
Homocysteine Triggers Inflammatory Responses in Macrophages through Inhibiting CSE-H2S Signaling via DNA Hypermethylation of CSE Promoter

Abstract
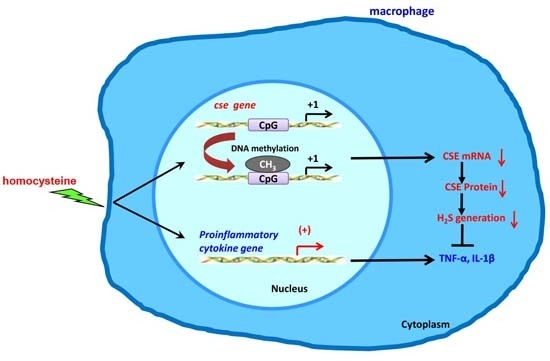
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
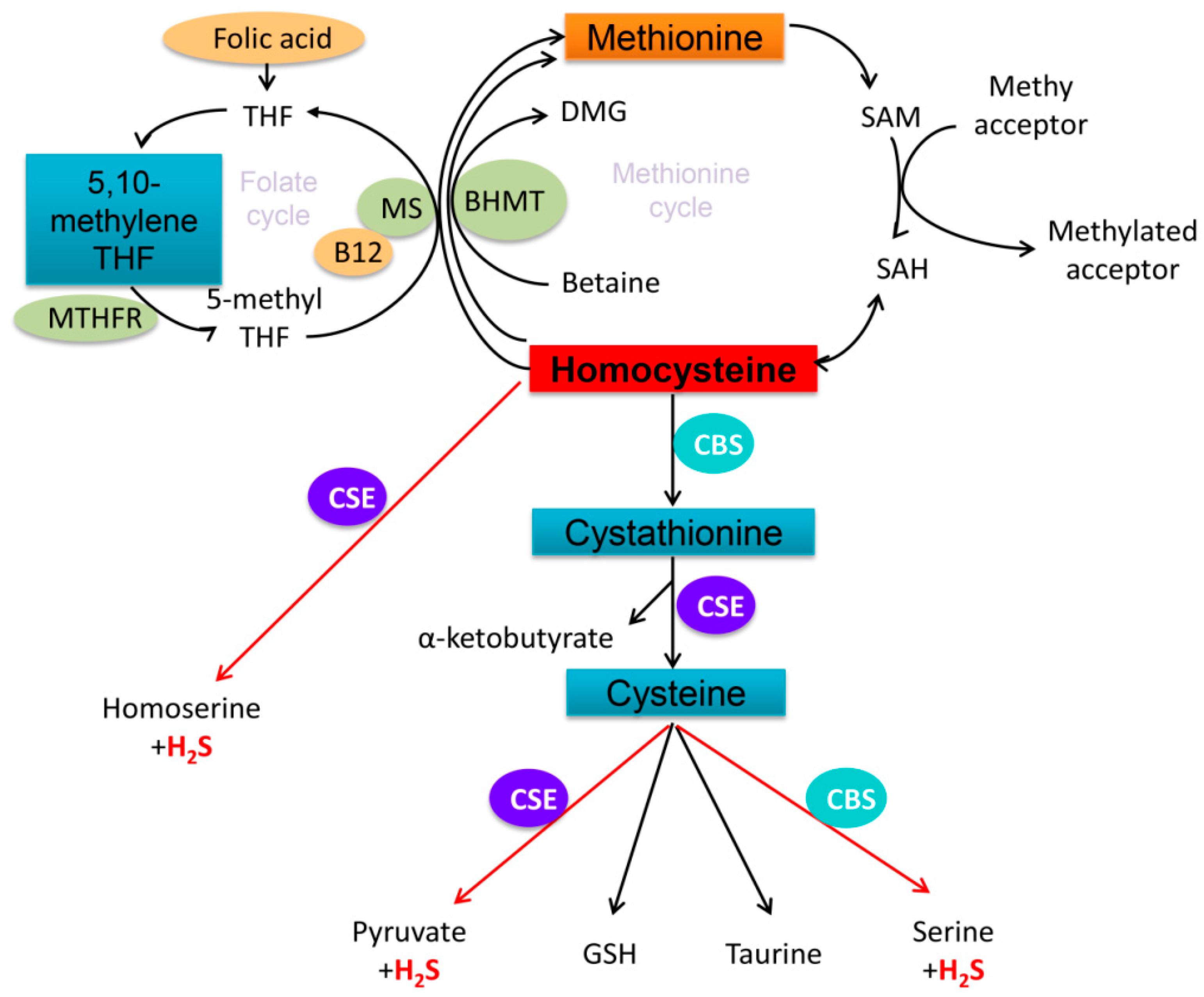
1. Introduction
2. Results and Discussion
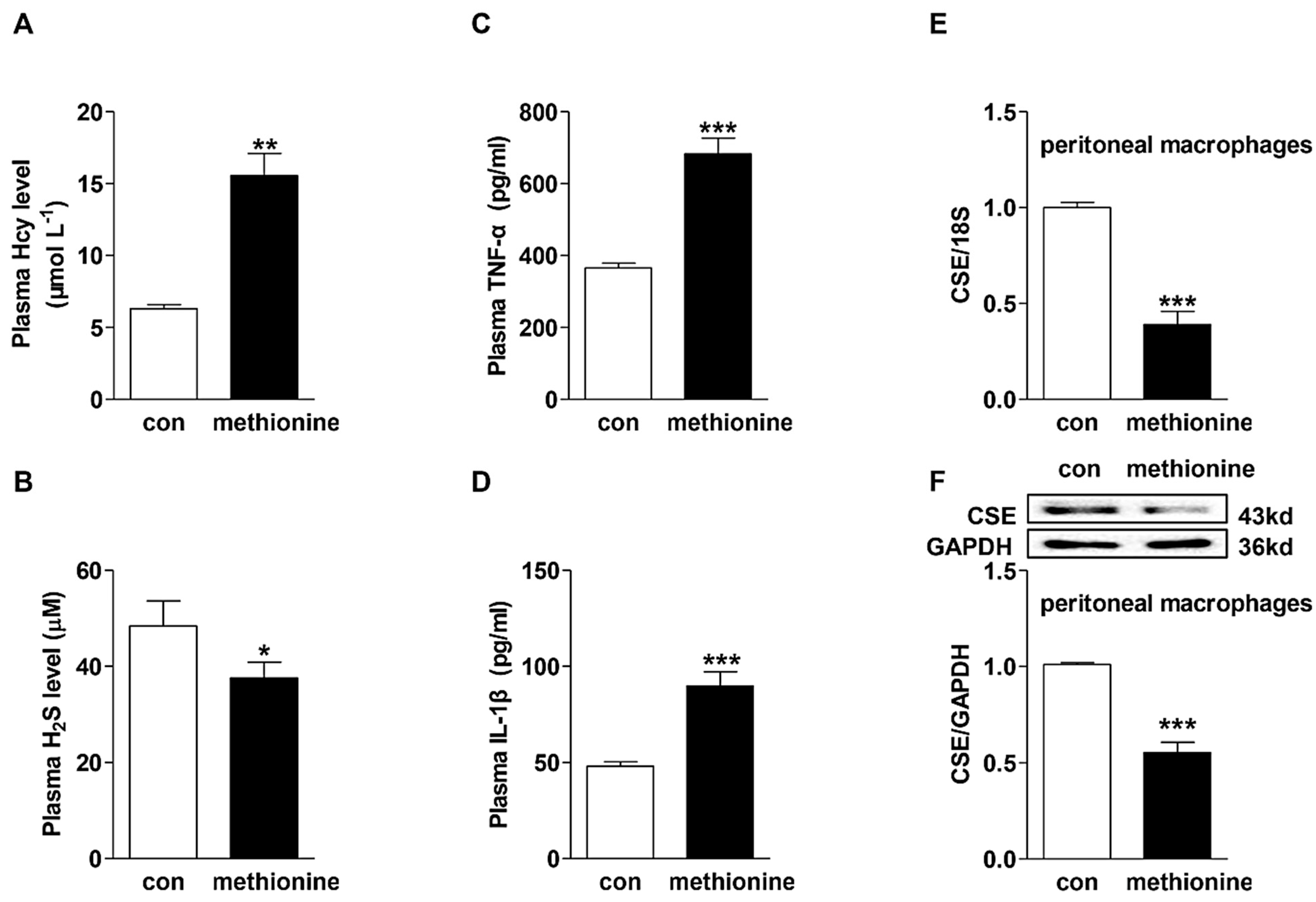
2.1. Effects of Methionine Administration on Plasma Hcy, H2S and Pro-Inflammatory Cytokine Levels in C57BL/6 Mice and CSE Expression in Peritoneal Macrophages
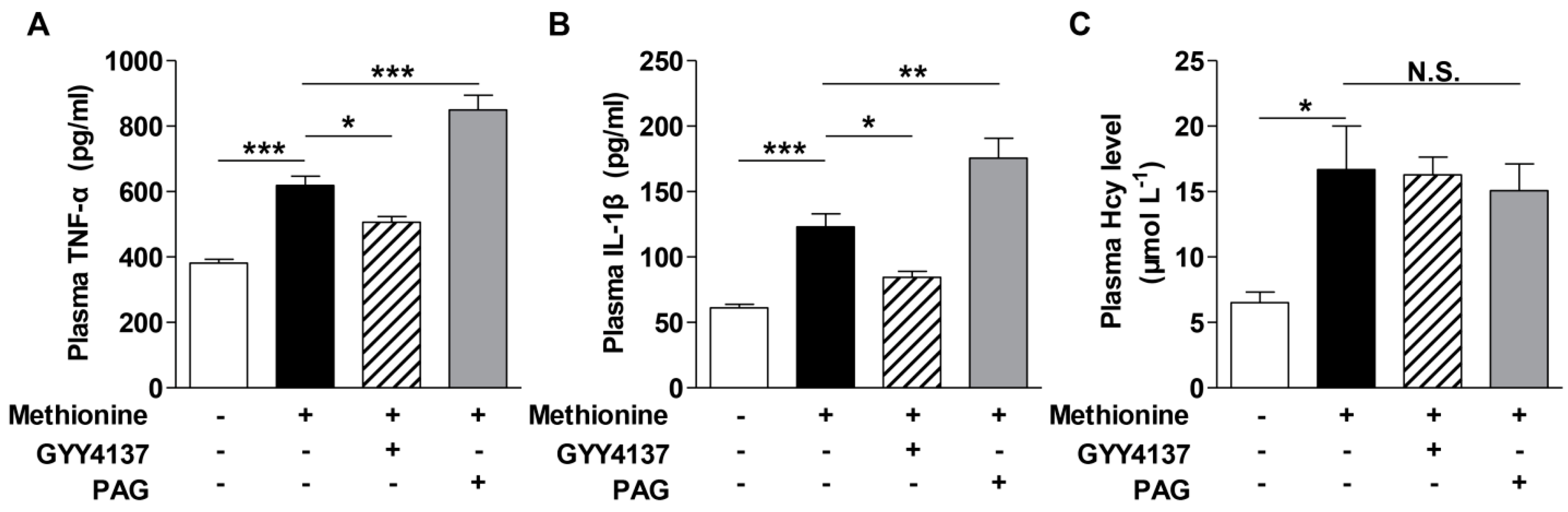
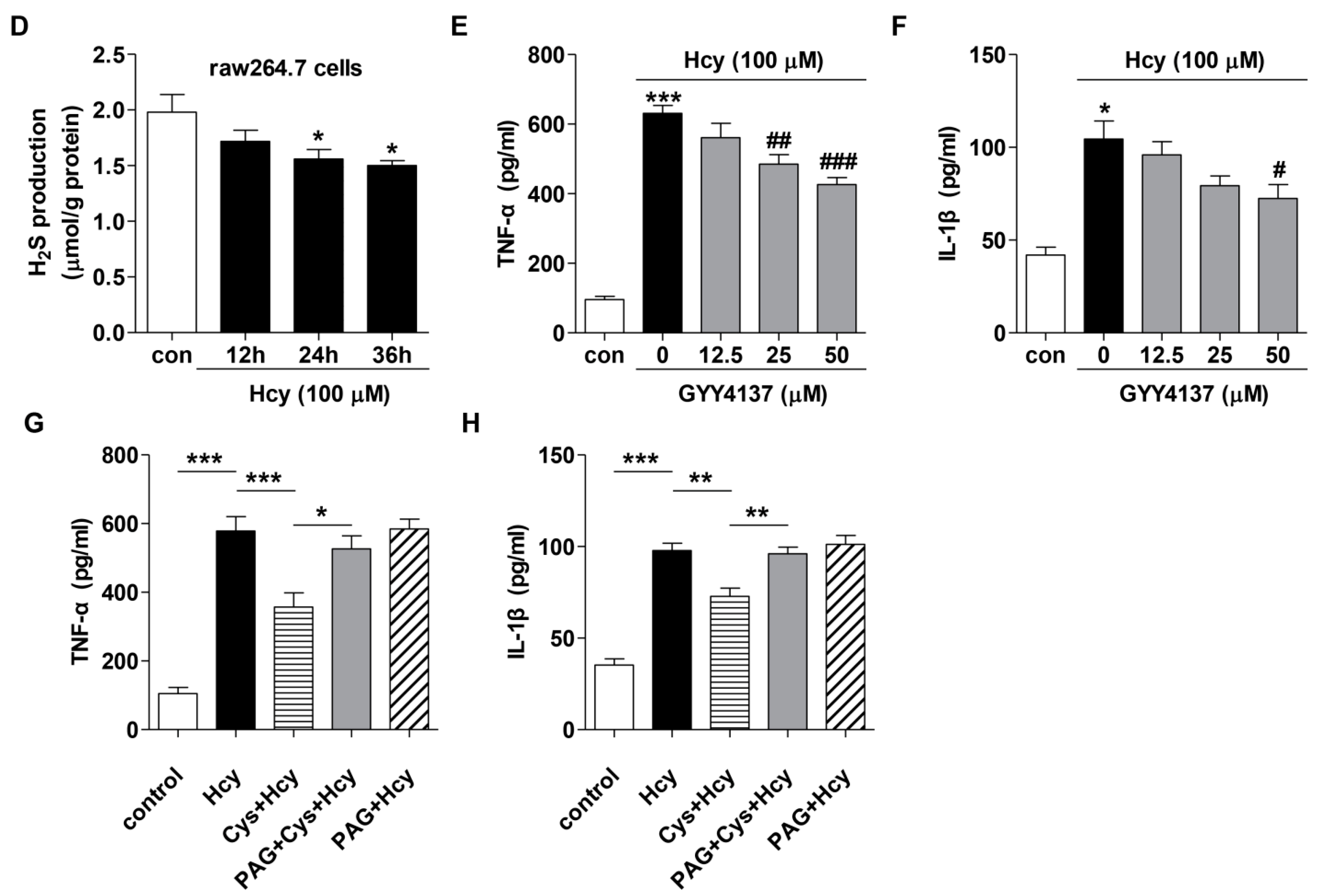
2.2. Effects of H2S on Cytokines Level in the Plasma of Methionine Diet-Treated Mice and Hcy-Treated Raw264.7 Macrophages



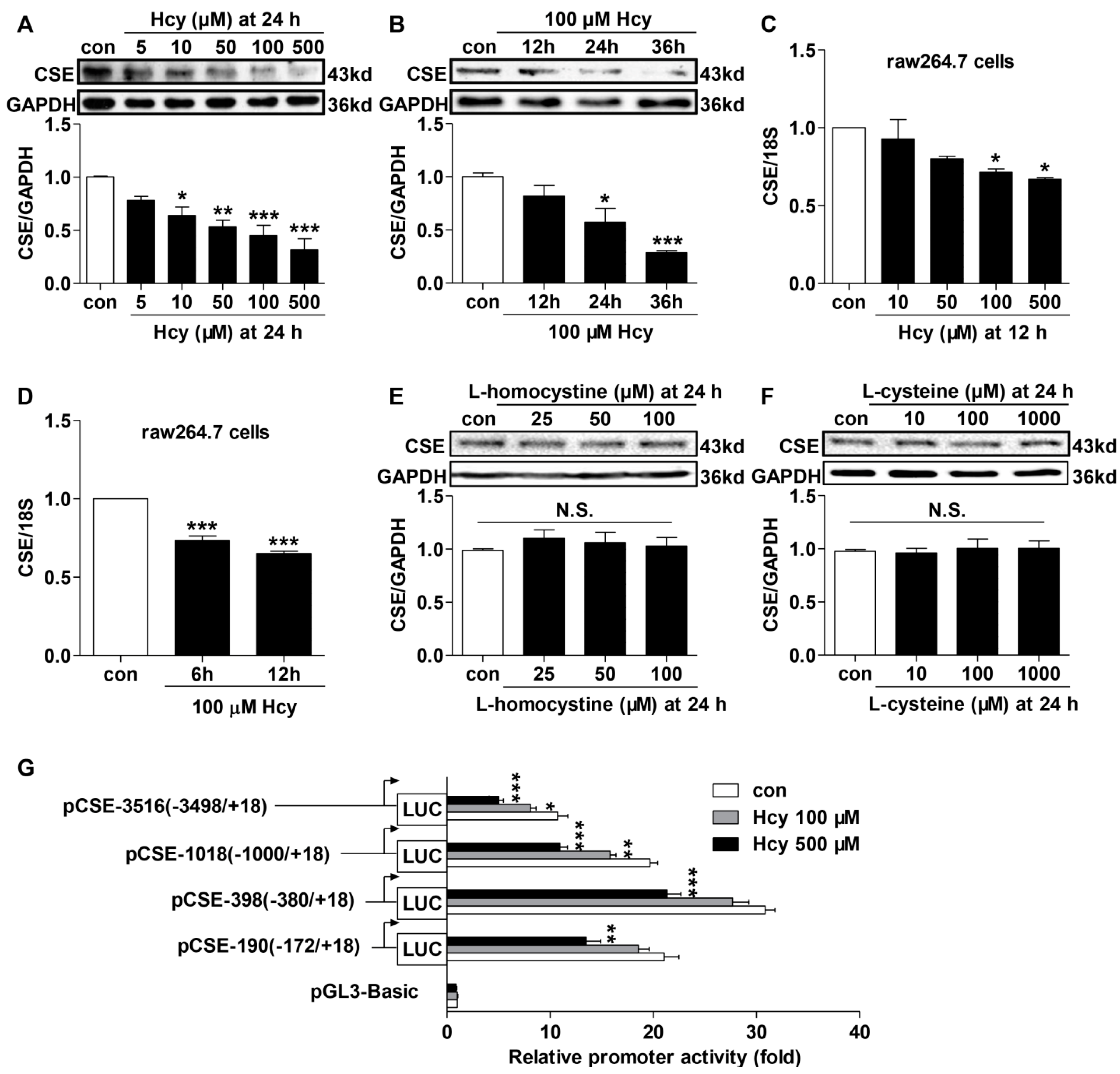
2.3. Hcy Suppressed CSE Expression in Hcy-Treated Raw264.7 Cells

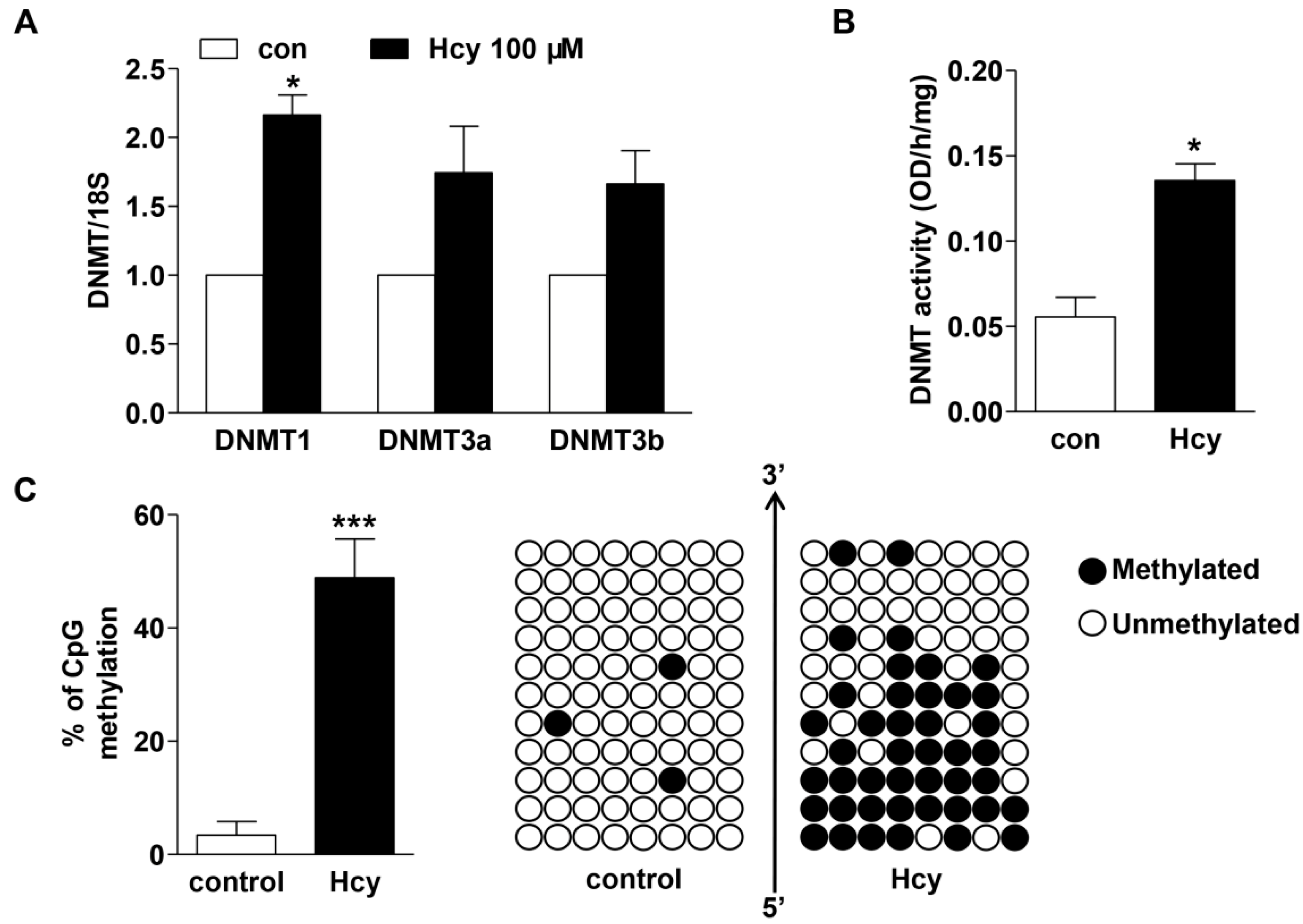
2.4. Hcy up-Regulated DNMT Expression and Enhanced DNA Hypermethylation in cse Promoter

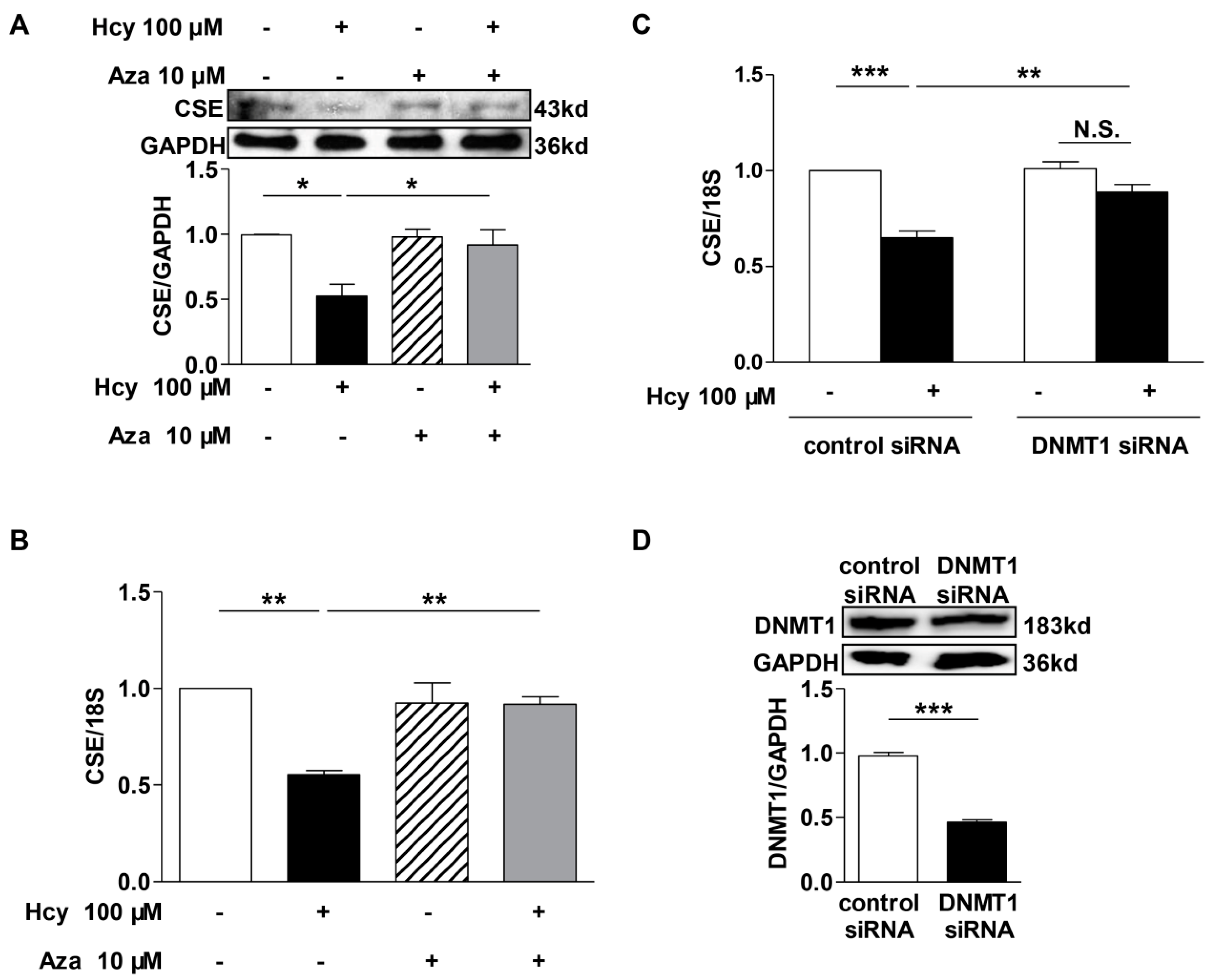
2.5. DNMT1 Inhibition Reversed the Repression of Hcy on CSE Transcription

2.6. Discussion

3. Experimental Section
3.1. Reagents and Antibodies
3.2. Cell Culture
3.3. Animals
3.4. H2S Measurement
3.5. Immunoblotting Analysis
3.6. Quantitative Polymerase Chain Reaction (PCR)
3.7. Cse Promoter Activity Analyses
3.8. Bisulfite Sequencing (BSP) Assay
3.9. Dnmt Activity
3.10. siRNA Transfection
3.11. Cytokine Production Assay
3.12. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Castro, R.; Rivera, I.; Blom, H.J.; Jakobs, C.; Tavares de Almeida, I. Homocysteine metabolism, hyperhomocysteinaemia and vascular disease: An overview. J. Inherit. Metab. Dis. 2006, 29, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Ansari, R.; Mahta, A.; Mallack, E.; Luo, J.J. Hyperhomocysteinemia and neurologic disorders: A review. J. Clin. Neurol. 2014, 10, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Morrison, L.D.; Smith, D.D.; Kish, S.J. Brain S-adenosylmethionine levels are severely decreased in Alzheimerʼs disease. J. Neurochem. 1996, 67, 1328–1331. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S.; et al. H2S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine γ-lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.F.; Lu, M.; Hon Wong, P.T.; Bian, J.S. Hydrogen sulfide: Neurophysiology and neuropathology. Antioxid. Redox Signal. 2011, 15, 405–419. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Geng, B.; Yu, F.; Zhao, J.; Jiang, H.; Du, J.; Tang, C. Hydrogen sulfide inhibits myocardial injury induced by homocysteine in rats. Amino Acids 2008, 34, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Sen, U.; Basu, P.; Abe, O.A.; Givvimani, S.; Tyagi, N.; Metreveli, N.; Shah, K.S.; Passmore, J.C.; Tyagi, S.C. Hydrogen sulfide ameliorates hyperhomocysteinemia-associated chronic renal failure. Am. J. Physiol. Ren. Physiol. 2009, 297, F410–F419. [Google Scholar] [CrossRef] [PubMed]
- Kamat, P.K.; Kalani, A.; Givvimani, S.; Sathnur, P.B.; Tyagi, S.C.; Tyagi, N. Hydrogen sulfide attenuates neurodegeneration and neurovascular dysfunction induced by intracerebral-administered homocysteine in mice. Neuroscience 2013, 252, 302–319. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.M.; Dear, A.; Craig, J.M.; Norman, P.E.; Golledge, J. The potential role of homocysteine mediated DNA methylation and associated epigenetic changes in abdominal aortic aneurysm formation. Atherosclerosis 2013, 228, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Lonn, E.; Yusuf, S.; Arnold, M.J.; Sheridan, P.; Pogue, J.; Micks, M.; McQueen, M.J.; Probstfield, J.; Fodor, G.; Held, C.; et al. Homocysteine lowering with folic acid and B vitamins in vascular disease. N. Engl. J. Med. 2006, 354, 1567–1577. [Google Scholar] [PubMed]
- Mani, S.; Untereiner, A.; Wu, L.; Wang, R. Hydrogen sulfide and the pathogenesis of atherosclerosis. Antioxid. Redox Signal. 2014, 20, 805–817. [Google Scholar] [CrossRef] [PubMed]
- Meng, S.; Ciment, S.; Jan, M.; Tran, T.; Pham, H.; Cueto, R.; Yang, X.F.; Wang, H. Homocysteine induces inflammatory transcriptional signaling in monocytes. Front. Biosci. 2013, 18, 685–695. [Google Scholar]
- Lazzerini, P.E.; Capecchi, P.L.; Selvi, E.; Lorenzini, S.; Bisogno, S.; Galeazzi, M.; Laghi Pasini, F. Hyperhomocysteinemia, inflammation and autoimmunity. Autoimmun. Rev. 2007, 6, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.H.; Wang, F.; You, S.J.; Cao, Y.J.; Cao, L.D.; Han, Q.; Liu, C.F.; Hu, L.F. Dysregulation of cystathionine γ-lyase (CSE)/hydrogen sulfide pathway contributes to ox-LDL-induced inflammation in macrophage. Cell. Signal. 2013, 25, 2255–2262. [Google Scholar] [CrossRef] [PubMed]
- Beard, R.S.; Bearden, S.E. Vascular complications of cystathionine β-synthase deficiency: Future directions for homocysteine-to-hydrogen sulfide research. Am. J. Physiol. Heart C 2011, 300, H13–H26. [Google Scholar] [CrossRef] [PubMed]
- Sen, U.; Givvimani, S.; Abe, O.A.; Lederer, E.D.; Tyagi, S.C. Cystathionine β-synthase and cystathionine γ-lyase double gene transfer ameliorate homocysteine-mediated mesangial inflammation through hydrogen sulfide generation. Am. J. Physiol. Cell. Physiol. 2011, 300, C155–C163. [Google Scholar] [CrossRef] [PubMed]
- Flannigan, K.L.; Agbor, T.A.; Blackler, R.W.; Kim, J.J.; Khan, W.I.; Verdu, E.F.; Ferraz, J.G.; Wallace, J.L. Impaired hydrogen sulfide synthesis and IL-10 signaling underlie hyperhomocysteinemia-associated exacerbation of colitis. Proc. Natl. Acad. Sci. USA 2014, 111, 13559–13564. [Google Scholar] [CrossRef] [PubMed]
- Hine, C.; Harputlugil, E.; Zhang, Y.; Ruckenstuhl, C.; Lee, B.C.; Brace, L.; Longchamp, A.; Trevino-Villarreal, J.H.; Mejia, P.; Ozaki, C.K.; et al. Endogenous hydrogen sulfide production is essential for dietary restriction benefits. Cell 2015, 160, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Physiological implications of hydrogen sulfide: A whiff exploration that blossomed. Physiol. Rev. 2012, 92, 791–896. [Google Scholar] [CrossRef] [PubMed]
- Ishii, I.; Akahoshi, N.; Yu, X.N.; Kobayashi, Y.; Namekata, K.; Komaki, G.; Kimura, H. Murine cystathionine γ-lyase: Complete cDNA and genomic sequences, promoter activity, tissue distribution and developmental expression. Biochem. J. 2004, 381, 113–123. [Google Scholar] [PubMed]
- Lund, G.; Andersson, L.; Lauria, M.; Lindholm, M.; Fraga, M.F.; Villar-Garea, A.; Ballestar, E.; Esteller, M.; Zaina, S. DNA methylation polymorphisms precede any histological sign of atherosclerosis in mice lacking apolipoprotein E. J. Biol. Chem. 2004, 279, 29147–29154. [Google Scholar] [CrossRef] [PubMed]
- Jamaluddin, M.D.; Chen, I.; Yang, F.; Jiang, X.; Jan, M.; Liu, X.; Schafer, A.I.; Durante, W.; Yang, X.; Wang, H. Homocysteine inhibits endothelial cell growth via DNA hypomethylation of the cyclin A gene. Blood 2007, 110, 3648–3655. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.H.; Hu, J.; Zhou, Y.L.; Hu, S.; Wang, Y.M.; Chen, W.; Xiao, Y.; Huang, L.Y.; Jiang, X.; Xu, G.Y. Promoted interaction of nuclear factor-κB with demethylated cystathionine-β-synthetase gene contributes to gastric hypersensitivity in diabetic rats. J. Neurosci.: Off. J. Soc. Neurosci. 2013, 33, 9028–9038. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.P. Epigenetic variation and human disease. J. Nutr. 2002, 132, 2388S–2392S. [Google Scholar] [PubMed]
- Zhou, S.; Zhang, Z.; Xu, G. Notable epigenetic role of hyperhomocysteinemia in atherogenesis. Lipids Health Dis. 2014, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Yi-Deng, J.; Tao, S.; Hui-Ping, Z.; Jian-Tuan, X.; Jun, C.; Gui-Zhong, L.; Shu-Ren, W. Folate and ApoE DNA methylation induced by homocysteine in human monocytes. DNA Cell Biol. 2007, 26, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.; Li, H.; Untereiner, A.; Wu, L.; Yang, G.; Austin, R.C.; Dickhout, J.G.; Lhotak, S.; Meng, Q.H.; Wang, R. Decreased endogenous production of hydrogen sulfide accelerates atherosclerosis. Circulation 2013, 127, 2523–2534. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.Y.; Lu, S.C.; Lee, C.M.; Chen, Y.J.; Dugan, T.A.; Huang, W.H.; Chang, S.F.; Liao, W.S.; Chen, C.H.; Lee, Y.T. Homocysteine inhibits arterial endothelial cell growth through transcriptional downregulation of fibroblast growth factor-2 involving G protein and DNA methylation. Circ. Res. 2008, 102, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Curro, M.; Trovato-Salinaro, A.; Gugliandolo, A.; Koverech, G.; Lodato, F.; Caccamo, D.; Calabrese, V.; Ientile, R. Resveratrol protects against homocysteine-induced cell damage via cell stress response in neuroblastoma cells. J. Neurosci. Res. 2015, 93, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Kamat, P.K.; Kalani, A.; Tyagi, S.C.; Tyagi, N. Hydrogen Sulfide epigenetically attenuates homocysteine-induced mitochondrial toxicity mediated through nmda receptor in mouse brain endothelial (bEnd3) Cells. J. Cell. Physiol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; He, Y.; Zong, Y.; Guo, J.; Sun, L.; Ma, Y.; Dong, W.; Gui, L. 17β-estradiol attenuates homocysteine-induced oxidative stress and inflammatory response as well as MAPKs cascade via activating PI3-K/Akt signal transduction pathway in Raw264.7 cells. Acta Biochim. Biophys. Sin. 2015, 47, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Du, H.P.; Li, J.; Xu, R.; Wang, Y.L.; You, S.J.; Liu, H.; Wang, F.; Cao, Y.J.; Liu, C.F.; et al. Statins upregulate cystathionine γ-lyase transcription and H2S generation via activating Akt signaling in macrophage. Pharmacol. Res. 2014, 87, 18–25. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.-J.; Li, Q.; Du, H.-P.; Wang, Y.-L.; You, S.-J.; Wang, F.; Xu, X.-S.; Cheng, J.; Cao, Y.-J.; Liu, C.-F.; et al. Homocysteine Triggers Inflammatory Responses in Macrophages through Inhibiting CSE-H2S Signaling via DNA Hypermethylation of CSE Promoter. Int. J. Mol. Sci. 2015, 16, 12560-12577. https://doi.org/10.3390/ijms160612560
Li J-J, Li Q, Du H-P, Wang Y-L, You S-J, Wang F, Xu X-S, Cheng J, Cao Y-J, Liu C-F, et al. Homocysteine Triggers Inflammatory Responses in Macrophages through Inhibiting CSE-H2S Signaling via DNA Hypermethylation of CSE Promoter. International Journal of Molecular Sciences. 2015; 16(6):12560-12577. https://doi.org/10.3390/ijms160612560
Chicago/Turabian StyleLi, Jiao-Jiao, Qian Li, Hua-Ping Du, Ya-Li Wang, Shou-Jiang You, Fen Wang, Xing-Shun Xu, Jian Cheng, Yong-Jun Cao, Chun-Feng Liu, and et al. 2015. "Homocysteine Triggers Inflammatory Responses in Macrophages through Inhibiting CSE-H2S Signaling via DNA Hypermethylation of CSE Promoter" International Journal of Molecular Sciences 16, no. 6: 12560-12577. https://doi.org/10.3390/ijms160612560