
Rational Design of Diketopyrrolopyrrole-Based Small Moleculesas Donating Materials for Organic Solar Cells

Abstract
:
1. Introduction

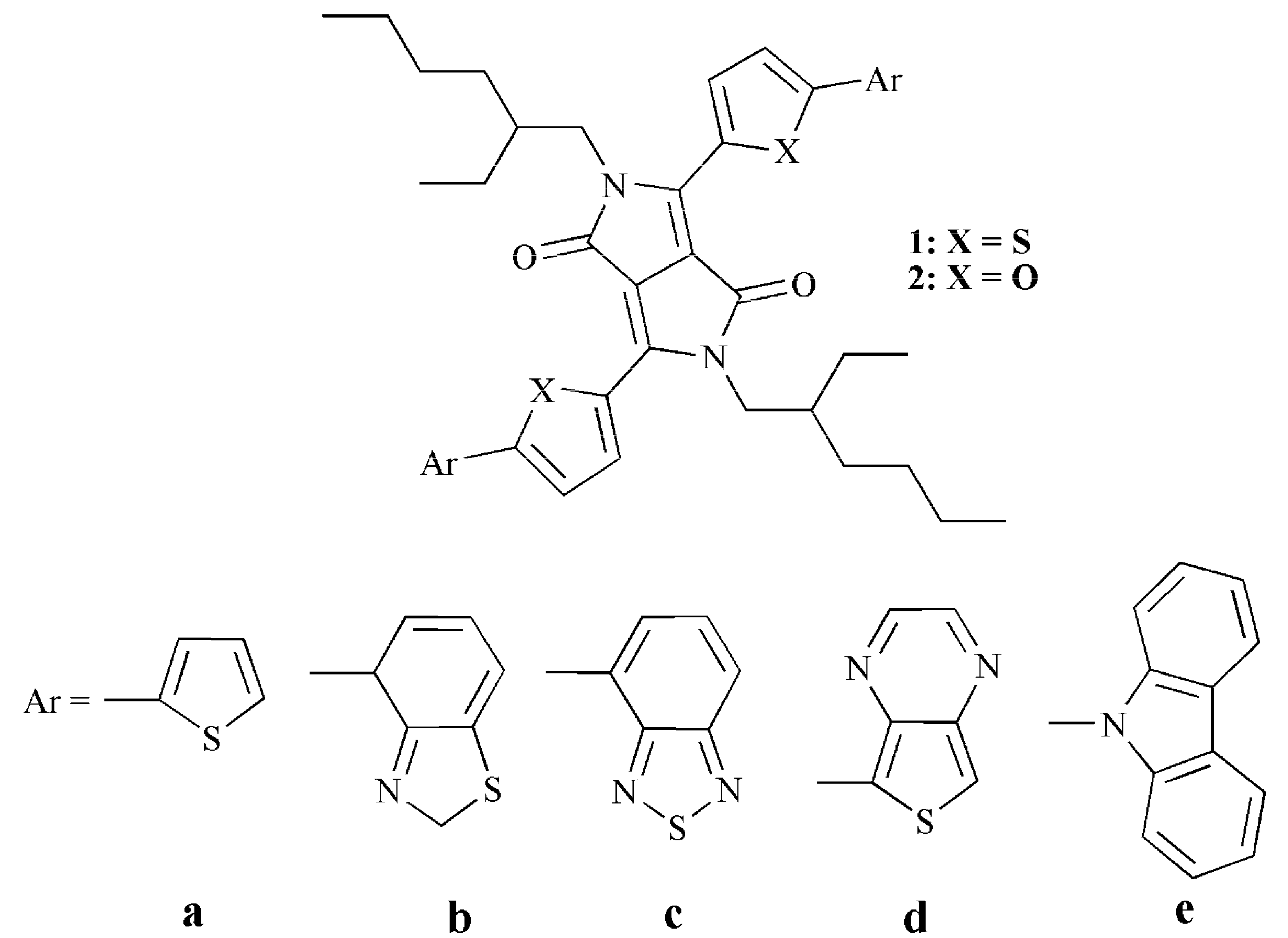
2. Results and Discussion
2.1. Frontier Molecular Orbitals


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | HOMOs | LUMOs | ||||
|---|---|---|---|---|---|---|
| BEDPP a | CB b | Ar c | BEDPP a | CB b | Ar c | |
| 1a | 55.3 | 30.2 | 14.5 | 46.8 | 37.8 | 15.4 |
| 1b | 61.1 | 29.2 | 9.7 | 48.3 | 37.5 | 14.2 |
| 1c | 58.7 | 29.0 | 12.4 | 28.0 | 25.3 | 46.8 |
| 1d | 52.1 | 28.5 | 19.4 | 23.4 | 23.3 | 53.3 |
| 1e | 55.2 | 28.2 | 16.6 | 55.3 | 40.2 | 4.50 |
| 2a | 54.4 | 30.7 | 14.9 | 52.6 | 33.1 | 14.3 |
| 2b | 57.9 | 30.2 | 11.9 | 52.7 | 32.7 | 14.6 |
| 2c | 56.6 | 29.8 | 13.6 | 31.0 | 21.1 | 47.9 |
| 2d | 54.6 | 27.7 | 17.7 | 26.4 | 19.3 | 54.4 |
| 2e | 51.4 | 29.2 | 19.4 | 60.0 | 34.2 | 5.80 |

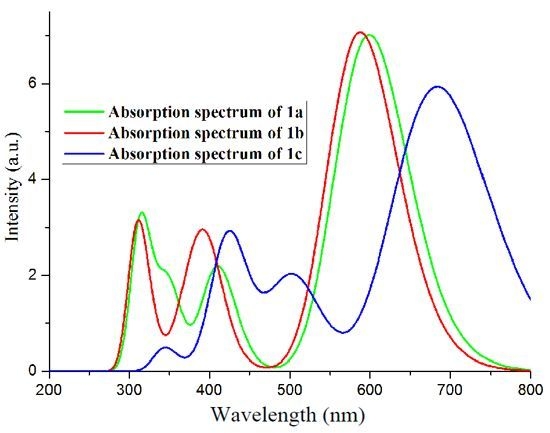
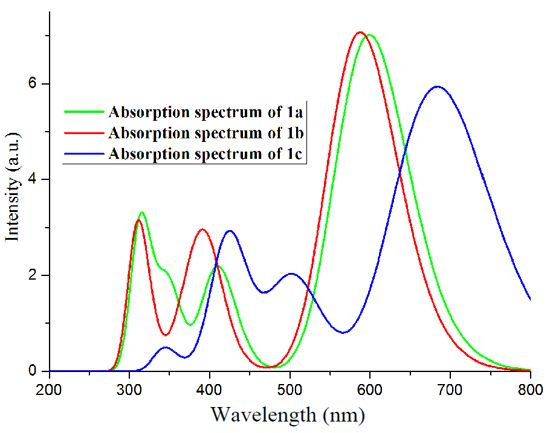
2.2. Absorption Spectra
| Species | λabs (nm) | f | Main configurations | R (nm) a |
|---|---|---|---|---|
| 1a | 599 | 0.97 | H → L (0.71) | 295 |
| 1b | 588 | 0.98 | H → L (0.71) | 281 |
| 1c | 684 | 0.82 | H → L (0.70) | 340 |
| 1d | 746 | 0.80 | H → L (0.70) | 387 |
| 1e | 575 | 0.87 | H → L (0.70) | 261 |
| 2a | 583 | 0.74 | H → L (0.71) | 209 |
| 2b | 581 | 0.81 | H → L (0.71) | 288 |
| 2c | 669 | 0.61 | H → L (0.70) | 354 |
| 2d | 716 | 0.56 | H → L (0.70) | 357 |
| 2e | 574 | 0.73 | H → L (0.70) | 271 |
| Exp b | 590 |
2.3. Reorganization Energies
| Species | λh | λe |
|---|---|---|
| 1a | 0.293 | 0.160 |
| 1b | 0.295 | 0.199 |
| 1c | 0.266 | 0.154 |
| 1d | 0.258 | 0.171 |
| 1e | 0.489 | 0.285 |
| 2a | 0.301 | 0.153 |
| 2b | 0.291 | 0.159 |
| 2c | 0.273 | 0.143 |
| 2d | 0.287 | 0.191 |
| 2e | 0.426 | 0.210 |
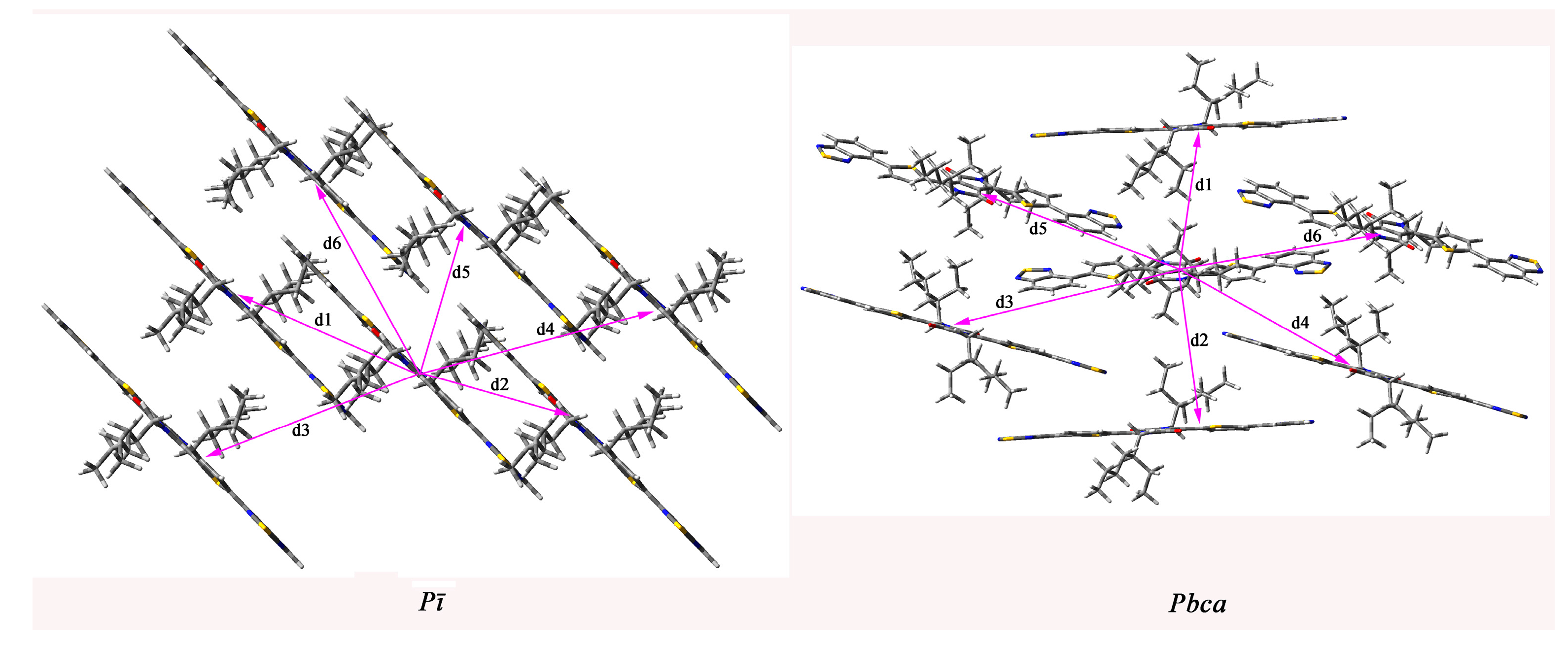
2.4. Calculated Crystal Structure and Transport Properties


| Space Groups | Pathway | Distance (Å) | Electron Coupling (eV) | Hole Coupling (eV) |
|---|---|---|---|---|
| Pī | 1 | 13.450 | −9.00 × 10−3 | −3.22 × 10−4 |
| 2 | 13.450 | −9.00 × 10−3 | −3.22× 10−4 | |
| 3 | 16.936 | −3.28 × 10−7 | 1.25 × 10−6 | |
| 4 | 16.936 | −3.28 × 10−7 | 1.25 × 10−6 | |
| 5 | 11.223 | 1.50 × 10−3 | −3.20 × 10−3 | |
| 6 | 13.509 | 1.50 × 10−8 | 1.04 × 10−8 | |
| Pbca | 1 | 13.373 | −5.86 × 10−6 | 8.19 × 10−6 |
| 2 | 13.373 | −5.86 × 10−6 | 8.19 × 10−6 | |
| 3 | 14.824 | −5.80 × 10−3 | 2.50 × 10−3 | |
| 4 | 14.824 | −5.80 × 10−3 | 2.50 × 10−3 | |
| 5 | 14.824 | −5.80 × 10−3 | 2.50 × 10−3 | |
| 6 | 15.591 | −6.50 × 10−3 | 5.79 × 10−4 |
| Species | Space Groups | Electron Mobility | Hole Mobility |
|---|---|---|---|
| 1a | P212121 | 1.05 × 10−2 | 8.18 × 10−3 |
| 1b | Pna21 | 2.21 × 10−4 | 1.18 × 10−4 |
| 1c | Pī | 8.97 × 10−2 | 2.00 × 10−3 |
| 1c | Pbca | 5.11 × 10−2 | 2.14 × 10−3 |
| 1d | C2/c | 0.136 | 9.57 × 10−3 |
| 1e | P21/c | 2.45 × 10−6 | 7.75 × 10−6 |
| 2a | P21 | 7.561 × 10−3 | 1.77 × 10−3 |
| 2b | P21 | 4.22 × 10−5 | 7.13 × 10−2 |
| 2c | Cc | 8.18 × 10−4 | 1.31 × 10−5 |
| 2d | P212121 | 3.14 × 10−5 | 2.56 × 10−5 |
| 2e | Pī | 2.55 × 10−5 | 1.78 × 10−5 |
3. Computational Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Service, R.F. Outlook brightens for plastic solar cells. Science 2011, 332, 293. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Li, Y.F.; Zhan, X.W. Small molecule semiconductors for high-efficiency organic photovoltaics. Chem. Soc. Rev. 2012, 41, 4245–4272. [Google Scholar] [CrossRef] [PubMed]
- Günes, S.; Neugebauer, H.; Sariciftci, N.S. Conjugated polymer-based organic solar cells. Chem. Rev. 2007, 107, 1324–1338. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.; Kim, C.; Nguyen, T.Q. Small molecule solution-processed bulk heterojunction solar cells. Chem. Mater. 2011, 23, 470–482. [Google Scholar] [CrossRef]
- Shang, H.X.; Fan, H.J.; Liu, Y.; Hu, W.P.; Li, Y.F.; Zhan, X.W. A solution-processable star-shaped molecule for high-performance organic solar cells. Adv. Mater. 2011, 23, 1554–1557. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.W. 2-Layer organic photovoltaic cell. Appl. Phys. Lett. 1986, 48, 183–185. [Google Scholar] [CrossRef]
- Liang, Y.; Yu, L. Development of semiconducting polymers for solar energy harvesting. Polym. Rev. 2010, 50, 454–473. [Google Scholar] [CrossRef]
- Montcada, N.F.; Pelado, B.; Viterisi, A.; Albero, J.; Coro, J.; Cruz, P.D.L.; Langa, F.; Palomares, E. High open circuit voltage in efficient thiophene-based small molecule solution processed organic solar cells. Org. Electron. 2013, 14, 2826–2832. [Google Scholar] [CrossRef]
- You, J.; Dou, L.; Yoshimura, K.; Kato, T.; Ohya, K.; Moriarty, T.; Emery, K.; Chen, C.C.; Gao, J.; Li, G.; et al. A polymer tandem solar cell with 10.6% power conversion efficiency. Nat. Commun. 2013, 4, 1446–1456. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhu, R.; Yang, Y. Polymer solar cells. Nat. Photonics 2012, 6, 153–161. [Google Scholar] [CrossRef]
- Brück, S.; Krause, C.; Turrisi, R.; Beverina, L.; Wilken, S.; Saak, W.; Lützen, A.; Borchert, H.; Schiek, M.; Parisi, J. Structure-property relationship of anilino-squaraines in organic solar cells. Phys. Chem. Chem. Phys. 2014, 16, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Roncali, J. Molecular bulk heterojunctions: An emerging approach to organic solar cells. Acc. Chem. Res. 2009, 42, 1719–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demeter, D.; Rousseau, T.; Leriche, P.; Cauchy, T.; Po, R.; Roncali, J. Manipulation of the open-circuit voltage of organic solar cells by desymmetrization of the structure of acceptoredonoreacceptor molecules. Adv. Funct. Mater. 2011, 21, 4379–4387. [Google Scholar] [CrossRef] [Green Version]
- Sahu, D.; Tsai, C.H.; Wei, H.Y.; Ho, K.C.; Chang, F.C.; Chu, C.W. Synthesis and applications of novel low bandgap star-burst molecules containing a triphenylamine core and dialkylated diketopyrrolopyrrole arms for organic photovoltaics. J. Mater. Chem. 2012, 22, 7945–7953. [Google Scholar] [CrossRef]
- Mishra, A.; Bäuerle, P. Small molecule organic semiconductors on the move: Promises for future solar energy technology. Angew. Chem. Int. Ed. 2012, 51, 2020–2067. [Google Scholar] [CrossRef] [PubMed]
- Leliège, A.; Régent, C.H.L.; Allain, M.; Blanchard, P.; Roncali, J. Structural modulation of internal charge transfer in small molecular donors for organic solar cells. Chem. Commun. 2012, 48, 8907–8909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Liu, M.; Zeng, Q.; Wu, Z.; Piao, L.; Zhao, S. Novel porphyrin–phthalocyanine heterodimers and heteropentamers: Synthesis, characterization and application in organic solar cells. RSC Adv. 2013, 3, 13259–13264. [Google Scholar] [CrossRef]
- Sun, Y.; Welch, G.C.; Leong, W.L.; Takacs, C.J.; Bazan, G.C.; Heeger, A.J. Solution-processed small-molecule solar cells with 6.7% efficiency. Nat. Mater. 2012, 11, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Roncali, J. Single material solar cells: The next frontier for organic photovoltaics? Adv. Energy. Mater. 2011, 1, 147–160. [Google Scholar] [CrossRef] [Green Version]
- Kyaw, A.K.K.; Wang, D.H.; Gupta, V.; Zhang, J.; Chand, S.; Bazan, G.C.; Heeger, A.J. Efficient solution-processed small-molecule solar cells with inverted structure. Adv. Mater. 2013, 25, 2397–2402. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zuo, Y.; Wan, X.; Long, G.; Zhang, Q.; Ni, W.; Liu, Y.; Li, Z.; He, G.; Li, C.; et al. Solution-processed and high-performance organic solar cells using small molecules with a benzodithiophene unit. J. Am. Chem. Soc. 2013, 135, 8484–8487. [Google Scholar] [CrossRef] [PubMed]
- Green, M.A.; Emery, K.; Hishikawa, Y.; Warta, W.; Dunlop, E.D. Solar cell efficiency tables (version 39). Prog. Photovolt. 2012, 20, 12–20. [Google Scholar] [CrossRef]
- Walker, B.; Liu, J.; Kim, C.; Welch, G.C.; Park, J.K.; Lin, J.; Zalar, P.; Proctor, C.M.; Seo, J.H.; Bazan, G.C.; et al. Optimization of energy levels by molecular design: Evaluation of bis-diketopyrrolopyrrole molecular donor materials for bulk heterojunction solar cells. Energ. Environ. Sci. 2013, 6, 952–962. [Google Scholar] [CrossRef] [Green Version]
- Janssen, R.A.J.; Nelson, J. Factors limiting device efficiency in organic photovoltaics. Adv. Mater. 2013, 25, 1847–1858. [Google Scholar] [CrossRef] [PubMed]
- He, C.; He, Q.; Yang, X.; Wu, G.; Yang, C.; Bai, F.; Shuai, Z.; Wang, L.; Li, Y. Synthesis and photovoltaic properties of a solution-processable organic molecule containing triphenylamine and DCM moieties. J. Phys. Chem. C 2007, 111, 8661–8666. [Google Scholar] [CrossRef]
- Jin, R.F.; Chang, Y.F. A theoretical study on photophysical properties of triphenylamine-cored molecules with naphthalimide arms and different p-conjugated bridges as organic solar cell materials. Phys. Chem. Chem. Phys. 2015, 17, 2094–2130. [Google Scholar] [CrossRef] [PubMed]
- Kungwan, N.; Khongpracha, P.; Namuangruk, S.; Meeprasert, J.; Chitpakdee, C.; Jungsuttiwong, S.; Promarak, V. Theoretical study of linker-type effect in carbazole–carbazole-based dyes on performances of dye-sensitized solar cells. Theor. Chem. Acc. 2014, 133, 1523–1536. [Google Scholar] [CrossRef]
- Martínez, J.P.; Osuna, S.; Solà, M.; Voityuk, A. Extent of charge separation and exciton delocalization for electronically excited states in a triphenylamine-C60 donor–acceptor conjugate: A combined molecular dynamics and TD-DFT study. Theor. Chem. Acc. 2015, 134, 12–18. [Google Scholar] [CrossRef]
- Mohamad, M.; Ahmed, R.; Shaari, A.; Goumri-Said, S. First principles investigations of vinazene molecule and molecular crystal: A prospective candidate for organic photovoltaic applications. J. Mol. Model. 2015, 21, 27–23. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhang, M.J.; Huo, L.J.; Cui, C.H.; Wu, Y.; Hou, J.H.; Li, Y.F. Poly(thieno[3, 2-b]thiophene-alt-bithiazole): A D–A copolymer donor showing improved photovoltaic performance with Indene-C60 bisadduct acceptor. Macromolecules 2012, 45, 6930–6937. [Google Scholar] [CrossRef]
- He, Z.; Zhong, C.; Huang, X.; Wong, W.Y.; Wu, H.; Chen, L.; Su, S.; Cao, Y. Simultaneous enhancement of open-circuit voltage, short-circuit current density, and fill factor in polymer solar cells. Adv. Mater. 2011, 23, 4636–4643. [Google Scholar] [CrossRef] [PubMed]
- Yin, B.; Yang, L.; Liu, Y.; Chen, Y.; Qi, Q.; Zhang, F.; Yin, S. Solution-processed bulk heterojunction organic solar cells based on an oligothiophene derivative. Appl. Phys. Lett. 2010, 97, 023303-1–023303-3. [Google Scholar] [CrossRef]
- Zhang, J.; Den, D.; He, C.; Zhang, M.; Zhang, Z.; Zhang, Z.; Li, Y. Star-shaped molecules with triphenylamine core and dicyanovinyl end-groups for organic solar cells. Chem. Mater. 2011, 23, 817–822. [Google Scholar] [CrossRef]
- Huang, J.; Jia, H.; Li, L.; Lu, Z.; Zhang, W.; He, W.; Jiang, B.; Tang, A.; Tan, Z.; Zhan, C.; et al. Fine-tuning device performances of small molecule solar cells via the more polarized DPP-attached donor units. Phys. Chem. Chem. Phys. 2012, 14, 14238–14242. [Google Scholar] [CrossRef] [PubMed]
- Murphy, L.; Hong, W.; Aziz, H.; Li, Y.N. Influences of using a high mobility donor polymer on solar cell performance. Org. Electron. 2013, 14, 3484–3492. [Google Scholar] [CrossRef]
- Lin, Y.Z.; Li, Y.F.; Zhan, X.W. A solution-processable electron acceptor based on dibenzosilole and diketopyrrolopyrrole for organic solar cells. Adv. Energy Mater. 2013, 3, 724–728. [Google Scholar] [CrossRef]
- Qiao, Y.; Guo, Y.L.; Yu, C.M.; Zhang, F.J.; Xu, W.; Liu, Y.Q.; Zhu, D.B. Diketopyrrolopyrrole-containing quinoidal small molecules for highperformance, air-stable, and solution-processable n-channel organic field-effect transistors. J. Am. Chem. Soc. 2012, 134, 4084–4087. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Song, C.E.; Cho, A.; Kim, J.; Eom, Y.; Ahn, J.; Moon, S.J.; Lim, E. Synthesis of diketopyrrolopyrrole (DPP)-based small molecule donors containing thiophene or furan for photovoltaic applications. Mater. Chem. Phys. 2014, 143, 825–829. [Google Scholar] [CrossRef]
- Lenes, M.; Wetzelaer, G.A.H.; Kooistra, F.B.; Veenstra, S.C.; Hummelen, J.C.; Blom, P.W.M. Fullerene bisadducts for enhanced open-circuit voltages and efficiencies in polymer solar cells. Adv. Mater. 2008, 20, 2116–2119. [Google Scholar] [CrossRef]
- Shaheen, S.E.; Brabec, C.J.; Serdar Sariciftci, N. 2.5% efficient organic plastic solar cells. Appl. Phys. Lett. 2001, 78, 841–843. [Google Scholar] [CrossRef]
- Wienk, M.M.; Kroon, J.M.; Verhees, W.J.H.; Knol, J.; Hummelen, J.C.; van Hal, P.A.; Janssen, R.A.J. Efficient methano[70]fullerene/MDMO-PPV bulk heterojunction photovoltaic cells. Angew. Chem. Int. Ed. 2003, 42, 3371–3375. [Google Scholar] [CrossRef] [PubMed]
- Schleyer, P.V.R.; Allinger, N.L.; Clark, T.; Gasteiger, J.; Kollman, P.A.; Schaefer, H.F., III; Schreiners, P.R. Encyclopedia of Computational Chemistry; Wiley: Chichester, UK, 1998; pp. 2646–2664. [Google Scholar]
- Marcus, R.A. Electron transfer reactions in chemistry. Theory and experiment. Rev. Mod. Phys. 1993, 65, 599–610. [Google Scholar] [CrossRef]
- Marcus, R.A. Chemical and electrochemical electron-transfer theory. Annu. Rev. Phys. Chem. 1964, 15, 155–196. [Google Scholar] [CrossRef]
- Gruhn, N.E.; Da Silva Filho, D.A.; Bill, T.G.; Malagoli, M.; Coropceanu, V.; Kahn, A.; Brédas, J.L. The vibrational reorganization energy in pentacene: Molecular influences on charge transport. J. Am. Chem. Soc. 2002, 124, 7918–7919. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.C.; Cheng, C.P.; You, Z.Q.; Hsu, C.P. Charge transport properties of tris(8-hydroxyquinolinato)aluminum(III): Why it is an electron transporter. J. Am. Chem. Soc. 2005, 127, 66–67. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, F.; Yang, X.; Li, Q.; Shuai, Z. Theoretical comparative studies of charge mobilities for molecular materials: Pet versus bnpery. Org. Electron. 2008, 9, 635–640. [Google Scholar] [CrossRef]
- Shizu, K.; Sato, T.; Ito, A.; Tanaka, K.; Kaji, H. Theoretical design of a hole-transporting molecule: Hexaaza[16]parabiphenylophane. J. Mater. Chem. 2011, 21, 6375–6382. [Google Scholar] [CrossRef]
- Gaussian 09. Available online: http://www.gaussian.com/g_prod/g09.htm (accessed on 25 August 2015).
- Cheung, D.L.; Troisi, A. Theoretical study of the organic photovoltaic electron acceptor PCBM: morphology, electronic structure, and charge localization. J. Phys. Chem. C 2010, 114, 20479–20488. [Google Scholar] [CrossRef]
- McMahon, D.P.; Trois, A. Evaluation of the external reorganization energy of polyacenes. J. Phys. Chem. Lett. 2010, 1, 941–946. [Google Scholar] [CrossRef]
- Di Motta, S.; Di Donato, E.; Negri, F.; Orlandi, G.; Fazzi, D.; Castiglioni, C. Resistive molecular memories: Influence of molecular parameters on the electrical bistability. J. Am. Chem. Soc. 2009, 131, 6591–6598. [Google Scholar] [CrossRef] [PubMed]
- Köse, M.E.; Long, H.; Kim, K.; Graf, P.; Ginley, D. Charge transport simulations in conjugated dendrimers. J. Phys. Chem. A 2010, 114, 4388–4393. [Google Scholar] [CrossRef] [PubMed]
- Köse, M.E.; Mitchell, W.J.; Kopidakis, N.; Chang, C.H.; Shaheen, S.E.; Kim, K.; Rumbles, G. Theoretical studies on conjugated phenyl-cored thiophene dendrimers for photovoltaic applications. J. Am. Chem. Soc. 2007, 129, 14257–14270. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Nakai, H.; Nakatsuji, H. Ab initio molecular orbital model of scanning tunneling microscopy. J. Chem. Phys. 1996, 104, 2410. [Google Scholar] [CrossRef]
- Troisi, A.; Orlandi, G. Dynamics of the intermolecular transfer integral in crystalline organic semiconductors. J. Phys. Chem. A 2006, 110, 4065–4070. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Chen, X.; Zou, Y.; Xiao, L.; Xu, X.; He, Y.; Li, L.; Li, Y. Benzo[1,2-b:4,5-b′]difuran-based donor–acceptor copolymers for polymer solar cells. Macromolecules 2012, 45, 6898–6905. [Google Scholar] [CrossRef]
- BIOVIA Materials Studio. Available online: http://accelrys.com/products/collaborative-science/biovia-materials-studio/ (accessed on 25 August 2015).
- Deng, W.Q.; Goddard, W.A., III. Predictions of hole mobilities in oligoacene organic semiconductors from quantum mechanical calculations. J. Phys. Chem. B 2004, 108, 8614–8621. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, R.; Wang, K. Rational Design of Diketopyrrolopyrrole-Based Small Moleculesas Donating Materials for Organic Solar Cells. Int. J. Mol. Sci. 2015, 16, 20326-20343. https://doi.org/10.3390/ijms160920326
Jin R, Wang K. Rational Design of Diketopyrrolopyrrole-Based Small Moleculesas Donating Materials for Organic Solar Cells. International Journal of Molecular Sciences. 2015; 16(9):20326-20343. https://doi.org/10.3390/ijms160920326
Chicago/Turabian StyleJin, Ruifa, and Kai Wang. 2015. "Rational Design of Diketopyrrolopyrrole-Based Small Moleculesas Donating Materials for Organic Solar Cells" International Journal of Molecular Sciences 16, no. 9: 20326-20343. https://doi.org/10.3390/ijms160920326