
Brain-Specific Cytoskeletal Damage Markers in Cerebrospinal Fluid: Is There a Common Pattern between Amyotrophic Lateral Sclerosis and Primary Progressive Multiple Sclerosis?

 ,
,
Abstract
:1. Introduction
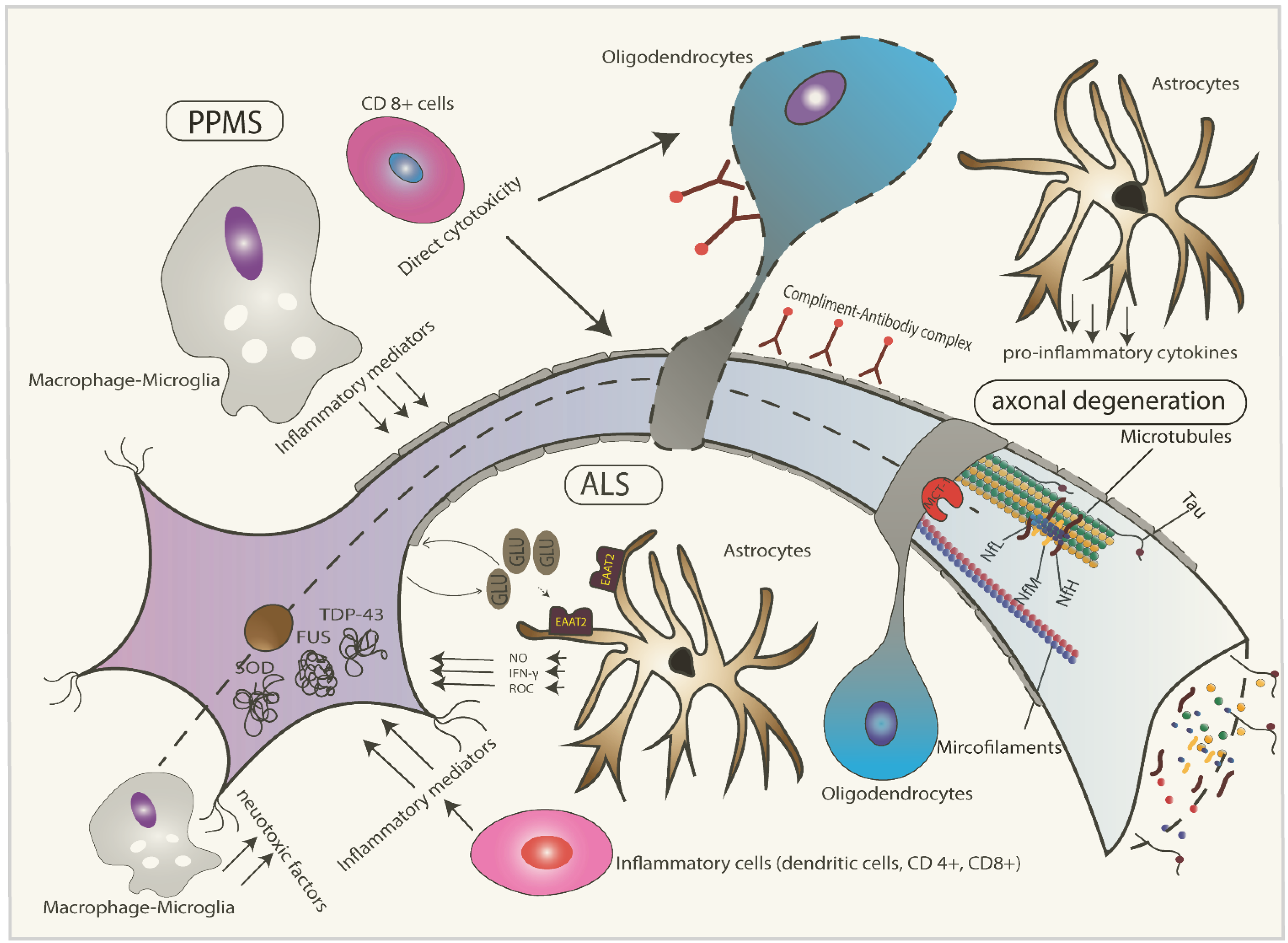
2. Pathophysiology of ALS
3. Pathophysiology of Progressive Multiple Sclerosis (MS)
4. Physiology of Cerebrospinal Fluid (CSF) and Its Relationship with Brain Specific Proteins in CSF
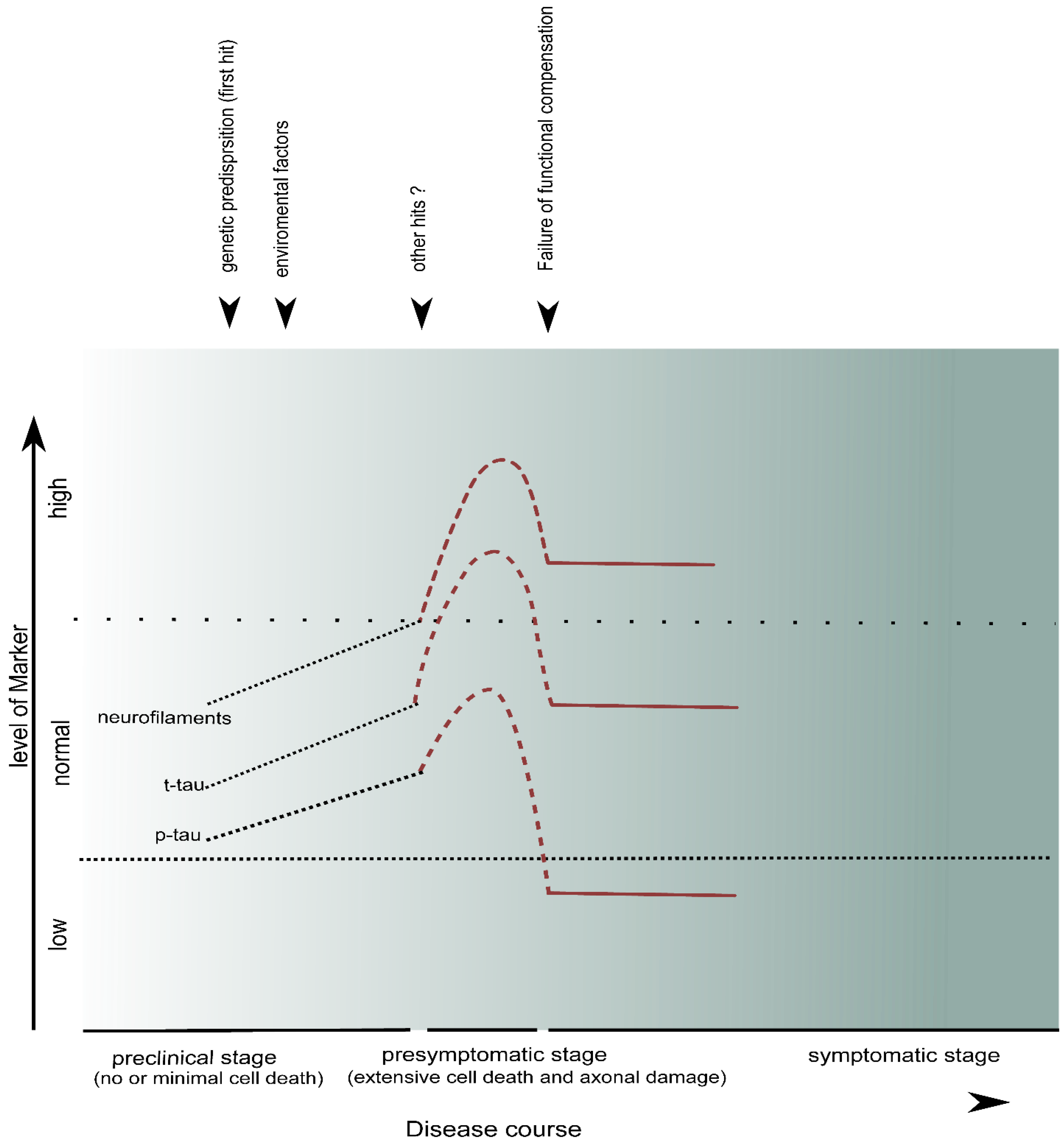
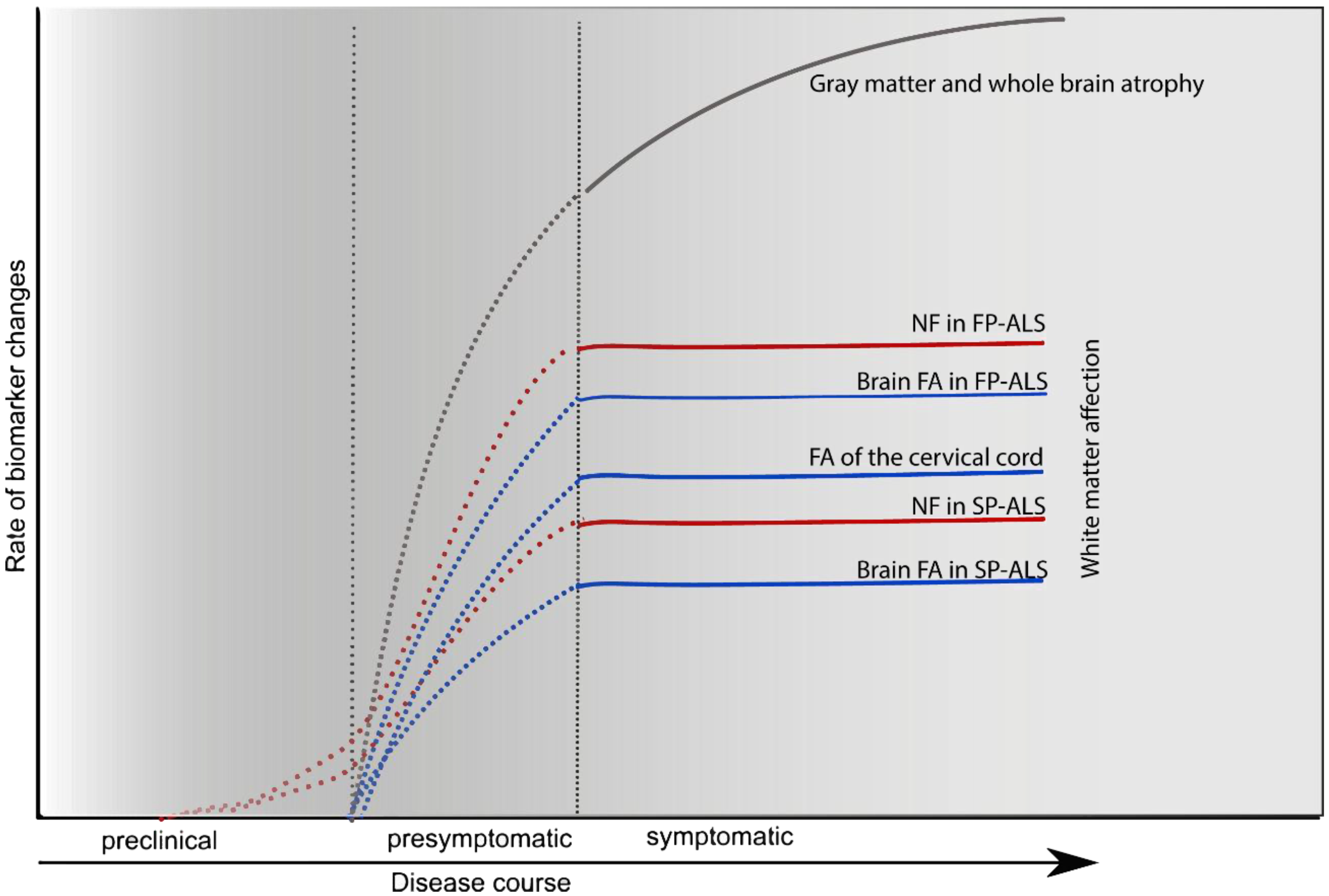
5. CSF Markers of Neuroaxonal Damage
5.1. Neurofilaments
5.1.1. Neurofilaments (NF) in ALS

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Marker | Method | Sample Size | Finding | Correlation with Progression Rate | Reference |
|---|---|---|---|---|---|
| NfL | ELISA | 12 ALS, 11 AD, 34 control | ALS > AD | not reported | [68] |
| ELISA | 60 sALS, 19 fALS, 206 OND | ALS > reference > control | yes | [71] | |
| ELISA | 32 ALS, 26 ALS-mimic | ALS > ALS-mimic | not reported | [70] | |
| ELISA | 37 ALS, 25 CIDP, 21 OND | ALS > CIDP and OND | yes | [72] | |
| ECL | 38 ALS, 20 control | ALS > control | not reported | [74] | |
| NfH | ELISA | ALS 69, 33 Control | ALS > control | yes | [69] |
| ELISA | 32 ALS, 26 ALS-mimic | ALS > ALS-mimic | not reported | [70] | |
| ECL | 50 ALS, 73 controls | ALS > control | not reported | [75] | |
| ELISA | 20 ALS | Higher level is associated with rapid progression | yes | [73] |
5.1.2. Neurofilaments (NF) in Primary Progressive Multiple Sclerosis (PPMS)
| Marker | Method | Sample Size | Finding | Correlation with Disability | Reference |
|---|---|---|---|---|---|
| NfL | Dot Blot | 16 PPMS, 13 SPMS, 6 PPMS, 36 IND and NIND | PMS > RRMS > control | yes | [57] |
| ELISA | 21 RRMS, 20 SPMS, 10 PPMS | Non-measurable | no | [78] | |
| ELISA | 65 RRMS, 10 SPMS, 20 PPMS | Sig. correlation between NfL and MSSS at follow up | not reported | [79] | |
| ELISA | 33 RRMS, 7 SPMS, 1 PPMS | PMS > RRMS > OND | yes | [80] | |
| ELISA | 40 SPMS, 21 PPMS, 26 RRMS, 20 NIND | RRMS > PMS > NIND | not reported | [77] | |
| ELISA | 38 RRMS, 25 SPMS, 23 PPMS, 62 CIS, 72 healthy control | RRMS/SPMS/PPMS > control | no | [59] | |
| NfH | In-House ELISA technique | 23 SP/PP MS, 11 RRMS, 318 NIND | NfHSMI35 PMS > RRMS | yes | [76] |
| ELISA | 21 RRMS, 20 SPMS, 10 PPMS | RRMS = SPMS = PPMS > controls | no | [78] |
5.2. Tau-Protein (Tau)
5.2.1. Tau-Protein (Tau) in Amyotrophic Lateral Sclerosis (ALS)
| Marker | Sample | Finding | Relation to Clinical Severity | Reference |
|---|---|---|---|---|
| t-Tau | 20 sALS, 20 controls | 70% of ALS have high CSF-tau compared to controls | no | [86] |
| 18 sALS, 75 control | ALS = control | no | [83] | |
| 67 sALS, 2 fALS, 33 control | ALS > control | no | [69] | |
| 57 sALS, 110 controls | ALS = controls | no | [84] | |
| 51 ALS, 23 4R-tau, 23 control | ALS = 4R-Tau = control | yes | [85] |
5.2.2. Tau-Protein (Tau) in Primary Progressive Multiple Sclerosis (PPMS)
| Marker | Number | Finding | Correlation to Disability | Reference |
|---|---|---|---|---|
| t-Tau | 15 RRMS, 11 SPMS, 10 PPMS, 17 ALS, 29 healthy controls | MS > control and ALS 72% of PMS having highly elevated tau compared to 27% of RRMS | not reported | [87] |
| 84 RRMS, 21 SPMS, 9 PPMS, 60 NIND, 79 IND | RRMS > IND > PMS > NIND | not reported | [90] | |
| 43 defined MS, 20 CIS, 56 controls (OND) | MS = control | No | [92] | |
| 50 CIS, 35 RRMS, 8 SPMS, 9 PPMS, 46 control | MS > control | No | [88] | |
| 32 RRMS, 2 SPMS, 4 PPMS, 19 healthy controls | MS = Control | No | [91] | |
| 30 RRMS, 9 SPMS, 6 PPMS, 38 healthy controls | MS > control | No | [89] |
5.3. Phosphorylated Tau-Protein (p-Tau)
5.3.1. p-Tau in ALS
5.3.2. p-Tau in PPMS
5.4. Tubulin and Actin
6. Discussion
| CSF-Markers of Axonal Damage | PPMS | Reference | ALS | Reference |
|---|---|---|---|---|
| Neurofilaments | PPMS > healthy controls | [75,76,77,78,79] | ALS > healthy controls | [68,69,70,71,72,73,74] |
| t-Tau | PPMS > or = healthy controls | [87,88,89,90,91] | ALS > or = healthy controls | [83,84,85,86] |
| p-Tau | PPMS < healthy controls | [93] | ALS < 4-repeat tauopathy | [85] |




7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
References
- Luessi, F.; Siffrin, V.; Zipp, F. Neurodegeneration in multiple sclerosis: Novel treatment strategies. Expert Rev. Neurother. 2012, 12, 1061–1076. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.; Kingwell, E.; Rieckmann, P.; Tremlett, H.; Neurologists, U.M.C. The natural history of secondary progressive multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2010, 81, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.J.; Polman, C.H.; Miller, D.H.; McDonald, W.I.; Brochet, B.; Filippi, M.M.X.; de Sa, J. Primary progressive multiple sclerosis. Brain 1997, 120, 1085–1096. [Google Scholar] [CrossRef] [PubMed]
- Frischer, J.M.; Bramow, S.; Dal-Bianco, A.; Lucchinetti, C.F.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Lassmann, H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009, 132, 1175–1189. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, A.; Freeman, J.; Lo, A.C. Treatment of progressive multiple sclerosis: What works, what does not, and what is needed. Lancet neurol. 2015, 14, 194–207. [Google Scholar] [CrossRef]
- Ferguson, T.A.; Elman, L.B. Clinical presentation and diagnosis of amyotrophic lateral sclerosis. NeuroRehabilitation 2007, 22, 409–416. [Google Scholar] [PubMed]
- Vucic, S.; Rothstein, J.D.; Kiernan, M.C. Advances in treating amyotrophic lateral sclerosis: Insights from pathophysiological studies. Trends Neurosci. 2014, 37, 443–442. [Google Scholar] [CrossRef] [PubMed]
- Traxinger, K.; Kelly, C.; Johnson, B.A.; Lyles, R.H.; Glass, J.D. Prognosis and epidemiology of amyotrophic lateral sclerosis: Analysis of a clinic population, 1997–2011. Neurol. Clin. Pract. 2013, 3, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Van Den Bosch, L.; van Damme, P.; Bogaert, E.; Robberecht, W. The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2006, 1762, 1068–1082. [Google Scholar] [CrossRef] [PubMed]
- Rice, C.M.; Cottrell, D.; Wilkins, A.; Scolding, N.J. Primary progressive multiple sclerosis: Progress and challenges. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1100–1106. [Google Scholar] [CrossRef] [PubMed]
- Martola, J.; Stawiarz, L.; Fredrikson, S.; Hillert, J.; Bergstrom, J.; Flodmark, O.; Kristoffersen Wiberg, M. Progression of non-age-related callosal brain atrophy in multiple sclerosis: A 9-year longitudinal MRI study representing four decades of disease development. J. Neurol. Neurosurg. Psychiatry 2007, 78, 375–380. [Google Scholar] [PubMed]
- El Mendili, M.M.; Cohen-Adad, J.; Pelegrini-Issac, M.; Rossignol, S.; Morizot-Koutlidis, R.; Marchand-Pauvert, V.; Iglesias, C.; Sangari, S.; Katz, R.; Lehericy, S.; et al. Multi-parametric spinal cord MRI as potential progression marker in amyotrophic lateral sclerosis. PLoS ONE 2014, 9, e95516. [Google Scholar] [CrossRef] [PubMed]
- Faber, I.; Servelhere, K.R.; Martinez, A.R.; D’Abreu, A.; Lopes-Cendes, I.; Franca-Jr, M.C. Clinical features and management of hereditary spastic paraplegia. Arq. Neuropsiquiatr. 2014, 72, 219–226. [Google Scholar] [PubMed]
- Gordon, P.H.; Cheng, B.; Katz, I.B.; Pinto, M.; Hays, A.P.; Mitsumoto, H.; Rowland, L.P. The natural history of primary lateral sclerosis. Neurology 2006, 66, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.H.; Leary, S.M. Primary-progressive multiple sclerosis. Lancet Neurol. 2007, 6, 903–912. [Google Scholar] [CrossRef]
- Gotkine, M.; Argov, Z. Clinical differentiation between primary lateral sclerosis and upper motor neuron predominant amyotrophic lateral sclerosis—Author reply. Arch. Neurol. 2007, 64, 1545. [Google Scholar] [CrossRef] [PubMed]
- Gordon, P.H. Amyotrophic lateral sclerosis: An update for 2013 clinical features, pathophysiology, management and therapeutic trials. Aging Dis. 2013, 4, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Blokhuis, A.M.; Groen, E.J.; Koppers, M.; van den Berg, L.H.; Pasterkamp, R.J. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 2013, 125, 777–794. [Google Scholar] [CrossRef] [PubMed]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Muller, K.; Marroquin, N.; Nordin, F.; Hubers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.B.; Lee, V.M.; Trojanowski, J.Q. Gains or losses: Molecular mechanisms of TDP43-mediated neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Dormann, D.; Rodde, R.; Edbauer, D.; Bentmann, E.; Fischer, I.; Hruscha, A.; Than, M.E.; Mackenzie, I.R.; Capell, A.; Schmid, B.; et al. ALS-associated fused in sarcoma (FUS) mutations disrupt transportin-mediated nuclear import. EMBO J. 2010, 29, 2841–2857. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, M.; Furukawa, Y. A seeded propagation of Cu, Zn-superoxide dismutase aggregates in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Lagier-Tourenne, C.; Polymenidou, M.; Cleveland, D.W. TDP-43 and FUS/TLS: Emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 2010, 19, R46–R64. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, N.J.; Zhang, Y.J.; Baker, M.; Gass, J.M.; Finch, N.A.; Xu, Y.F.; Stewart, H.; Kelley, B.J.; Kuntz, K.; Crook, R.J.; et al. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008, 4, e1000193. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.R.; King, O.D.; Shorter, J.; Gitler, A.D. Stress granules as crucibles of als pathogenesis. J. Cell Biol. 2013, 201, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Zinszner, H.; Sok, J.; Immanuel, D.; Yin, Y.; Ron, D. TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J. Cell Sci. 1997, 110, 1741–1750. [Google Scholar] [PubMed]
- Dormann, D.; Haass, C. TDP-43 and FUS: A nuclear affair. Trends Neurosci. 2011, 34, 339–348. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; McGeer, E.G. Inflammatory processes in amyotrophic lateral sclerosis. Muscle Nerve 2002, 26, 459–470. [Google Scholar]
- Valori, C.F.; Brambilla, L.; Martorana, F.; Rossi, D. The multifaceted role of glial cells in amyotrophic lateral sclerosis. Cell. Mol. Life Sci. 2014, 71, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Beers, D.R.; Appel, S.H. Immune-mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis. J. Neuroimmune Pharmacol. 2013, 8, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Brites, D.; Vaz, A.R. Microglia centered pathogenesis in ALS: Insights in cell interconnectivity. Front. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.W.; et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, S.; Garbelli, S.; Pasini, A.; Alimonti, D.; Perotti, C.; Melazzini, M.; Bendotti, C.; Mora, G. Immune system alterations in sporadic amyotrophic lateral sclerosis patients suggest an ongoing neuroinflammatory process. J. Neuroimmunol. 2009, 210, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Neymotin, A.; Petri, S.; Calingasan, N.Y.; Wille, E.; Schafer, P.; Stewart, C.; Hensley, K.; Beal, M.F.; Kiaei, M. Lenalidomide (Revlimid) administration at symptom onset is neuroprotective in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2009, 220, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Rivest, S. MyD88-deficient bone marrow cells accelerate onset and reduce survival in a mouse model of amyotrophic lateral sclerosis. J. Cell Biol. 2007, 179, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Brettschneider, J.; del Tredici, K.; Toledo, J.B.; Robinson, J.L.; Irwin, D.J.; Grossman, M.; Suh, E.; van Deerlin, V.M.; Wood, E.M.; Baek, Y.; et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann. Neurol. 2013, 74, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Ravits, J.M.; La Spada, A.R. ALS motor phenotype heterogeneity, focality, and spread: Deconstructing motor neuron degeneration. Neurology 2009, 73, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Ravits, J.; Appel, S.; Baloh, R.H.; Barohn, R.; Brooks, B.R.; Elman, L.; Floeter, M.K.; Henderson, C.; Lomen-Hoerth, C.; Macklis, J.D.; et al. Deciphering amyotrophic lateral sclerosis: What phenotype, neuropathology and genetics are telling us about pathogenesis. Amyotroph. Lateral Scler. Front. Degener. 2013, 14 (Suppl. 1), 5–18. [Google Scholar] [CrossRef] [PubMed]
- Bruck, W. The pathology of multiple sclerosis is the result of focal inflammatory demyelination with axonal damage. J. Neurol. 2005, 252 (Suppl. 5), v3–v9. [Google Scholar] [CrossRef] [PubMed]
- Munzel, E.J.; Williams, A. Promoting remyelination in multiple sclerosis—Recent advances. Drugs 2013, 73, 2017–2029. [Google Scholar] [CrossRef] [PubMed]
- Patani, R.; Balaratnam, M.; Vora, A.; Reynolds, R. Remyelination can be extensive in multiple sclerosis despite a long disease course. Neuropathol. Appl. Neurobiol. 2007, 33, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Patrikios, P.; Stadelmann, C.; Kutzelnigg, A.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Bruck, W.; Lucchinetti, C.; Lassmann, H. Remyelination is extensive in a subset of multiple sclerosis patients. Brain 2006, 129, 3165–3172. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; van Horssen, J.; Mahad, D. Progressive multiple sclerosis: Pathology and pathogenesis. Nat. Rev. Neurol. 2012, 8, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Lucchinetti, C.F.; Bruck, W.; Rodriguez, M.; Lassmann, H. Distinct patterns of multiple sclerosis pathology indicates heterogeneity on pathogenesis. Brain Pathol. 1996, 6, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Lucchinetti, C.; Bruck, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination. Ann. Neurol. 2000, 47, 707–717. [Google Scholar] [CrossRef]
- Hauser, S.L.; Oksenberg, J.R. The neurobiology of multiple sclerosis: Genes, inflammation, and neurodegeneration. Neuron 2006, 52, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H. Multiple sclerosis: Is there neurodegeneration independent from inflammation? J. Neurol. Sci. 2007, 259, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Matthews, P.M.; de Stefano, N.; Worsley, K.J.; Narayanan, S.; Francis, G.S.; Antel, J.P.; Wolfson, C.; Arnold, D.L. Imaging axonal damage of normal-appearing white matter in multiple sclerosis. Brain 1998, 121, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Tumani, H.; Hartung, H.P.; Hemmer, B.; Teunissen, C.; Deisenhammer, F.; Giovannoni, G.; Zettl, U.K.; BioMS Study Group. Cerebrospinal fluid biomarkers in multiple sclerosis. Neurobiol. Dis. 2009, 35, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H. Mechanisms of white matter damage in multiple sclerosis. Glia 2014, 62, 1816–1830. [Google Scholar] [CrossRef] [PubMed]
- Davson, H.; Hollingsworth, G.; Segal, M.B. The mechanism of drainage of the cerebrospinal fluid. Brain 1970, 93, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Felgenhauer, K.; Schliep, G.; Rapic, N. Evaluation of the blood-CSF barrier by protein gradients and the humoral immune response within the central nervous system. J. Neurol. Sci. 1976, 30, 113–128. [Google Scholar] [CrossRef]
- Reiber, H. Flow rate of cerebrospinal fluid (CSF)—A concept common to normal blood-CSF barrier function and to dysfunction in neurological diseases. J. Neurol. Sci. 1994, 122, 189–203. [Google Scholar] [CrossRef]
- Felgenhauer, K. The filtration concept of the blood-CSF-barrier as basis for the differentiation of CSF proteins. In New Concepts of A Blood—Brain Barrier; Springer US: New York, NY, USA, 1995; pp. 209–217. [Google Scholar]
- Reiber, H. Dynamics of brain-derived proteins in cerebrospinal fluid. Clin. Chim. Acta 2001, 310, 173–186. [Google Scholar] [CrossRef]
- Semra, Y.K.; Seidi, O.A.; Sharief, M.K. Heightened intrathecal release of axonal cytoskeletal proteins in multiple sclerosis is associated with progressive disease and clinical disability. J. Neuroimmunol. 2002, 122, 132–139. [Google Scholar] [CrossRef]
- Teunissen, C.E.; Khalil, M. Neurofilaments as biomarkers in multiple sclerosis. Mult. Scler. 2012, 18, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Kuhle, J.; Plattner, K.; Bestwick, J.P.; Lindberg, R.L.; Ramagopalan, S.V.; Norgren, N.; Nissim, A.; Malaspina, A.; Leppert, D.; Giovannoni, G.; et al. A comparative study of CSF neurofilament light and heavy chain protein in MS. Mult. Scler. 2013, 19, 1597–1603. [Google Scholar] [CrossRef] [PubMed]
- Yabe, J.T.; Chylinski, T.; Wang, F.S.; Pimenta, A.; Kattar, S.D.; Linsley, M.D.; Chan, W.K.H.; Shea, T.B. Neurofilaments consist of distinct populations that can be distinguished by C-terminal phosphorylation, bundling, and axonal transport rate in growing axonal neurites. J. Neurosci. 2001, 21, 2195–2205. [Google Scholar] [PubMed]
- Strong, M.J. Neurofilament metabolism in sporadic amyotrophic lateral sclerosis. J. Neurol. Sci. 1999, 169, 170–177. [Google Scholar] [CrossRef]
- Fuchs, E.; Cleveland, D.W. A structural scaffolding of intermediate filaments in health and disease. Science 1998, 279, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Dujmovic, I. Cerebrospinal fluid and blood biomarkers of neuroaxonal damage in multiple sclerosis. Mult. Scler. Int. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Gresle, M.M.; Shaw, G.; Jarrott, B.; Alexandrou, E.N.; Friedhuber, A.; Kilpatrick, T.J.; Butzkueven, H. Validation of a novel biomarker for acute axonal injury in experimental autoimmune encephalomyelitis. J. Neurosci. Res. 2008, 86, 3548–3555. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Xie, F.; Siedlak, S.L.; Nunomura, A.; Honda, K.; Moreira, P.I.; Zhua, X.; Smith, M.A.; Perry, G. Neurofilament proteins in neurodegenerative diseases. Cell. Mol. Life Sci. 2004, 61, 3057–3075. [Google Scholar] [CrossRef] [PubMed]
- Munoz, D.G.; Greene, C.; Perl, D.P.; Selkoe, D.J. Accumulation of phosphorylated neurofilaments in anterior horn motoneurons of amyotrophic lateral sclerosis patients. J. Neuropathol. Exp. Neurol. 1988, 47, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Figlewicz, D.A.; Krizus, A.; Martinoli, M.G.; Meininger, V.; Dib, M.; Rouleau, G.A.; Julien, J.P. Variants of the heavy neurofilament subunit are associated with the development of amyotrophic lateral sclerosis. Hum. Mol. Genet. 1994, 3, 1757–1761. [Google Scholar] [CrossRef] [PubMed]
- Rosengren, L.E.; Karlsson, J.E.; Karlsson, J.O.; Persson, L.I.; Wikkelso, C. Patients with amyotrophic lateral sclerosis and other neurodegenerative diseases have increased levels of neurofilament protein in CSF. J. Neurochem. 1996, 67, 2013–2018. [Google Scholar] [CrossRef] [PubMed]
- Brettschneider, J.; Petzold, A.; Sussmuth, S.D.; Ludolph, A.C.; Tumani, H. Axonal damage markers in cerebrospinal fluid are increased in ALS. Neurology 2006, 66, 852–856. [Google Scholar] [CrossRef] [PubMed]
- Reijn, T.S.; Abdo, W.F.; Schelhaas, H.J.; Verbeek, M.M. CSF neurofilament protein analysis in the differential diagnosis of ALS. J. Neurol. 2009, 256, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H.; Jacobsson, J.; Rosengren, L.; Blennow, K.; Andersen, P.M. Cerebrospinal fluid neurofilament light levels in amyotrophic lateral sclerosis: Impact of SOD1 genotype. Eur. J. Neurol. 2007, 14, 1329–1333. [Google Scholar] [CrossRef] [PubMed]
- Tortelli, R.; Ruggieri, M.; Cortese, R.; D'Errico, E.; Capozzo, R.; Leo, A.; Mastrapasqua, M.; Zoccolella, S.; Leante, R.; Livrea, P.; et al. Elevated cerebrospinal fluid neurofilament light levels in patients with amyotrophic lateral sclerosis: A possible marker of disease severity and progression. Eur.J. Neurol 2012, 19, 1561–1567. [Google Scholar] [CrossRef] [PubMed]
- Boylan, K.B.; Glass, J.D.; Crook, J.E.; Yang, C.; Thomas, C.S.; Desaro, P.; Johnston, A.; Overstreet, K.; Kelly, C.; Polak, M.; et al. Phosphorylated neurofilament heavy subunit (pNF-H) in peripheral blood and csf as a potential prognostic biomarker in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2013, 84, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.H.; Macdonald-Wallis, C.; Gray, E.; Pearce, N.; Petzold, A.; Norgren, N.; Giovannoni, G.; Fratta, P.; Sidle, K.; Fish, M.; et al. Neurofilament light chain: A prognostic biomarker in amyotrophic lateral sclerosis. Neurology 2015, 84, 2247–2257. [Google Scholar] [CrossRef] [PubMed]
- Kuhle, J.; Regeniter, A.; Leppert, D.; Mehling, M.; Kappos, L.; Lindberg, R.L.; Petzold, A. A highly sensitive electrochemiluminescence immunoassay for the neurofilament heavy chain protein. J. Neuroimmunol. 2010, 220, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Petzold, A.; Eikelenboom, M.J.; Keir, G.; Grant, D.; Lazeron, R.H.; Polman, C.H.; Uitdehaag, B.M.; Thompson, E.J.; Giovannoni, G. Axonal damage accumulates in the progressive phase of multiple sclerosis: Three year follow up study. J. Neurol. Neurosurg. Psychiatry 2005, 76, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Romme Christensen, J.; Bornsen, L.; Khademi, M.; Olsson, T.; Jensen, P.E.; Sorensen, P.S.; Sellebjerg, F. CSF inflammation and axonal damage are increased and correlate in progressive multiple sclerosis. Mult. Scler. 2013, 19, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Eikelenboom, M.J.; Petzold, A.; Lazeron, R.H.; Silber, E.; Sharief, M.; Thompson, E.J.; Barkhof, F.; Giovannoni, G.; Polman, C.H.; Uitdehaag, B.M. Multiple sclerosis: Neurofilament light chain antibodies are correlated to cerebral atrophy. Neurology 2003, 60, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Salzer, J.; Svenningsson, A.; Sundstrom, P. Neurofilament light as a prognostic marker in multiple sclerosis. Mult. Scler. 2010, 16, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Madeddu, R.; Farace, C.; Tolu, P.; Solinas, G.; Asara, Y.; Sotgiu, M.A.; Delogu, L.G.; Prados, J.C.; Sotgiu, S.; Montella, A. Cytoskeletal proteins in the cerebrospinal fluid as biomarker of multiple sclerosis. Neurol. Sci. 2013, 34, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Shahani, N.; Brandt, R. Functions and malfunctions of the tau proteins. Cell. Mol. Life Sci. 2002, 59, 1668–1680. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Jimenez, F.J.; Hernanz, A.; Medina-Acebron, S.; de Bustos, F.; Zurdo, J.M.; Alonso, H.; Puertas, I.; Barcenilla, B.; Sayed, Y.; Cabrera-Valdivia, F. Tau protein concentrations in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neurol. Scand. 2005, 111, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Paladino, P.; Valentino, F.; Piccoli, T.; Piccoli, F.; La Bella, V. Cerebrospinal fluid tau protein is not a biological marker in amyotrophic lateral sclerosis. Eur.J. Neurol. 2009, 16, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Grossman, M.; Elman, L.; McCluskey, L.; McMillan, C.T.; Boller, A.; Powers, J.; Rascovsky, K.; Hu, W.; Shaw, L.; Irwin, D.J.; et al. Phosphorylated tau as a candidate biomarker for amyotrophic lateral sclerosis. JAMA Neurol. 2014. [Google Scholar] [CrossRef]
- Sussmuth, S.D.; Tumani, H.; Ecker, D.; Ludolph, A.C. Amyotrophic lateral sclerosis: Disease stage related changes of tau protein and S100 β in cerebrospinal fluid and creatine kinase in serum. Neurosci. Lett. 2003, 353, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Kapaki, E.; Paraskevas, G.P.; Michalopoulou, M.; Kilidireas, K. Increased cerebrospinal fluid tau protein in multiple sclerosis. Eur. Neurol. 2000, 43, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Brettschneider, J.; Maier, M.; Arda, S.; Claus, A.; Sussmuth, S.D.; Kassubek, J.; Tumani, H. Tau protein level in cerebrospinal fluid is increased in patients with early multiple sclerosis. Mult. Scler. 2005, 11, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Terzi, M.; Birinci, A.; Cetinkaya, E.; Onar, M.K. Cerebrospinal fluid total tau protein levels in patients with multiple sclerosis. Acta Neurol. Scand. 2007, 115, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Bartosik-Psujek, H.; Archelos, J.J. Tau protein and 14-3-3 are elevated in the cerebrospinal fluid of patients with multiple sclerosis and correlate with intrathecal synthesis of IgG. J. Neurol. 2004, 251, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, I.; Cardoso, M.I.; Sa, M.J. Tau protein seems not to be a useful routine clinical marker of axonal damage in multiple sclerosis. Mult. Scler. 2006, 12, 354–356. [Google Scholar] [CrossRef] [PubMed]
- Colucci, M.; Roccatagliata, L.; Capello, E.; Narciso, E.; Latronico, N.; Tabaton, M.; Mancardi, G.L. The 14-3-3 protein in multiple sclerosis: A marker of disease severity. Mult. Scler. 2004, 10, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.M.; Patani, R.; Reynolds, R.; Nicholas, R.; Compston, A.; Spillantini, M.G.; Chandran, S. Abnormal tau phosphorylation in primary progressive multiple sclerosis. Acta Neuropathol. 2010, 119, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Downing, K.H. Structural basis for the interaction of tubulin with proteins and drugs that affect microtubule dynamics. Annu. Rev. Cell Dev. Biol. 2000, 16, 89–111. [Google Scholar] [CrossRef] [PubMed]
- Laferriere, N.B.; MacRae, T.H.; Brown, D.L. Tubulin synthesis and assembly in differentiating neurons. Biochem. Cell Biol. 1997, 75, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Gunning, P.; Hardeman, E.; Jeffrey, P.; Weinberger, R. Creating intracellular structural domains: Spatial segregation of actin and tropomyosin isoforms in neurons. Bioessays 1998, 20, 892–900. [Google Scholar] [CrossRef]
- Doussau, F.; Augustine, G.J. The actin cytoskeleton and neurotransmitter release: An overview. Biochimie 2000, 82, 353–363. [Google Scholar] [CrossRef]
- Tumani, H.; Teunissen, C.; Süssmuth, S.; Otto, M.; Ludolph, A.C.; Brettschneider, J. Cerebrospinal fluid biomarkers of neurodegeneration in chronic neurological diseases. Expert Rev. Mol. Diagn. 2008, 8, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Vinsant, S.; Mansfield, C.; Jimenez-Moreno, R.; del Gaizo Moore, V.; Yoshikawa, M.; Hampton, T.G.; Prevette, D.; Caress, J.; Oppenheim, R.W.; Milligan, C. Characterization of early pathogenesis in the SOD1(G93A) mouse model of ALS: Part II, results and discussion. Brain Behav. 2013, 3, 431–457. [Google Scholar] [CrossRef] [PubMed]
- Vinsant, S.; Mansfield, C.; Jimenez-Moreno, R.; del Gaizo Moore, V.; Yoshikawa, M.; Hampton, T.G.; Prevette, D.; Caress, J.; Oppenheim, R.W.; Milligan, C. Characterization of early pathogenesis in the SOD1(G93A) mouse model of ALS: Part I, background and methods. Brain Behav. 2013, 3, 335–350. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, M.; Swash, M. The onset of amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2006, 77, 388–389. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, A.; Nicholson, G. Normal complement of motor units in asymptomatic familial (SOD1 mutation) amyotrophic lateral sclerosis carriers. J. Neurol. Neurosurg. Psychiatry 2001, 71, 478–481. [Google Scholar] [CrossRef] [PubMed]
- Magnus, T.; Beck, M.; Giess, R.; Puls, I.; Naumann, M.; Toyka, K.V. Disease progression in amyotrophic lateral sclerosis: Predictors of survival. Muscle Nerve 2002, 25, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Agosta, F.; Valsasina, P.; Riva, N.; Copetti, M.; Messina, M.J.; Prelle, A.; Comi, G.; Filippi, M. The cortical signature of amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e42816. [Google Scholar] [CrossRef] [PubMed]
- Verstraete, E.; van den Heuvel, M.P.; Veldink, J.H.; Blanken, N.; Mandl, R.C.; Hulshoff Pol, H.E.; van den Berg, L.H. Motor network degeneration in amyotrophic lateral sclerosis: A structural and functional connectivity study. PLoS ONE 2010, 5, e13664. [Google Scholar] [CrossRef] [PubMed]
- Mezzapesa, D.M.; D’Errico, E.; Tortelli, R.; Distaso, E.; Cortese, R.; Tursi, M.; Federico, F.; Zoccolella, S.; Logroscino, G.; Dicuonzo, F.; et al. Cortical thinning and clinical heterogeneity in amyotrophic lateral sclerosis. PLoS ONE 2013, 8, e80748. [Google Scholar] [CrossRef] [PubMed]
- Menke, R.A.; Korner, S.; Filippini, N.; Douaud, G.; Knight, S.; Talbot, K.; Turner, M.R. Widespread grey matter pathology dominates the longitudinal cerebral MRI and clinical landscape of amyotrophic lateral sclerosis. Brain 2014, 137, 2546–2555. [Google Scholar] [CrossRef] [PubMed]
- Kwan, J.Y.; Meoded, A.; Danielian, L.E.; Wu, T.; Floeter, M.K. Structural imaging differences and longitudinal changes in primary lateral sclerosis and amyotrophic lateral sclerosis. Neuroimage Clin. 2012, 2, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Fisher, E.; Lee, J.C.; Nakamura, K.; Rudick, R.A. Gray matter atrophy in multiple sclerosis: A longitudinal study. Ann. Neurol. 2008, 64, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Lukas, C.; Knol, D.L.; Sombekke, M.H.; Bellenberg, B.; Hahn, H.K.; Popescu, V.; Weier, K.; Radue, E.W.; Gass, A.; Kappos, L.; et al. Cervical spinal cord volume loss is related to clinical disability progression in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2014. [Google Scholar] [CrossRef]
- Agosta, F.; Absinta, M.; Sormani, M.P.; Ghezzi, A.; Bertolotto, A.; Montanari, E.; Comi, G.; Filippi, M. In vivo assessment of cervical cord damage in MS patients: A longitudinal diffusion tensor MRI study. Brain 2007, 130, 2211–2219. [Google Scholar] [CrossRef] [PubMed]
- Okuda, D.T.; Siva, A.; Kantarci, O.; Inglese, M.; Katz, I.; Tutuncu, M.; Keegan, B.M.; Donlon, S.; le Hua, H.; Vidal-Jordana, A.; et al. Radiologically isolated syndrome: 5-Year risk for an initial clinical event. PLoS ONE 2014, 9, e90509. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdelhak, A.; Junker, A.; Brettschneider, J.; Kassubek, J.; Ludolph, A.C.; Otto, M.; Tumani, H. Brain-Specific Cytoskeletal Damage Markers in Cerebrospinal Fluid: Is There a Common Pattern between Amyotrophic Lateral Sclerosis and Primary Progressive Multiple Sclerosis? Int. J. Mol. Sci. 2015, 16, 17565-17588. https://doi.org/10.3390/ijms160817565
Abdelhak A, Junker A, Brettschneider J, Kassubek J, Ludolph AC, Otto M, Tumani H. Brain-Specific Cytoskeletal Damage Markers in Cerebrospinal Fluid: Is There a Common Pattern between Amyotrophic Lateral Sclerosis and Primary Progressive Multiple Sclerosis? International Journal of Molecular Sciences. 2015; 16(8):17565-17588. https://doi.org/10.3390/ijms160817565
Chicago/Turabian StyleAbdelhak, Ahmed, Andreas Junker, Johannes Brettschneider, Jan Kassubek, Albert C. Ludolph, Markus Otto, and Hayrettin Tumani. 2015. "Brain-Specific Cytoskeletal Damage Markers in Cerebrospinal Fluid: Is There a Common Pattern between Amyotrophic Lateral Sclerosis and Primary Progressive Multiple Sclerosis?" International Journal of Molecular Sciences 16, no. 8: 17565-17588. https://doi.org/10.3390/ijms160817565