LVESV (µL) | 15.1 ± 5.7 | 24.6 ± 5.4 * | 29.5 ± 8.6 $ | 0.0004 |

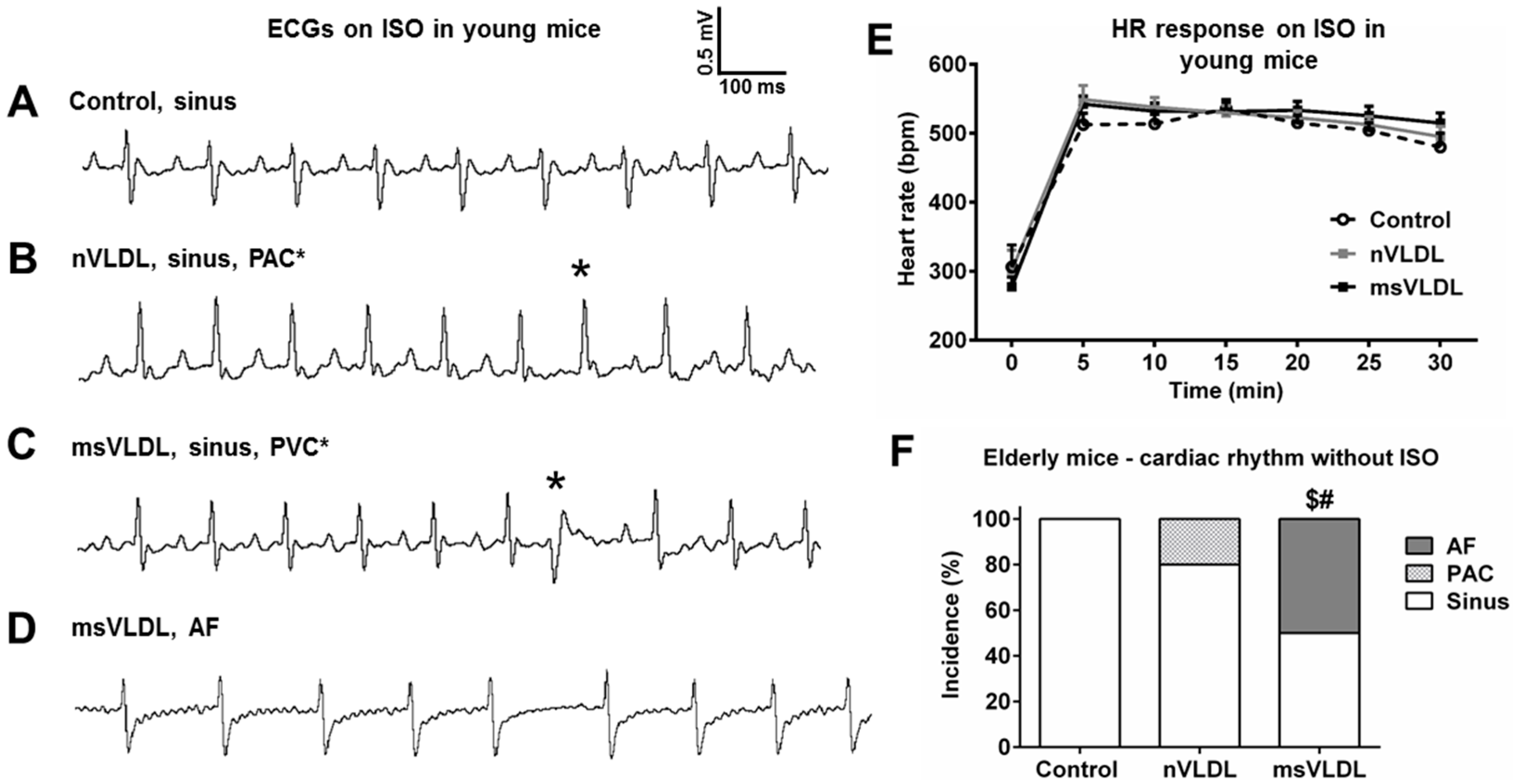
2.4. Isoproterenol Challenge Induced Atrial Fibrillation (AF) in msVLDL Mice

2.5. Unprovoked Electrocardiography Showed AF in msVLDL Mice
2.6. MetS-VLDL Caused Atrial Tissue Apoptosis

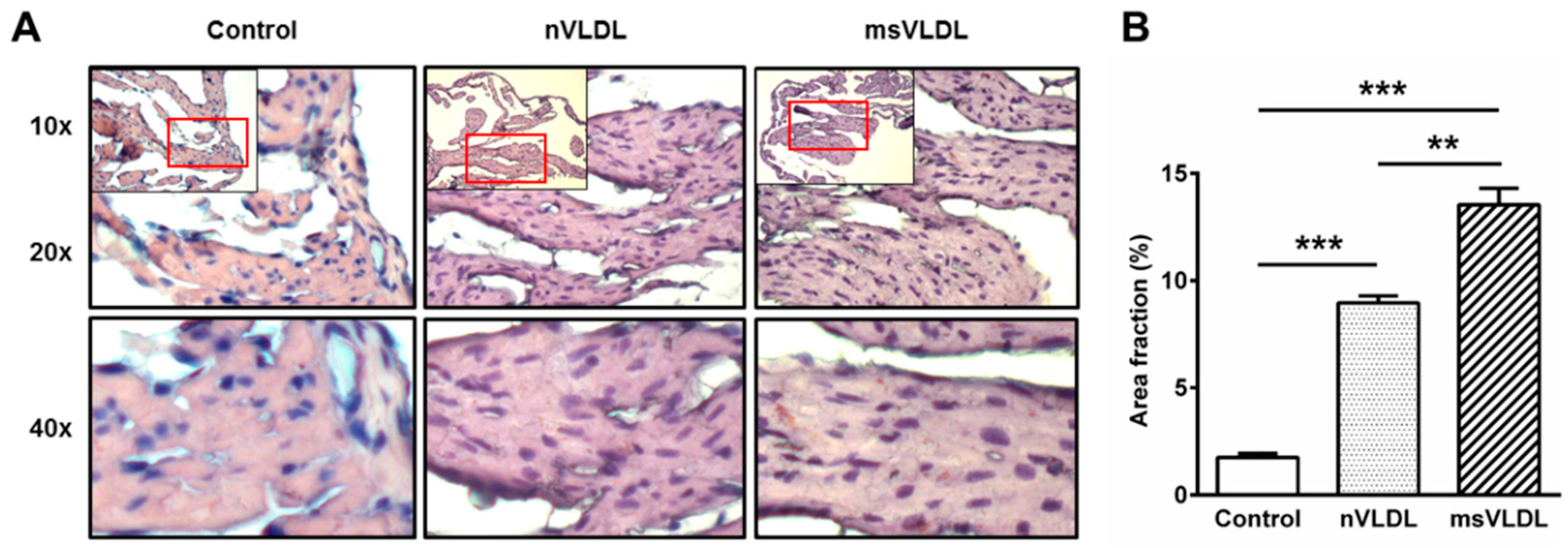
2.7. Increased Lipid Accumulation in Atrial Tissue of msVLDL Mice

3. Discussion
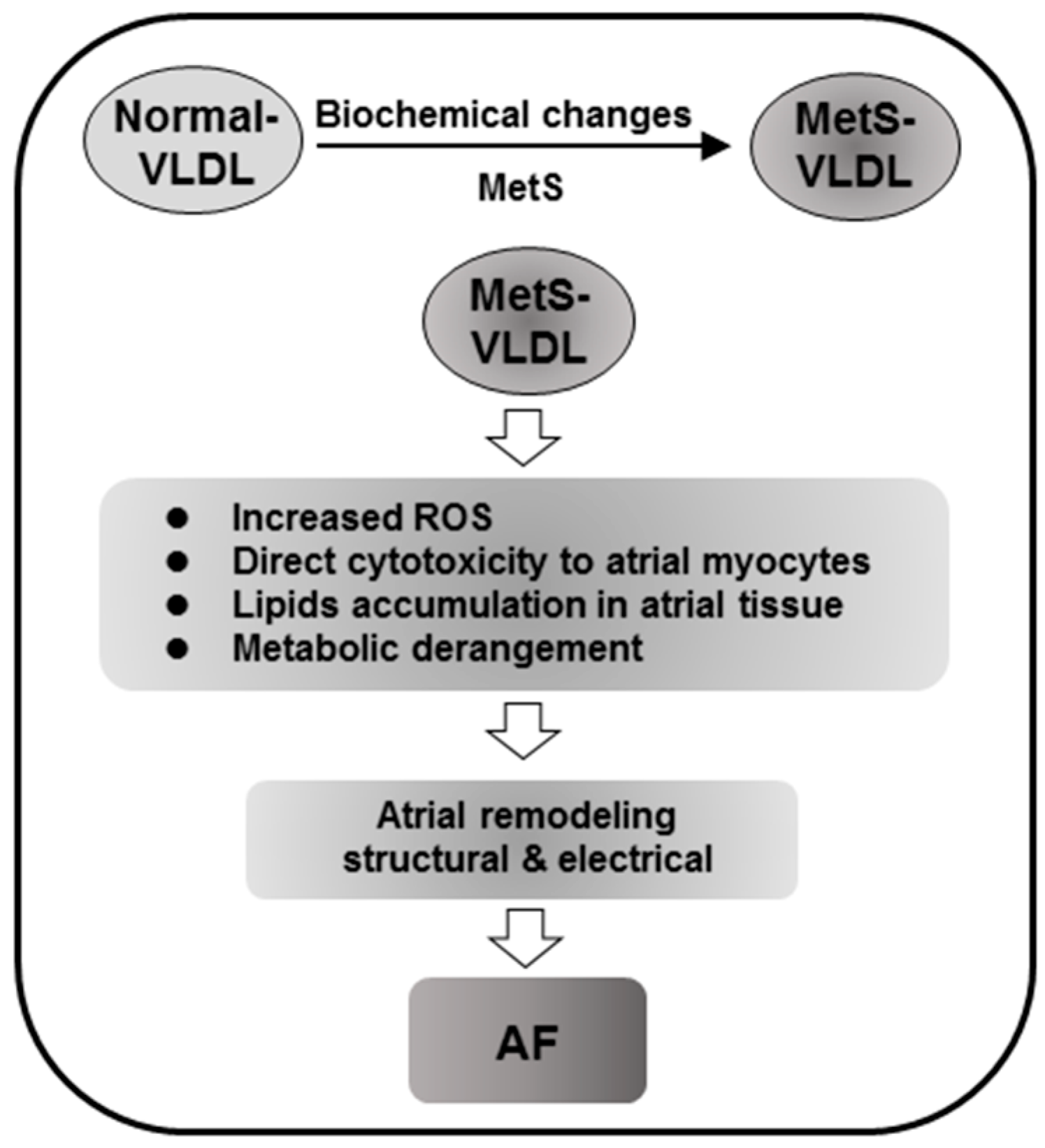
3.1. Increased Lipid Accumulation and in Vivo and in Vitro Cytotoxicity of MetS-VLDL to Atrium
3.2. MetS-VLDL Causes Cardiac Remodeling
3.3. Clinical Implications

4. Material and Methods
4.1. VLDL Isolation
4.2. HL-1 Atrial Myocyte Culture
4.3. Cytotoxicity of VLDL to HL-1 Cells
4.4. Reactive Oxygen Species (ROS)
4.5. VLDL Uptake by HL-1 Cells
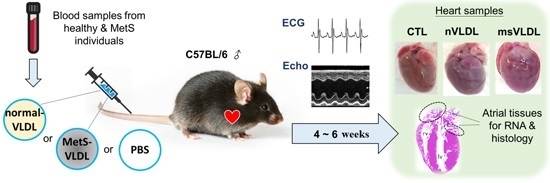
4.6. Mice and Diet
4.7. Mouse Echocardiography
4.8. Isoproterenol-Challenged Electrocardiography for Young Mice
4.9. Unprovoked Electrocardiography for Elderly Mice
4.10. Oil Red O (ORO) Staining of Atrial Tissue and Quantification
4.11. In Situ Terminal Deoxynucleotidyl Transferase (TUNEL) Assay of Atrial Tissues
4.12. Data Analysis and Statistics
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| The following abbreviations are used in this manuscript: |
| MetS: | metabolic syndrome |
| AF: | atrial fibrillation |
| VLDL: | very-low-density lipoproteins |
| nVLDL: | normal-VLDL treated group |
| msVLDL: | MetS-VLDL treated group |
| ORD: | oil red O |
| ISO: | isoproterenol |
References
- Go, A.S.; Mozaffarian, D.; Roger, V.L.; Benjamin, E.J.; Berry, J.D.; Borden, W.B.; Bravata, D.M.; Dai, S.; Ford, E.S.; Fox, C.S.; et al. Executive summary: Heart disease and stroke statistics—2013 update: A report from the American Heart Association. Circulation 2013, 127, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Tanner, R.M.; Baber, U.; Carson, A.P.; Voeks, J.; Brown, T.M.; Soliman, E.Z.; Howard, V.J.; Muntner, P. Association of the metabolic syndrome with atrial fibrillation among United States adults (from the REasons for Geographic and Racial Differences in Stroke [REGARDS] Study). Am. J. Cardiol. 2011, 108, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Tadic, M.; Ivanovic, B.; Cuspidi, C. What do we currently know about metabolic syndrome and atrial fibrillation? Clin. Cardiol. 2013, 36, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, S.; Mohanty, P.; di Biase, L.; Bai, R.; Trivedi, C.; Santangeli, P.; Santoro, F.; Hongo, R.; Hao, S.; Beheiry, S.; et al. Long-term outcome of catheter ablation in atrial fibrillation patients with coexistent metabolic syndrome and obstructive sleep apnea: Impact of repeat procedures vs. lifestyle changes. J. Cardiovasc. Electrophysiol. 2014, 25, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.J.; Cho, S.I.; Tiwari, N.; Bergman, M.; Kizer, J.R.; Palma, E.C.; Taub, C.C. Impact of metabolic syndrome on the risk of atrial fibrillation recurrence after catheter ablation: Systematic review and meta-analysis. J. Interv. Card. Electrophysiol. 2014, 39, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Lopez, F.L.; Agarwal, S.K.; Maclehose, R.F.; Soliman, E.Z.; Sharrett, A.R.; Huxley, R.R.; Konety, S.; Ballantyne, C.M.; Alonso, A. Blood lipid levels, lipid-lowering medications, and the incidence of atrial fibrillation: The atherosclerosis risk in communities study. Circ. Arrhythm. Electrophysiol. 2012, 5, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Tanabe, N.; Yagihara, N.; Watanabe, T.; Aizawa, Y.; Kodama, M. Association between lipid profile and risk of atrial fibrillation. Circ. J. 2011, 75, 2767–2774. [Google Scholar] [CrossRef] [PubMed]
- Fauchier, L.; Clementy, N.; Babuty, D. Statin therapy and atrial fibrillation: Systematic review and updated meta-analysis of published randomized controlled trials. Curr. Opin. Cardiol. 2013, 28, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Bang, C.N.; Greve, A.M.; Abdulla, J.; Kober, L.; Gislason, G.H.; Wachtell, K. The preventive effect of statin therapy on new-onset and recurrent atrial fibrillation in patients not undergoing invasive cardiac interventions: A systematic review and meta-analysis. Int. J. Cardiol. 2013, 167, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Hanna, I.R.; Heeke, B.; Bush, H.; Brosius, L.; King-Hageman, D.; Dudley, S.C., Jr.; Beshai, J.F.; Langberg, J.J. Lipid-lowering drug use is associated with reduced prevalence of atrial fibrillation in patients with left ventricular systolic dysfunction. Heart Rhythm 2006, 3, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.Y.; Lin, C.H.; Loh el, W.; Ting, C.T.; Wu, T.J. CHADS2 score, statin therapy, and risks of atrial fibrillation. Am. J. Med. 2013, 126, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Veronese, G.; Montomoli, J.; Schmidt, M.; Horvath-Puho, E.; Sorensen, H.T. Statin use and risk of atrial fibrillation or flutter: A population-based case-control study. Am. J. Ther. 2015, 22, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Du, J.L.; Yuan, J.; Chen, Y.Q. Statin therapy is beneficial for the prevention of atrial fibrillation in patients with coronary artery disease: A meta-analysis. Eur. J. Pharmacol. 2013, 707, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Kim, J.M.; Shin, D.G.; Kim, J.R.; Cho, K.H. Relation of atrial fibrillation (AF) and change of lipoproteins: Male patients with AF exhibited severe pro-inflammatory and pro-atherogenic properties in lipoproteins. Clin. Biochem. 2014, 47, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Krauss, R.M. Lipids and lipoproteins in patients with type 2 diabetes. Diabetes Care 2004, 27, 1496–1504. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Kwon, S.; Shaughnessy, S.; Wallace, P.; Hutto, A.; Jenkins, A.J.; Klein, R.L.; Garvey, W.T. Critical evaluation of adult treatment panel III criteria in identifying insulin resistance with dyslipidemia. Diabetes Care 2004, 27, 978–983. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Lu, J.; Chen, S.H.; Huang, R.Y.; Yilmaz, H.R.; Dong, J.; Elayda, M.A.; Dixon, R.A.; Yang, C.Y. Effects of electronegative VLDL on endothelium damage in metabolic syndrome. Diabetes Care 2012, 35, 648–653. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Lee, J.H.; Kim, J.R.; Shin, D.G.; Lee, S.H.; Cho, K.H. Female patients with atrial fibrillation have increased oxidized and glycated lipoprotein properties and lower apolipoprotein A–I expression in HDL. Int. J. Mol. Med. 2011, 27, 841–849. [Google Scholar] [PubMed]
- Brooks, W.W.; Conrad, C.H. Isoproterenol-induced myocardial injury and diastolic dysfunction in mice: Structural and functional correlates. Comp. Med. 2009, 59, 339–343. [Google Scholar] [PubMed]
- Isa-Param, R.; Perez-Castellano, N.; Villacastin, J.; Moreno, J.; Salinas, J.; Alonso, R.; Ruiz, E.; Doblado, M.; Morales, R.; Macaya, C. Inducibility of atrial arrhythmias after adenosine and isoproterenol infusion in patients referred for atrial fibrillation ablation. Rev. Esp. Cardiol. 2006, 59, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Son, N.H.; Yu, S.; Tuinei, J.; Arai, K.; Hamai, H.; Homma, S.; Shulman, G.I.; Abel, E.D.; Goldberg, I.J. PPARγ-induced cardiolipotoxicity in mice is ameliorated by PPARα deficiency despite increases in fatty acid oxidation. J. Clin. Investig. 2010, 120, 3443–3454. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, J.E. Lipotoxicity: When tissues overeat. Curr. Opin. Lipidol. 2003, 14, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Van de Weijer, T.; Schrauwen-Hinderling, V.B.; Schrauwen, P. Lipotoxicity in type 2 diabetic cardiomyopathy. Cardiovasc. Res. 2011, 92, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Abdurrachim, D.; Luiken, J.J.; Nicolay, K.; Glatz, J.F.; Prompers, J.J.; Nabben, M. Good and bad consequences of altered fatty acid metabolism in heart failure: Evidence from mouse models. Cardiovasc. Res. 2015, 106, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Cal, R.; Castellano, J.; Revuelta-Lopez, E.; Aledo, R.; Barriga, M.; Farre, J.; Vilahur, G.; Nasarre, L.; Hove-Madsen, L.; Badimon, L.; et al. Low-density lipoprotein receptor-related protein 1 mediates hypoxia-induced very low density lipoprotein-cholesteryl ester uptake and accumulation in cardiomyocytes. Cardiovasc. Res. 2012, 94, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Perman, J.C.; Bostrom, P.; Lindbom, M.; Lidberg, U.; StAhlman, M.; Hagg, D.; Lindskog, H.; Scharin Tang, M.; Omerovic, E.; Mattsson Hulten, L.; et al. The VLDL receptor promotes lipotoxicity and increases mortality in mice following an acute myocardial infarction. J. Clin. Investig. 2011, 121, 2625–2640. [Google Scholar] [CrossRef] [PubMed]
- Pulinilkunnil, T.; Rodrigues, B. Cardiac lipoprotein lipase: Metabolic basis for diabetic heart disease. Cardiovasc. Res. 2006, 69, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, A.; Aikawa, M.; Libby, P.; Alcaide, P.; Luscinskas, F.W.; Sacks, F.M. Apolipoprotein CIII in apolipoprotein B lipoproteins enhances the adhesion of human monocytic cells to endothelial cells. Circulation 2006, 113, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, A.; Osaka, M.; Aikawa, M.; Uematsu, S.; Akira, S.; Libby, P.; Shimokado, K.; Sacks, F.M.; Yoshida, M. Toll-like receptor 2 mediates apolipoprotein CIII-induced monocyte activation. Circ. Res. 2008, 103, 1402–1409. [Google Scholar] [CrossRef] [PubMed]
- Asai, T.; Okumura, K.; Takahashi, R.; Matsui, H.; Numaguchi, Y.; Murakami, H.; Murakami, R.; Murohara, T. Combined therapy with PPARα agonist and l-carnitine rescues lipotoxic cardiomyopathy due to systemic carnitine deficiency. Cardiovasc. Res. 2006, 70, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Heijman, J.; Voigt, N.; Nattel, S.; Dobrev, D. Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ. Res. 2014, 114, 1483–1499. [Google Scholar] [CrossRef] [PubMed]
- Castellano, J.; Farre, J.; Fernandes, J.; Bayes-Genis, A.; Cinca, J.; Badimon, L.; Hove-Madsen, L.; Llorente-Cortes, V. Hypoxia exacerbates Ca2+-handling disturbances induced by very low density lipoproteins (VLDL) in neonatal rat cardiomyocytes. J. Mol. Cell. Cardiol. 2011, 50, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Pugh, K.G.; Wei, J.Y. Clinical implications of physiological changes in the aging heart. Drugs Aging 2001, 18, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA 2001, 285, 2486–2497. [Google Scholar] [CrossRef] [PubMed]
- Beisiegel, U.; Weber, W.; Ihrke, G.; Herz, J.; Stanley, K.K. The LDL-receptor-related protein, LRP, is an apolipoprotein E-binding protein. Nature 1989, 341, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Ge, F.; Hu, C.; Hyodo, E.; Arai, K.; Zhou, S.; Lobdell, H.T.; Walewski, J.L.; Homma, S.; Berk, P.D. Cardiomyocyte triglyceride accumulation and reduced ventricular function in mice with obesity reflect increased long chain fatty acid uptake and de novo fatty acid synthesis. J. Obes. 2012, 2012, 205648. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.-C.; Lin, H.-T.; Ke, L.-Y.; Wei, C.; Hsiao, Y.-L.; Chu, C.-S.; Lai, W.-T.; Shin, S.-J.; Chen, C.-H.; Sheu, S.-H.; et al. VLDL from Metabolic Syndrome Individuals Enhanced Lipid Accumulation in Atria with Association of Susceptibility to Atrial Fibrillation. Int. J. Mol. Sci. 2016, 17, 134. https://doi.org/10.3390/ijms17010134
Lee H-C, Lin H-T, Ke L-Y, Wei C, Hsiao Y-L, Chu C-S, Lai W-T, Shin S-J, Chen C-H, Sheu S-H, et al. VLDL from Metabolic Syndrome Individuals Enhanced Lipid Accumulation in Atria with Association of Susceptibility to Atrial Fibrillation. International Journal of Molecular Sciences. 2016; 17(1):134. https://doi.org/10.3390/ijms17010134
Chicago/Turabian StyleLee, Hsiang-Chun, Hsin-Ting Lin, Liang-Yin Ke, Chi Wei, Yi-Lin Hsiao, Chih-Sheng Chu, Wen-Ter Lai, Shyi-Jang Shin, Chu-Huang Chen, Sheng-Hsiung Sheu, and et al. 2016. "VLDL from Metabolic Syndrome Individuals Enhanced Lipid Accumulation in Atria with Association of Susceptibility to Atrial Fibrillation" International Journal of Molecular Sciences 17, no. 1: 134. https://doi.org/10.3390/ijms17010134