
Critical Role of Endoplasmic Reticulum Stress in Cognitive Impairment Induced by Microcystin-LR

Abstract
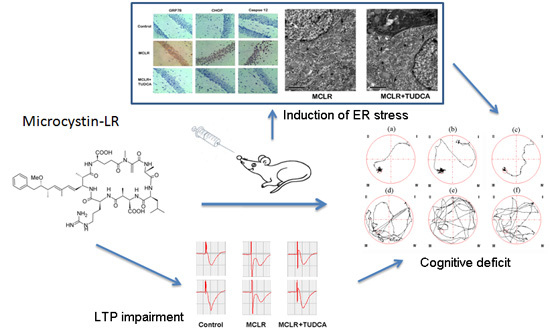
:
1. Introduction
2. Results
2.1. ER Stress Inhibitor Rescued MCLR-Induced Memory Impairment

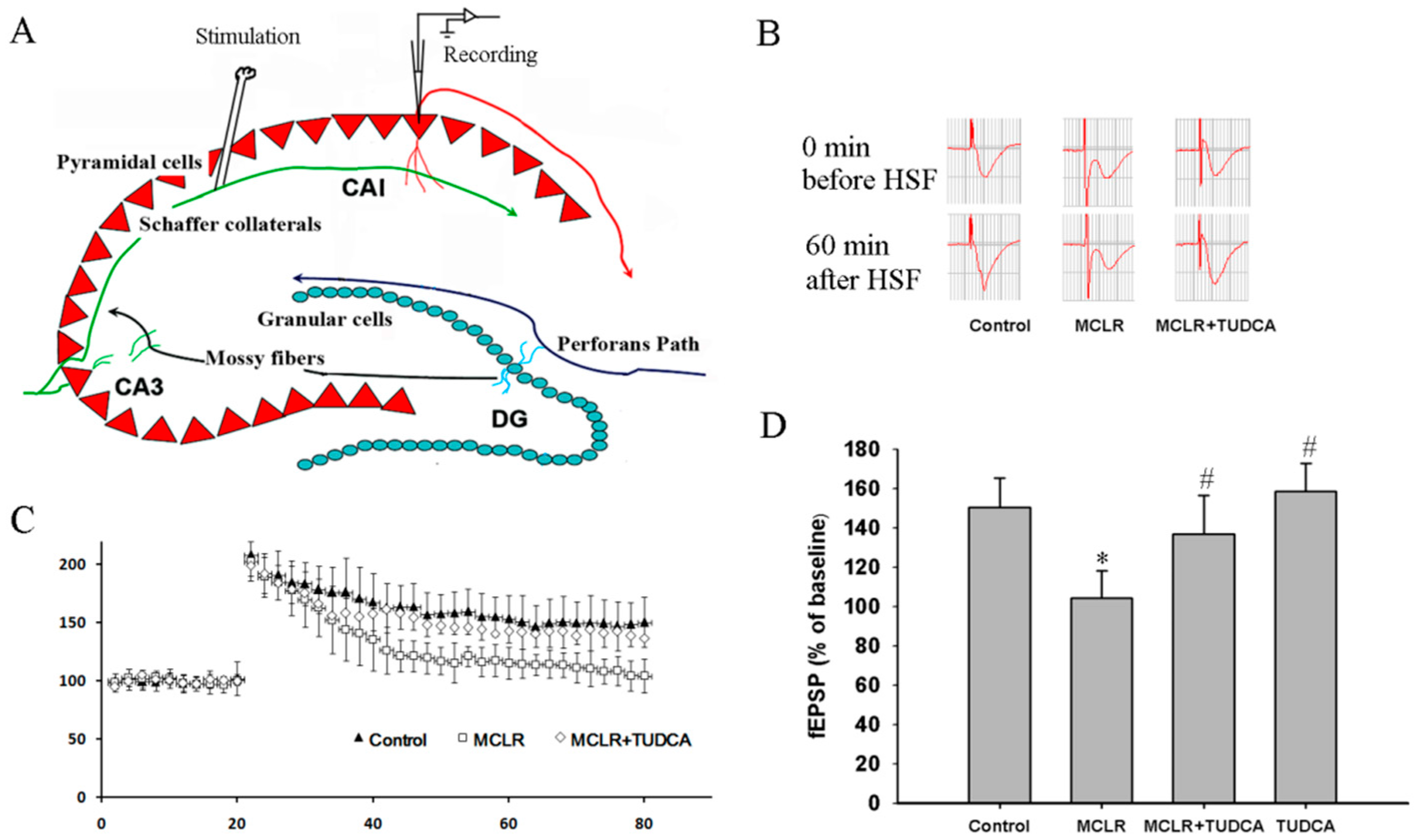
2.2. MCLR-Caused ER Stress Impaired Hippocampal Synaptic Plasticity

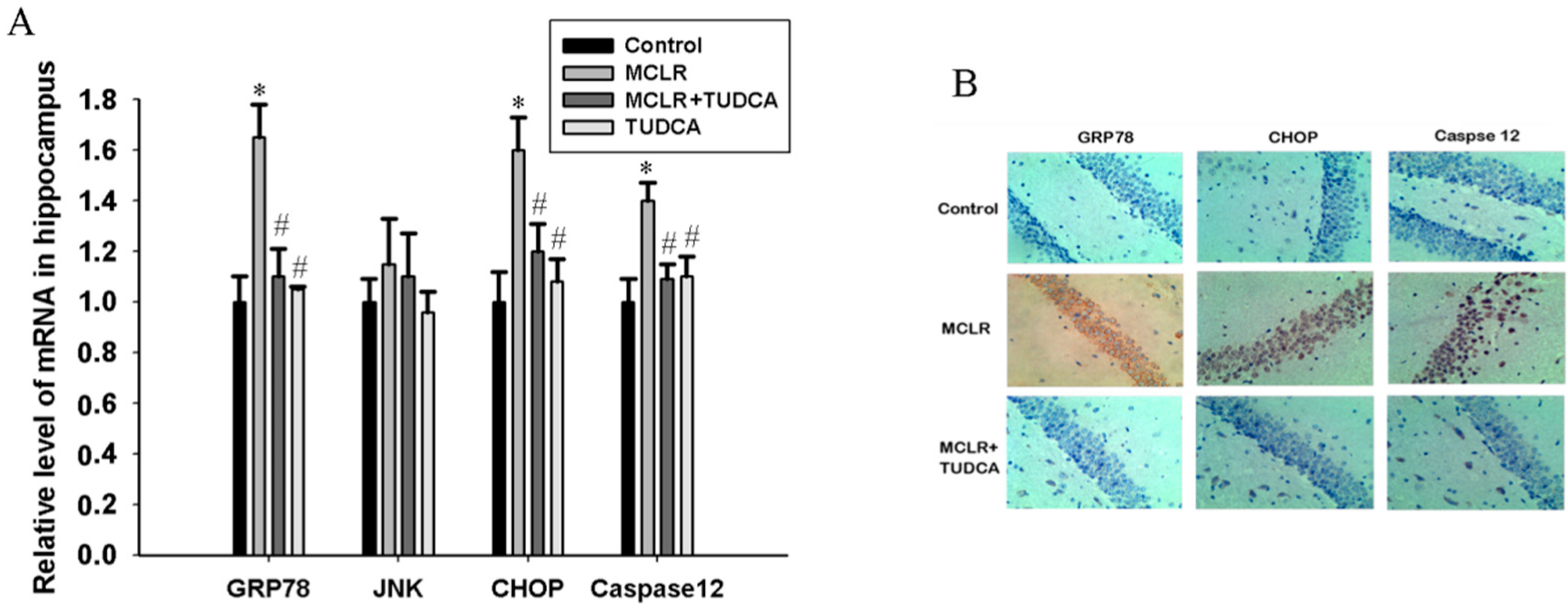
2.3. MCLR induced ER Stress in Hippocampus of Rats

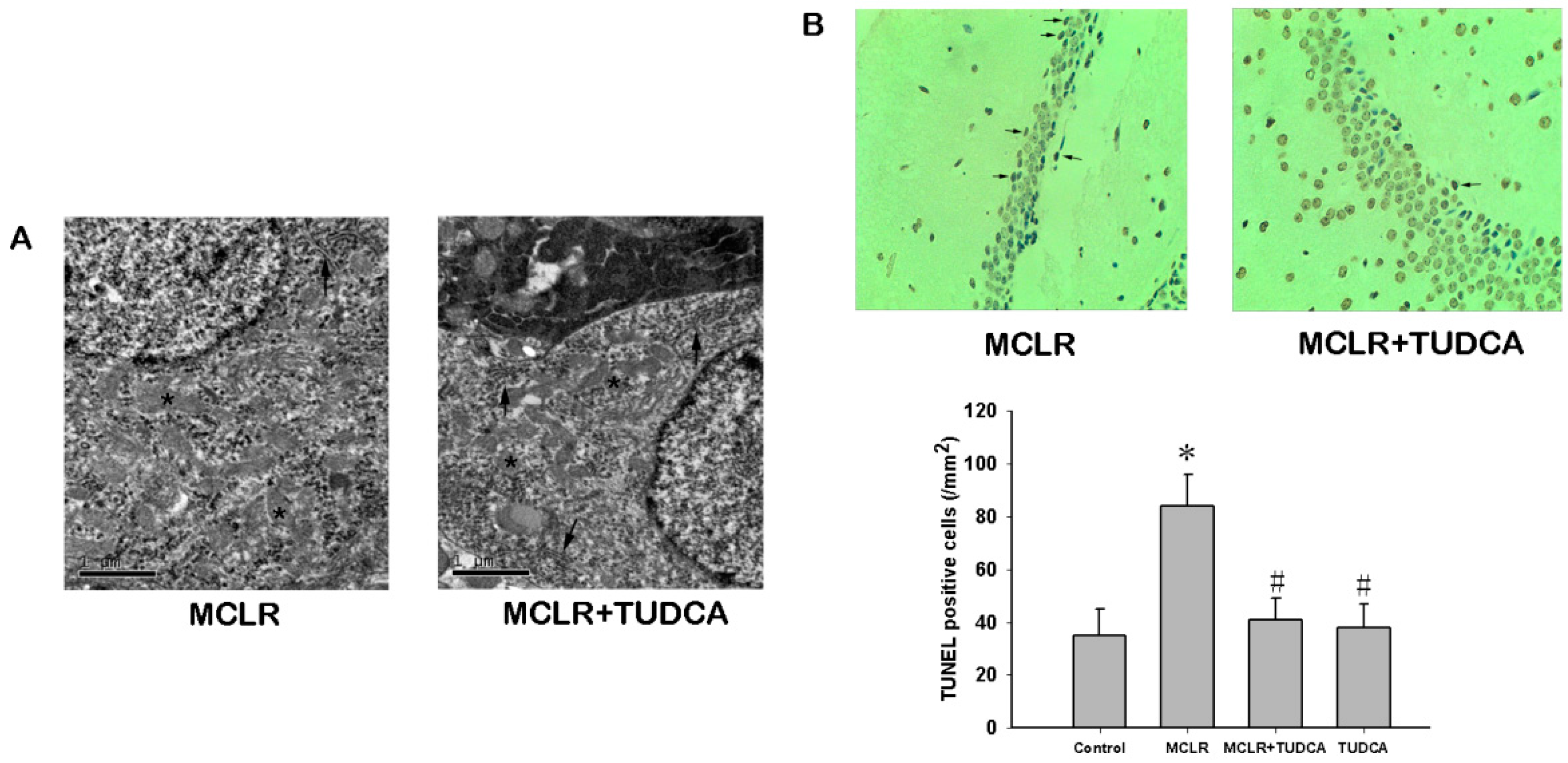
2.4. MCLR Induced Ultrastructural Changes and Apoptosis in Hippocampus

3. Discussion
4. Experimental Section
4.1. Animals
4.2. Chemicals
4.3. Behavioral Studies
4.4. LTP Measurements
4.5. Real-Time Polymerase Chain Reaction
4.6. Immunohistochemistry Assay

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Primer Sequence (5′–3′) | Accession No. | Melting Temperature (°C) | Product Size (bp) |
|---|---|---|---|---|
| GRP78 | F-AACCCAGATGAGGCTGTAGCA | NM022310 | 60 | 91 |
| R-ACATCAAGCAGAACCAGGTCAC | ||||
| JNK | F-TGATGACGCCTTACGTGGTA | XM341399 | 60 | 114 |
| R-GGCAAACCATTTCTCCCATA | ||||
| CHOP | F-CCAGCAGAGGTCACAAGCAC | NM007837 | 60 | 126 |
| R-CGCACTGACCACTCTGTTTC | ||||
| Caspase12 | F-CACTGCTGATACAGATGAGG | NM130442 | 60 | 138 |
| R-CCACTCTTGCCTACCTTCC |
4.7. TUNEL Assay
4.8. Ultrastructural Observation
4.9. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rantala, A.; Fewer, D.P.; Hisbergues, M.; Rouhiainen, L.; Vaitomaa, J.; Börner, T.; Sivonen, K. Phylogenetic evidence for the early evolution of microcystin synthesis. Proc. Natl. Acad. Sci. USA 2004, 101, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Fiore, M.F.; Genuario, D.B.; da Silva, C.S.P.; Shishido, T.K.; Moraes, L.A.B.; CantusioNeto, R.; Silva-Stenico, M.E. Microcystin production by a freshwater spring cyanobacterium of the genus Fischerella. Toxicon 2009, 53, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Wiedner, C.; Visser, P.M.; Fastner, J.; Metcalf, J.S.; Codd, G.A.; Mur, L.R. Effects of light on the microcystin content of Microcystis strain PCC 7806. Appl. Environ. Microbiol. 2003, 69, 1475–1481. [Google Scholar] [CrossRef] [PubMed]
- Fastner, J.; Codd, G.A.; Metcalf, J.S.; Woitke, P.; Wiedner, C.; Utkilen, H. An international comparison exercise for the determination of purified microcystin-LR and microcystins in field material. Anal. Bioanal. Chem. 2002, 374, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W.J.; Dietrich, D.R. Pathological and biochemical characterization of microcystin-induced hepatopancreas and kiney damage in carp (Cyprinus Carpio). Toxicol. Appl. Pharmacol. 2002, 164, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Fischera, W.J.; Altheimara, S.; Cattorib, V.; Meierb, P.J.; Dietricha, D.R.; Hagenbuchb, B. Organic anion transporting polypeptides expressed in liver and brain mediate uptake of microcystin. Toxicol. Appl. Pharmacol. 2005, 203, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Cazenave, J.; Nores, M.L.; Miceli, M.; Díaz, M.P.; Wunderlin, D.A.; Bistoni, M.A. Changes in the swimming activity and the glutathione-S-transferase activity of Jenynsia multidentata fed with microcystin-RR. Water Res. 2008, 42, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Nishiwaki, R.; Ohta, T.; Sueoka, E.; Suganuma, M.; Harada, K.; Watanabe, M.F.; Fujike, H. Two significant aspects of microcystin-LR: Specific binding and liver specificity. Cancer Lett. 1994, 83, 283–289. [Google Scholar] [CrossRef]
- Feurstein, D.; Holst, K.; Fischer, A.; Dietrich, D.R. Oatp-associated uptake and toxicity of microcystins in primary murine whole brain cells. Toxicol. Appl. Pharmacol. 2009, 234, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Feurstein, D.; Kleinteich, J.; Heussner, A.H.; Stemmer, K.; Dietrich, D.R. Investigation of microcystin congerner-dependent uptake into primary murine neurons. Environ. Health Perspect. 2010, 118, 1370–1375. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, W.W.; Azevedo, S.M.; An, J.S.; Molica, R.J.; Jochimsen, E.M.; Lau, S.; Rinehart, K.L.; Shaw, G.R.; Eaglesham, G.K. Human fatalities from cyanobacteria: Chemical and biological evidence for cyanotoxins. Environ. Health Perspect. 2001, 109, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Soares, R.M.; Yuan, M.; Servaites, J.C.; Delgado, A.; Magalhaes, V.F.; Hiborn, E.D.; Carmichael, W.W.; Azevedo, S.M. Sublethal exposure from microcystins to renal insufficiency patients in Rio de Janeiro, Brazil. Environ. Toxicol. 2006, 21, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Li, G.Y.; Cai, F.; Yan, W.; Li, C.R.; Wang, J.H. A proteomic analysis of MCLR-induced neurotoxicity: Implications for Alzheimer’s disease. Toxicol. Sci. 2012, 127, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Li, G.Y.; Yan, W.; Cai, F.; He, Y.; Chen, N.; Wang, J.H. Spatial learning impairment and pathological change in rats induced by acute exposure to microcystin-LR. Environ. Toxicol. 2014, 29, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, X.; Ju, J.; Li, Y.; Yin, L.; Pu, Y. Maternal repeated oral exposure to microcystin-LR affects neurobehaviors in developing rat. Environ. Toxicol. Chem. 2015, 34, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.J.; Grimwood, P.D.; Morris, R.D. Synaptic plasticity and memory: An evaluation of the hypothesis. Annu. Rev. Neurosci. 2000, 23, 649–711. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lin, F.; Cai, F.; Yan, W.; Zhou, Q.; Xie, L. Microcystin-LR inhibited hippocampal long-term potential via regulation of the glycogen synthase kinase-3β pathway. Chemosphere 2013, 93, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Mackintosh, C.; Beattie, K.A.; Klumpp, S.; Cohen, P.; Codd, G.A. Cyanobacterial microcystin-LR is a potent and specific inhibitor of protein phosphatase 1 and 2A from both mammals and higher plants. FEBS Lett. 1990, 264, 187–192. [Google Scholar] [CrossRef]
- Meng, G.; Sun, Y.; Fu, W.; Guo, Z.; Xu, L. Microcystin-LR induces cytoskeleton system reorganization through hyperphosphorylation of tau and HSP27 via PP2A inhibition and subsequent activation of the p38 MAPK signaling pathway in neuroendocrine (PC12) cells. Toxicology 2011, 290, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Liu, J.; Lin, S.; Guo, Z.; Xu, L. Microcystin-LR-caused ROS generation involved in p38 Activation and tau hyperphosphorylation in neuroendocrine (PC12) cells. Environ. Toxicol. 2015, 30, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.X.; Shen, H.M.; Ong, C.N. Critical role of reactive oxygen species and mitochondrial permeability transition in microcystin-induced rapid apoptosis in rat hepatocytes. Hepatology 2000, 32, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Boyce, M.; Yuan, J. Cellular response to endoplasmic reticulum stress: A matter of life or death. Cell Death Differ. 2006, 13, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Ackerman, S.L. Endoplasmic reticulum stress in health and disease. Curr. Opin. Cell Biol. 2006, 18, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 2005, 35, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Görgün, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Khaoustov, V.I.; Chung, C.C.; Sohn, J.; Krishnan, B.; Lewis, D.E.; Yoffe, B. Effect of tauroursodeoxycholic acid on endoplasmic reticulum stress-induced caspase-12 activation. Hepatoloty 2002, 36, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Sayal, A.J. Endoplasmic reticulum stress and the unfolded protein response. Clin. Liver Dis. 2009, 13, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Cai, C.; Wu, Y.; Shao, D.; Ye, B.; Zhang, Y.; Liu, J.; Wang, J.; Jia, X. Mitochondrial and endoplasmic reticulum pathways involved in microcystin-LR-induced apoptosis of the testes of male frog (Rana nigromaculata) in vivo. J. Hazard. Mater. 2013, 252–253, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Tu, W.W.; Xu, J. Mechanisms of microcystin-LR-induced cytoskeletal disruption in animal cells. Toxicon 2015, 101, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Xu, L.; Zhang, X.; Wang, Y.; Meng, X.; Miao, A.; Yang, L. Endoplasmic reticulum stress in murine liver and kidney exposed to microcystin-LR. Toxicon 2010, 56, 1334–1341. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.L.; Wang, X.F.; Xu, L.H. Alteration of proteins expression in apoptotic FL cells induced by MCLR. Environ. Toxicol. 2008, 23, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Li, G.; Chen, J. A proteomic analysis of prenatal transfer of microcystin-LR induced neurotoxicity in rat offspring. J. Proteom. 2015, 114, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Kaufman, R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Brandeis, R.; Brandys, Y.; Yehuda, S. The use of the Morris water maze in the study of memory and learning. Int. J. Neurosci. 1989, 48, 29–69. [Google Scholar] [CrossRef] [PubMed]
- Cai, F.; Wang, F.; Lin, F.K.; Liu, C.; Ma, L.Q.; Liu, J.; Wu, W.N.; Wang, W.; Wang, J.H.; Chen, J.G. Redox modulation of long-term potentiation in the hippocampus via regulation of the glycogen synthase kinase-3β pathway. Free Radic. Biol. Med. 2008, 45, 964–970. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Sun, Y.; Li, Z.; Song, T.; Wang, H.; Zhang, Y.; Ge, Z. Apoptosis induced by endoplasmic reticulum stress involved in diabetic kidney disease. Biochem. Biophys. Res. Commun. 2008, 370, 651–656. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, F.; Liu, J.; Li, C.; Wang, J. Critical Role of Endoplasmic Reticulum Stress in Cognitive Impairment Induced by Microcystin-LR. Int. J. Mol. Sci. 2015, 16, 28077-28086. https://doi.org/10.3390/ijms161226083
Cai F, Liu J, Li C, Wang J. Critical Role of Endoplasmic Reticulum Stress in Cognitive Impairment Induced by Microcystin-LR. International Journal of Molecular Sciences. 2015; 16(12):28077-28086. https://doi.org/10.3390/ijms161226083
Chicago/Turabian StyleCai, Fei, Jue Liu, Cairong Li, and Jianghua Wang. 2015. "Critical Role of Endoplasmic Reticulum Stress in Cognitive Impairment Induced by Microcystin-LR" International Journal of Molecular Sciences 16, no. 12: 28077-28086. https://doi.org/10.3390/ijms161226083