
Trianthema portulacastrum Linn. Displays Anti-Inflammatory Responses during Chemically Induced Rat Mammary Tumorigenesis through Simultaneous and Differential Regulation of NF-κB and Nrf2 Signaling Pathways

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
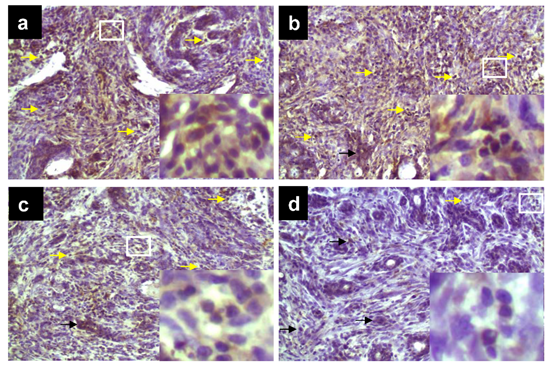
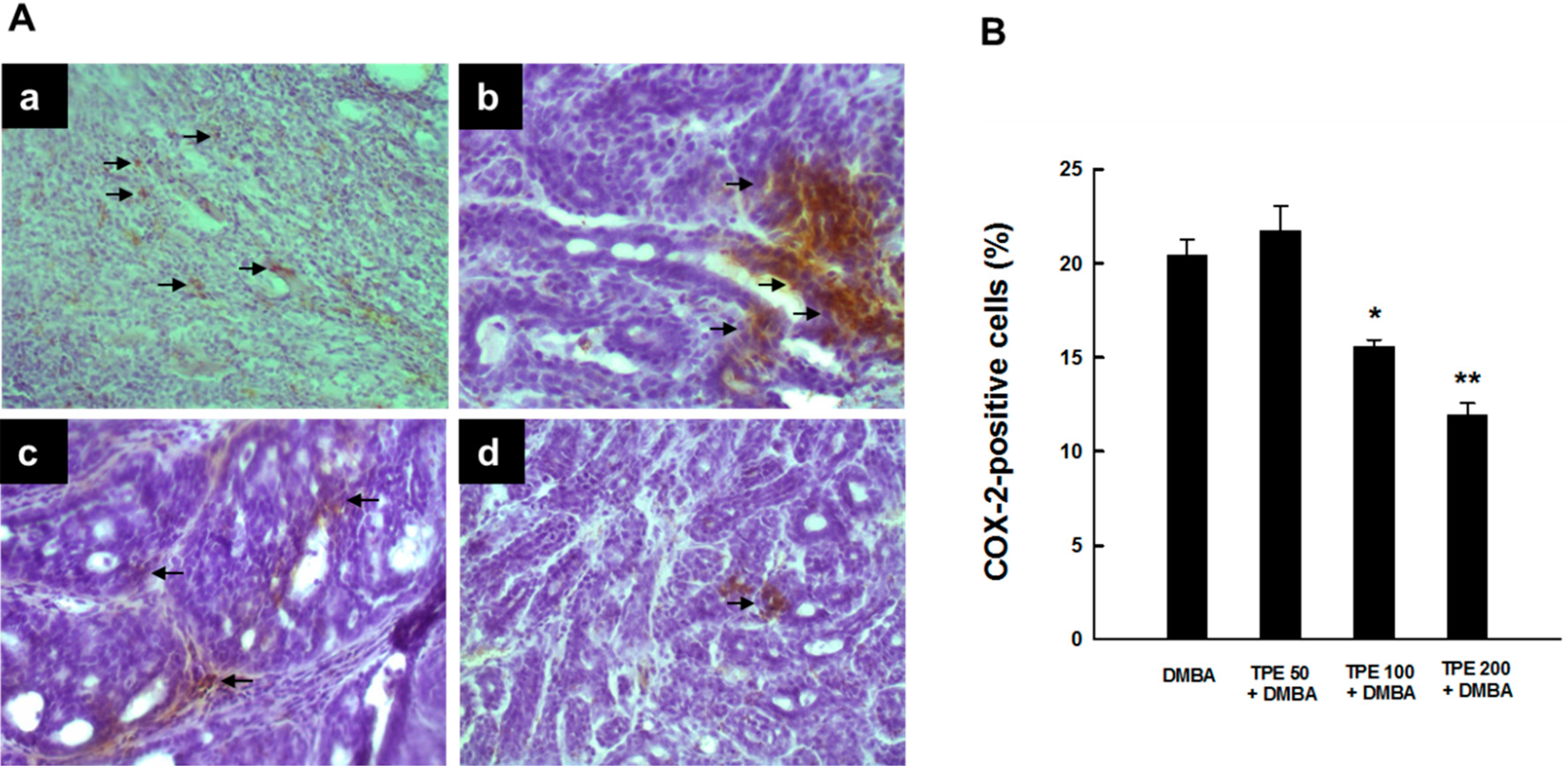
2.1. TPE Suppresses Elevated COX-2 Expression during DMBA-Induced Mammary Tumorigenesis

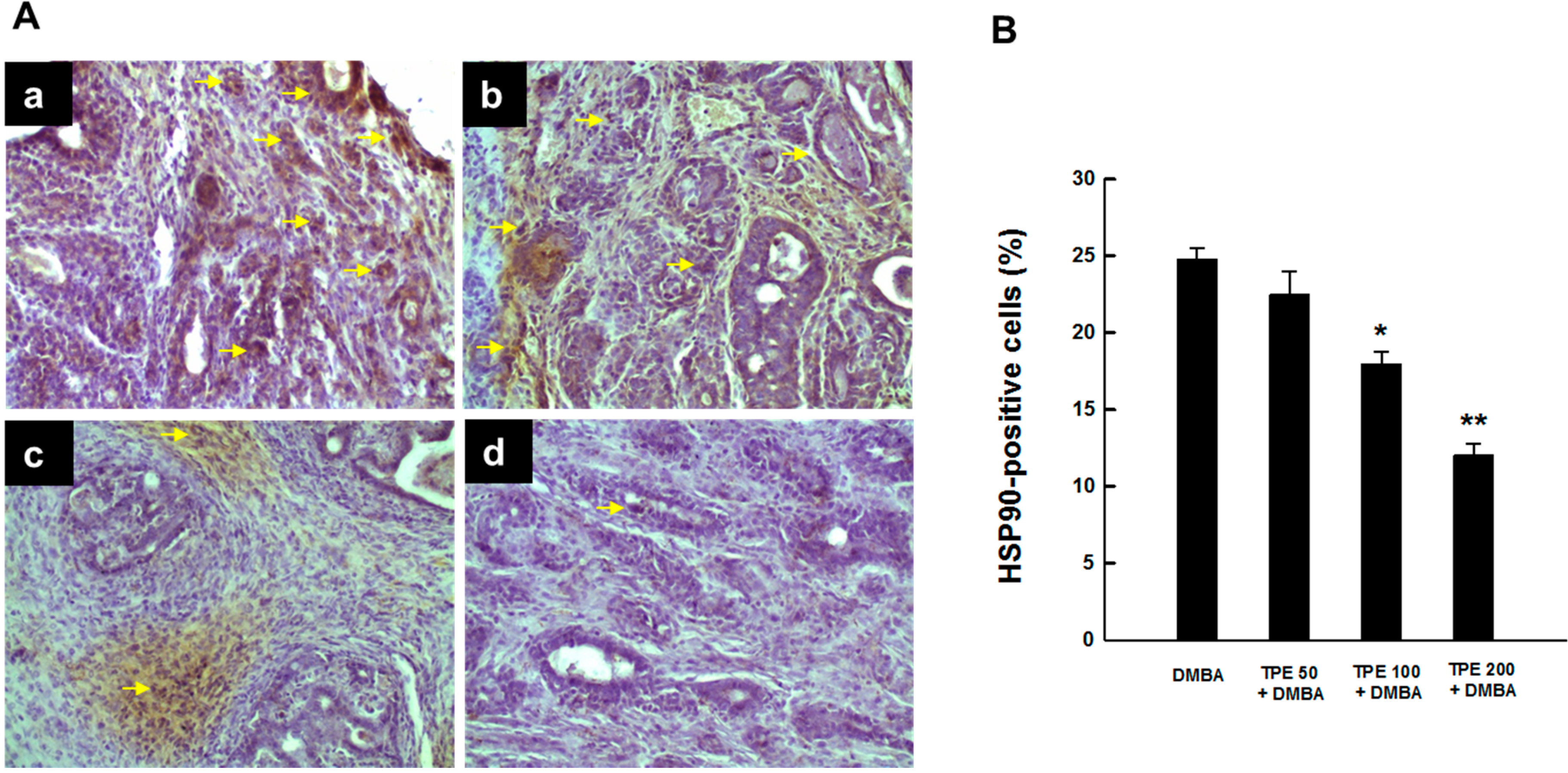
2.2. TPE Inhibits HSP90 Expression during DMBA Mammary Carcinogenesis

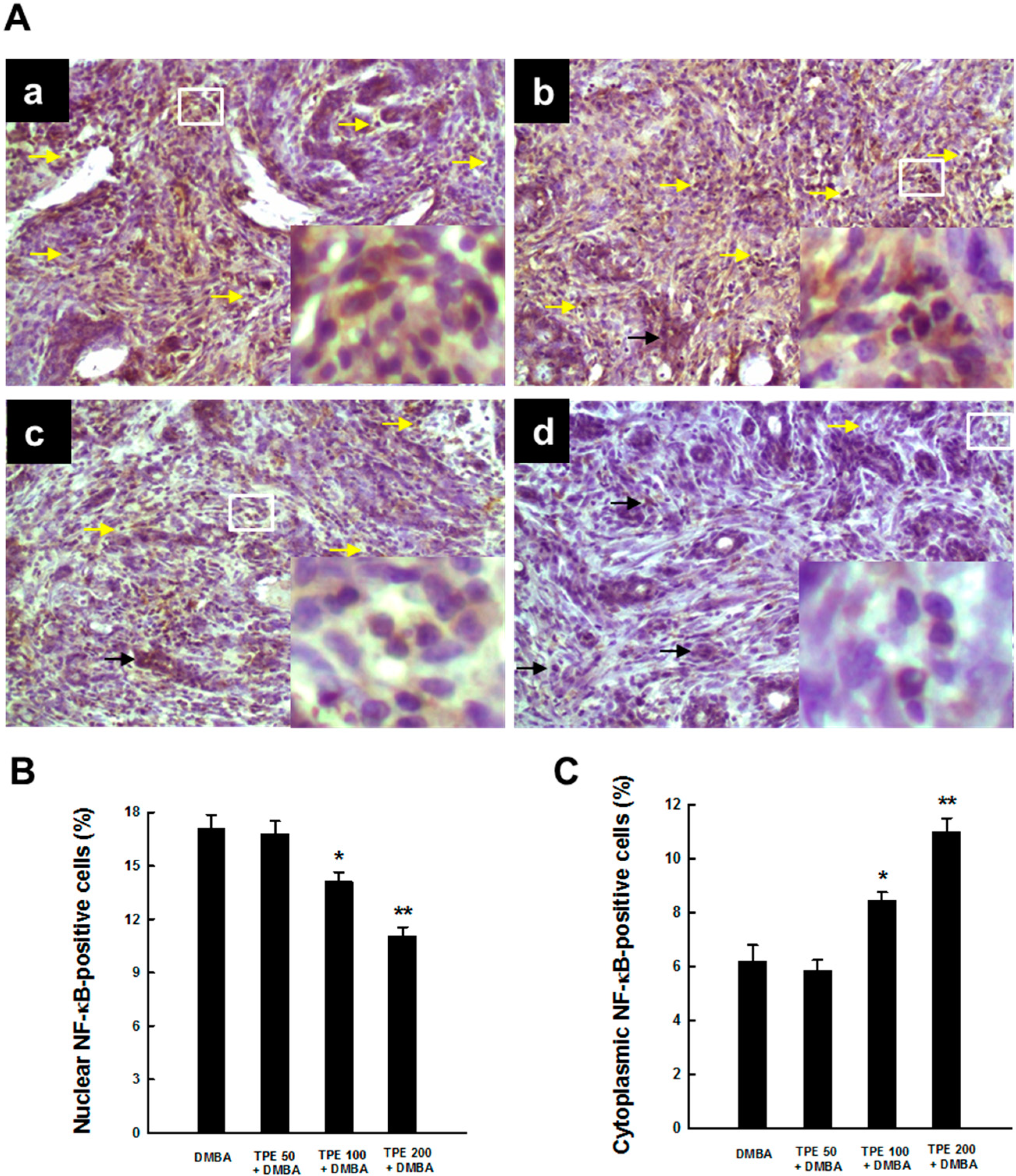
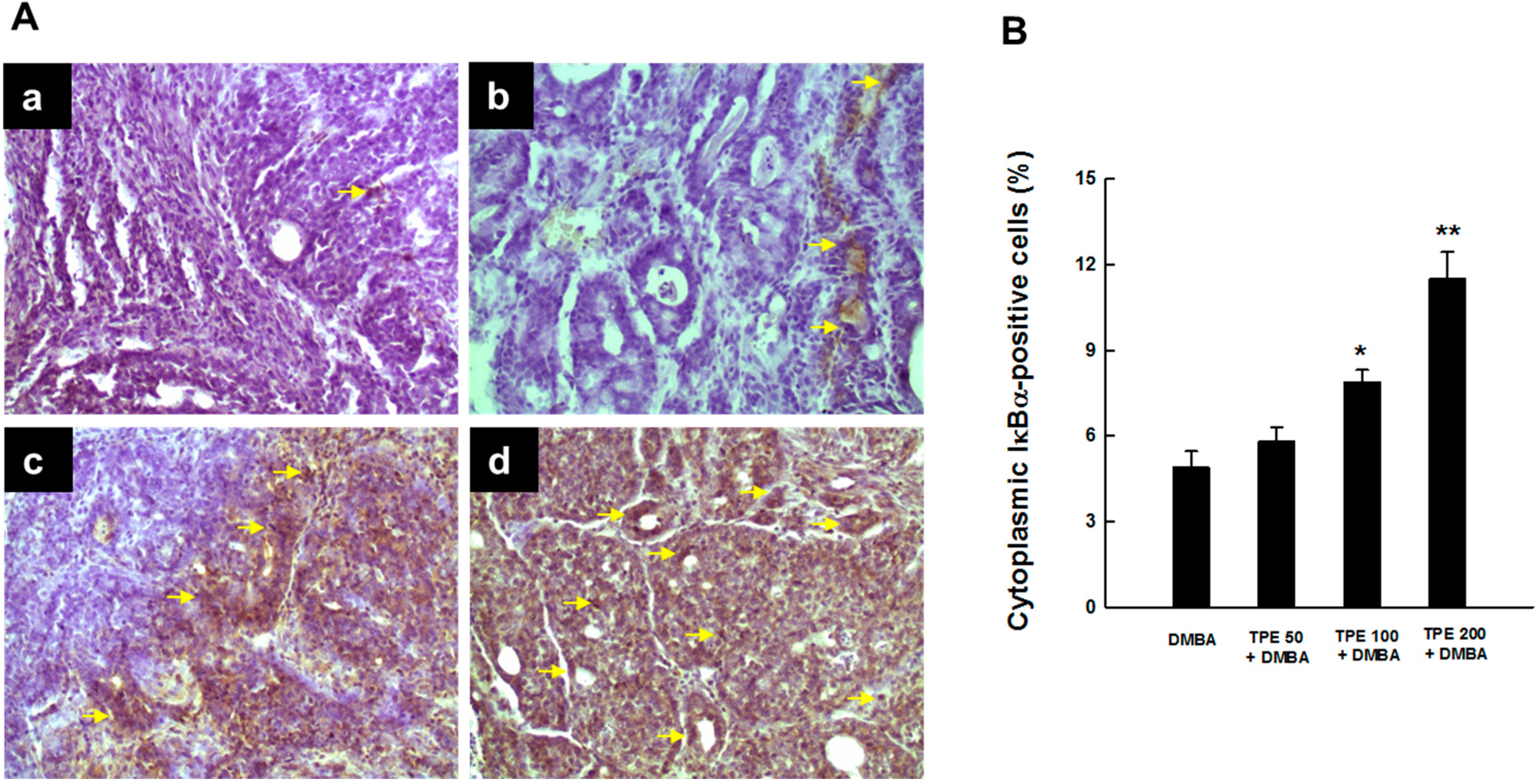
2.3. TPE Attenuates Activation of NF-κB during Mammary Tumorigenesis


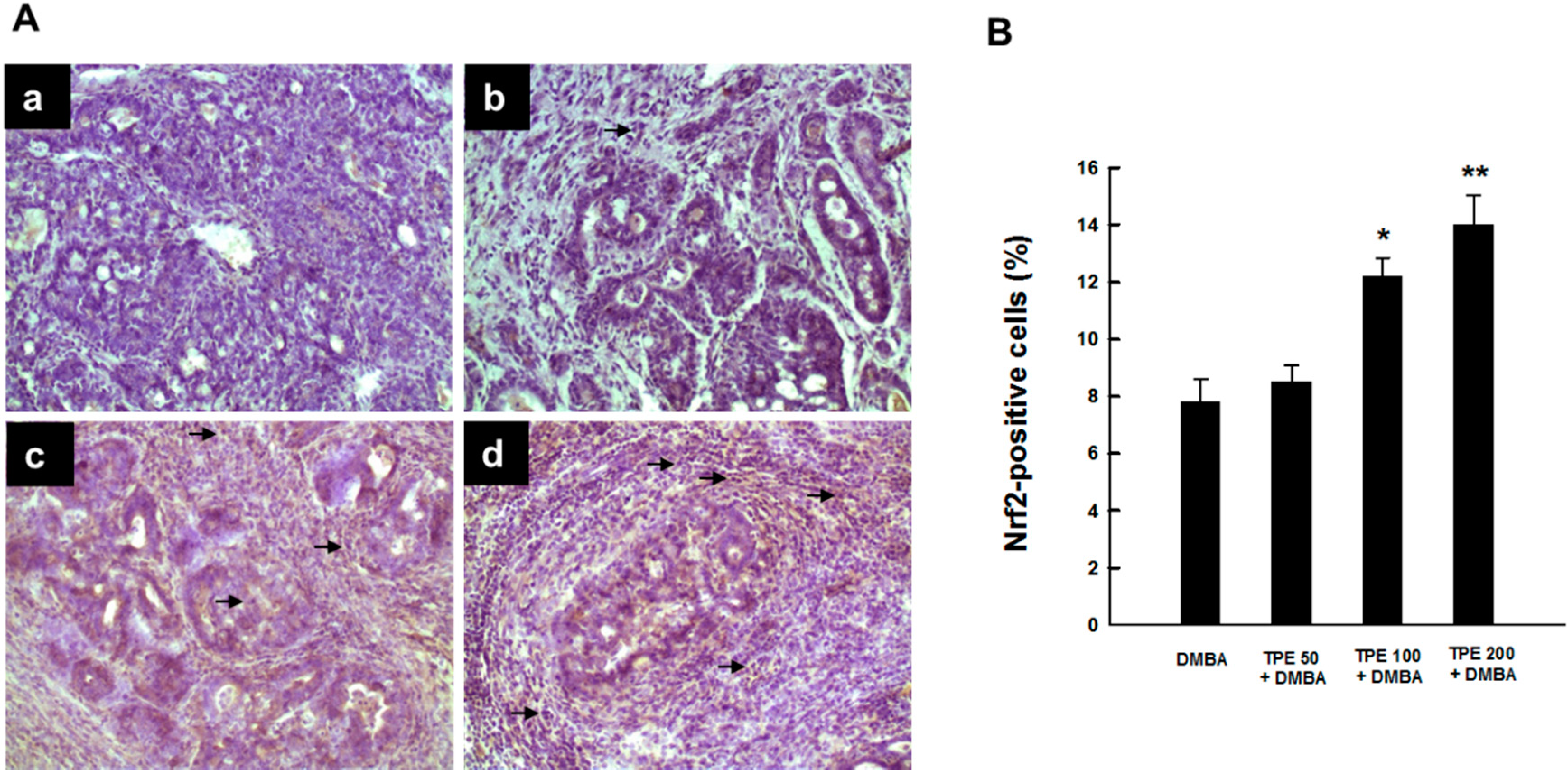
2.4. TPE Upregulates Nrf2 Expression during Mammary Tumorigenesis Induced by DMBA

3. Discussion
4. Experimental Section
4.1. Plant Material
4.2. Chemicals and Antibodies
4.3. Experimental Design and Tissue Harvesting
4.4. Immunohistochemical Analysis
4.5. Statistical Analyses
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kirtikar, K.R.; Basu, B.D. Indian Medicinal Plants; Lalit Mohan Basu: Allahabad, India, 1975; Volume 2. [Google Scholar]
- Jansen, P.C.M. Trianthema portulacastrum L.; Prota 2: Vegetables/Légumes. [CD-Rom]; Grubben, G.J.H., Denton, O.A., Eds.; PROTA: Wageningen, The Netherlands, 2004. [Google Scholar]
- Khan, N.; Sultana, A.; Tahir, N.; Jamila, N. Nutritional composition, vitamins, minerals and toxic heavy metals analysis of Trianthema portulacastrum L., a wild edible plant from Peshawar, Khyber Pakhtunkhwa. Pak. Afr. J. Biotechnol. 2013, 12, 6079–6085. [Google Scholar]
- Wahid, H.H.; Siddiqui, A. Survey of Drugs, 2nd ed.; Institute of History of Medicine and Medical Research: New Delhi, India, 1961. [Google Scholar]
- Fazal, U.; Razzack, M. A Handbook of Common Remedies in Unani Medicine; Central Council for Research in Unani Medicine: New Delhi, India, 1978. [Google Scholar]
- Khare, C.P. Indian Medicinal Plants, an Illustrated Dictionary; Springer-Verlag: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Vohora, S.B.; Shah, S.A.; Naqvi, S.A.H.; Ahmad, S.; Khan, M.S.Y. Studies on Trianthema portulacastrum. Planta Medica 1983, 47, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, H.R.; Malik, A.; Ali, M.S. Trianthenol: An antifungal tetraterpenoid from Trianthema portulacastrum (Aizoaceae). Phytochemistry 2001, 56, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.; Banu, G.S.; Pandian, M.R. Evaluation of the antioxidant activity of Trianthema portulacastrum L. Ind. J. Pharmacol. 2005, 37, 331–333. [Google Scholar] [CrossRef]
- Anreddy, R.N.R.; Porika, M.; Yellu, N.R.; Devarakonda, R.K. Hypogycemic and hypolipidemic activities of Trianthema portulacastrum Linn. plant in normal and alloxan induced diabetic rats. Int. J. Pharmacol. 2010, 6, 129–133. [Google Scholar] [CrossRef]
- Shivhare, M.K.; Singour, P.K.; Chaurasiya, P.K.; Pawar, R.S. Trianthema portulacastrum Linn. (Bishkhapra). Pharmacogn. Rev. 2012, 6, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, A.; Mandal, A.; Chatterjee, M. Prevention of alcohol-carbon tetrachloride-induced signs of early hepatotoxicity in mice by Trianthema portulacastrum L. Phytomedicine 1996, 3, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.; Bishayee, A.; Chatterjee, M. Trianthema portulacastrum affords antihepatotoxic activity against carbon tetrachloride-induced chronic liver damage in mice: Reflections in subcellular levels. Phytother. Res. 1997, 11, 216–221. [Google Scholar] [CrossRef]
- Mandal, A.; Karmakar, R.; Bandyopadhyay, S.; Chatterjee, M. Antihepatotoxic potential of Trianthema portulacastrum in carbon tetrachloride-induced chronic hepatocellular injury in mice: Reflection in haematological, histological and biochemical characteristics. Arch. Pharm. Res. 1998, 21, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.; Bandyopadhyay, S.; Chatterjee, M. Trianthema portulacastrum L. reverses hepatic lipid peroxidation, glutathione status and activities of related antioxidant enzymes in carbon tetrachloride-induced chronic liver damage in mice. Phytomedicine 1997, 4, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Pradhan, S.; Mukhopadhyay, I.; Bose, S.K.; Roy, S.; Chatterjee, M. Inhibition of early DNA-damage and chromosomal aberrations by Triantheme portulacastrum L. in carbon tetrachloride-induced mouse liver damage. Cell Biol. Int. 1999, 23, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.; Sharmila Banu, G.; Vanitha Pappa, P.; Sundararajan, M.; Rajasekara Pandian, M. Hepatoprotective activity of Trianthema portulacastrum L. against paracetamol and thioacetamide intoxication in albino rats. J. Ethnopharmacol. 2004, 92, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Sharmila Banu, G.; Kumar, G.; Murugesan, A.G. Ethanolic leaves extract of Trianthema portulacastrum L. ameliorates aflatoxin B1 induced hepatic damage in rats. Ind. J. Clin. Biochem. 2009, 24, 250–256. [Google Scholar] [CrossRef]
- Sharmila Banu, G.; Kumar, G.; Murugesan, A.G. Effect of ethanolic leaf extract of Trianthema portulacastrum L. on aflatoxin induced hepatic damage in rats. Ind. J. Clin. Biochem. 2009, 24, 414–418. [Google Scholar] [CrossRef]
- Shyam Sunder, A.; Rama Narsimha Reddy, A.; Rajeshwar, Y.; Kira, N.G.; Krishna Prasad, D.; Baburao, B.; Thirumurugu, S.; Karthik, A. Protective effect of methanolic extract of Trianthema portulacastrum in atherosclerotic diet induced renal and hepatic changes in rats. Der Pharm. Lett. 2010, 2, 540–545. [Google Scholar]
- Bhattacharya, S.; Chatterjee, M. Protective role of Trianthema portulacastrum against diethylnitrosamine-induced experimental hepatocarcinogenesis. Cancer Lett. 1998, 129, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Chatterjee, M. Trianthema portulacastrum restores the antioxidant defense enzyme levels and hepatic biotransformation patterns in experimental rat hepatocarcinogenesis. Ital. J. Biochem. 1998, 48, 225–232. [Google Scholar]
- Bhattacharya, S.; Chatterjee, M. Inhibitory effect of Trianthema portulacastrum L. diethylnitroso-amine-induced phenobarbital promoted hepatocarcinogenesis. Neoplasma 1999, 46, 105–111. [Google Scholar] [PubMed]
- Reuben, S.C.; Gopalan, A.; Petit, D.M.; Bishayee, A. Modulation of angiogenesis by dietary phytoconstituents in the prevention and intervention of breast cancer. Mol. Nutr. Food Res. 2012, 56, 14–29. [Google Scholar] [CrossRef] [PubMed]
- Vadodkar, A.S.; Suman, S.; Lakshmanaswamy, R.; Damodaran, C. Chemoprevention of breast cancer by dietary compounds. Anticancer Agents Med. Chem. 2012, 12, 1185–1202. [Google Scholar] [CrossRef] [PubMed]
- Parikh, N.R.; Mandal, A.; Bhatia, D.; Siveen, K.S.; Sethi, G.; Bishayee, A. Oleanane triterpenoids in the prevention and therapy of breast cancer: Current evidence and future perspectives. Phytochem. Rev. 2014, 13, 793–810. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, A.; Mandal, A. Trianthema portulacastrum Linn. exerts chemoprevention of 7,12-dimethylbenz(a)anthracene-induced mammary tumorigenesis in rats. Mutat. Res. 2014, 768, 107–118. [Google Scholar] [CrossRef]
- Macciò, A.; Madeddu, C. Obesity, inflammation, and postmenopausal breast cancer: Therapeutic implications. Sci. World J. 2011, 11, 2020–2036. [Google Scholar] [CrossRef]
- Shostak, K.; Chariot, A. NF-κB, stem cells and breast cancer: The links get stronger. Breast Cancer Res. 2011, 13. [Google Scholar] [CrossRef] [PubMed]
- Baumgarten, S.C.; Frasor, J. Minireview: Inflammation: An instigator of more aggressive estrogen receptor (ER) positive breast cancers. Mol. Endocrinol. 2012, 26, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.E.; Casto, B.C.; Harris, Z.M. Cyclooxygenase-2 and the inflammogenesis of breast cancer. World J. Clin. Oncol. 2014, 5, 677–692. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.A. Impact of obesity on mammary gland inflammation and local estrogen production. J. Mammary Gland Biol. Neoplasia 2014, 19, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Pick, E.; Kluger, Y.; Giltnane, J.M.; Moeder, C.; Camp, R.L.; Rimm, D.L.; Kluger, H.M. High HSP90 expression is associated with decreased survival in breast cancer. Cancer Res. 2007, 67, 2932–2937. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Chang, J.T.; Geradts, J.; Neckers, L.M.; Haystead, T.; Spector, N.L.; Lyerly, H.K. Amplification and high-level expression of heat shock protein 90 marks aggressive phenotypes of human epidermal growth factor receptor 2 negative breast cancer. Breast Cancer Res. 2012, 14. [Google Scholar] [CrossRef] [PubMed]
- Nakshatri, H.; Bhat-Nakshatri, P.; Martin, D.A.; Goulet, R.J., Jr.; Sledge, G.W., Jr. Constitutive activation of NF-κB during progression of breast cancer to hormone-independent growth. Mol. Cell. Biol. 1997, 17, 3629–3639. [Google Scholar]
- Zubair, A.; Frieri, M. Role of nuclear factor-κB in breast and colorectal cancer. Curr. Allergy Asthma Rep. 2013, 13, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, M.M.; Sung, B.; Yadav, V.R.; Kannappan, R.; Aggarwal, B.B. NF-κB addiction and its role in cancer: “One size does not fit all”. Oncogene 2011, 30, 1615–1630. [Google Scholar] [CrossRef] [PubMed]
- Bellezza, I.; Mierla, A.L.; Minelli, A. Nrf2 and NF-κB and their concerted modulation in cancer pathogenesis and progression. Cancers 2010, 2, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Doh, S.T.; Chan, J.Y.; Kong, A.N.; Cai, L. Regulatory potential for concerted modulation of Nrf2- and Nfkb1-mediated gene expression in inflammation and carcinogenesis. Br. J. Cancer 2008, 99, 2070–2082. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Fouad, T.M.; Kogawa, T.; Reuben, J.M.; Ueno, N.T. The role of inflammation in inflammatory breast cancer. Adv. Exp. Med. Biol. 2014, 816, 53–73. [Google Scholar] [PubMed]
- Johnson, J.J. Carnosol: A promising anti-cancer and anti-inflammatory agent. Cancer Lett. 2011, 305, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sinha, D.; Biswas, J.; Sung, B.; Aggarwal, B.B.; Bishayee, A. Chemopreventive and chemotherapeutic potential of curcumin in breast cancer. Curr. Drug Targets 2012, 13, 1799–1819. [Google Scholar] [CrossRef] [PubMed]
- Howe, L.R.; Subbaramaiah, K.; Hudis, C.A.; Dannenberg, A.J. Molecular pathways: Adipose inflammation as a mediator of obesity-associated cancer. Clin. Cancer Res. 2013, 19, 6074–6083. [Google Scholar] [CrossRef] [PubMed]
- Crawford, S. Anti-inflammatory/antioxidant use in long-term maintenance cancer therapy: A new therapeutic approach to disease progression and recurrence. Ther. Adv. Med. Oncol. 2014, 6, 52–68. [Google Scholar] [CrossRef] [PubMed]
- Subbaramaiah, K.; Sue, E.; Bhardwaj, P.; Du, B.; Hudis, C.A.; Giri, D.; Kopelovich, L.; Zhou, X.K.; Dannenberg, A.J. Dietary polyphenols suppress elevated levels of proinflammatory mediators and aromatase in the mammary gland of obese mice. Cancer Prev. Res. 2013, 6, 886–897. [Google Scholar] [CrossRef]
- Howe, L.R. Inflammation and breast cancer. Cyclooxygenase/prostaglandin signaling and breast cancer. Breast Cancer Res. 2007, 9. [Google Scholar] [CrossRef]
- Singh-Ranger, G.; Salhab, M.; Mokbel, K. The role of cyclooxygenase-2 in breast cancer: Review. Breast Cancer Res. Treat. 2008, 109, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.S.; Mann, M.; DuBois, R.N. The role of cyclooxygenases in inflammation, cancer, and development. Oncogene 1999, 18, 7908–7916. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.E. Cyclooxygenase-2 (cox-2) blockade in the chemoprevention of cancers of the colon, breast, prostate, and lung. Inflammopharmacology 2009, 17, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Glover, J.A.; Hughes, C.M.; Cantwell, M.M.; Murray, L.J. A systematic review to establish the frequency of cyclooxygenase-2 expression in normal breast epithelium, ductal carcinoma in situ, microinvasive carcinoma of the breast and invasive breast cancer. Br. J. Cancer 2011, 105, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Abou-Issa, H.M.; Alshafie, G.A.; Seibert, K.; Koki, A.T.; Masferrer, J.L.; Harris, R.E. Dose-response effects of the COX-2 inhibitor, celecoxib, on the chemoprevention of mammary carcinogenesis. Anticancer Res. 2001, 21, 3425–3432. [Google Scholar] [PubMed]
- Pandi, M.; Manikandan, R.; Muthumary, J. Anticancer activity of fungal taxol derived from Botryodiplodia theobromae Pat., an endophytic fungus, against 7,12 dimethyl benz(a)anthracene (DMBA)-induced mammary gland carcinogenesis in Sprague Dawley rats. Biomed. Pharmacother. 2010, 64, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.; George, J.; Srivastava, S.; Tyagi, S.; Shukla, Y. Inhibitory effects of tea polyphenols by targeting cyclooxygenase-2 through regulation of nuclear factor kappa B, Akt and p53 in rat mammary tumors. Investig. New Drugs 2011, 29, 225–231. [Google Scholar] [CrossRef]
- Nakatsugi, S.; Ohta, T.; Kawamori, T.; Mutoh, M.; Tanigawa, T.; Watanabe, K.; Sugie, S.; Sugimura, T.; Wakabayashi, K. Chemoprevention by nimesulide, a selective cyclooxygenase-2 inhibitor, of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP)-induced mammary gland carcinogenesis in rats. Jpn. J. Cancer Res. 2000, 91, 886–892. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Bueso-Ramos, C.; Aggarwal, B.B. Suppression of 7,12-dimethylbenz(a)anthracene-induced mammary carcinogenesis in rats by resveratrol: Role of nuclear factor-κB, cyclooxygenase 2, and matrix metalloprotease 9. Cancer Res. 2006, 62, 4945–4954. [Google Scholar]
- Shimp, S.K., 3rd; Parson, C.D.; Regna, N.L.; Thomas, A.N.; Chafin, C.B.; Reilly, C.M.; Nichole Rylander, M. HSP90 inhibition by 17-DMAG reduces inflammation in J774 macrophages through suppression of Akt and nuclear factor-κB pathways. Inflamm. Res. 2012, 61, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Tamura, Y.; Torigoe, T.; Kutomi, G.; Hirata, K.; Sato, N. New paradigm for intrinsic function of heat shock proteins as endogenous ligands in inflammation and innate immunity. Curr. Mol. Med. 2012, 12, 1198–1206. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kolls, J.K. Act1-hsp90 heats up TH17 inflammation. Nat. Immunol. 2013, 14, 16–17. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Thao, L.; Sensintaffar, J.; Zhang, L.; Boehm, M.F.; Fritz, L.C.; Burrows, FJ. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 2003, 425, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Sankhala, K.K.; Mita, M.M.; Mita, A.C.; Takimoto, C.H. Heat shock proteins: A potential anticancer target. Curr. Drug Targets 2011, 12, 2001–2008. [Google Scholar] [CrossRef] [PubMed]
- Barrott, J.J.; Haystead, T.A. Hsp90, an unlikely ally in the war on cancer. FEBS J. 2013, 280, 1381–1396. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Choi, K.; Kim, H.; Kim, K.; Lee, M.H.; Lee, J.H.; Kim Rim, J.C. Isoflavone-deprived soy peptide suppresses mammary tumorigenesis by inducing apoptosis. Exp. Mol. Med. 2009, 41, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.L.; Kim, S.A.; Choi, H.S.; Yoon, J.H.; Ahn, S.G. Epigallocatechin-3-gallate suppresses the expression of HSP70 and HSP90 and exhibits anti-tumor activity in vitro and in vivo. BMC Cancer 2010, 10. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, A.; Thoppil, R.J.; Mandal, A.; Darvesh, A.S.; Ohanyan, V.; Meszaros, J.G.; Háznagy-Radnai, E.; Hohmann, J.; Bhatia, D. Black currant phytoconstituents exert chemoprevention of diethylnitrosamine-initiated hepatocarcinogenesis by suppression of the inflammatory response. Mol. Carcinogenes. 2013, 52, 304–317. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-κB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed]
- Rayet, B.; Gélinas, C. Aberrant rel/nfkb genes and activity in human cancer. Oncogene 1999, 18, 6938–6947. [Google Scholar] [CrossRef] [PubMed]
- Viatour, P.; Merville, M.P.; Bours, V.; Chariot, A. Phosphorylation of NF-κB and IκB proteins: Implications in cancer and inflammation. Trends Biochem. Sci. 2005, 30, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.; Sung, B.; Aggarwal, B.B. Nuclear factor-κB activation: From bench to bedside. Exp. Biol. Med. 2008, 233, 21–31. [Google Scholar] [CrossRef]
- Sovak, M.A.; Bellas, R.E.; Kim, D.W.; Zanieski, G.J.; Rogers, A.E.; Traish, A.M.; Sonenshein, G.E. Aberrant nuclear factor-κB/Rel expression and the pathogenesis of breast cancer. J. Clin. Investig. 1997, 100, 2952–2960. [Google Scholar] [CrossRef] [PubMed]
- Van Laere, S.; van der Auwera, I.; van den Eynden, G.G.; Fox, S.B.; Bianchi, F.; Harris, A.L.; van Dam, P.; van Marck, E.A.; Vermeulen, P.B.; Dirix, L.Y. Distinct molecular signature of inflammatory breast cancer by cDNA microarray analysis. Breast Cancer Res. Treat. 2005, 93, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Jana, D.; Das, S.; Sarkar, D.K.; Mandal, S.; Maji, A.; Mukhopadhyay, M. Role of nuclear factor-κB in female breast cancer: A study in Indian patients. Asian Pac. J. Cancer Prev. 2012, 13, 5511–5515. [Google Scholar] [CrossRef] [PubMed]
- Ralhan, R.; Pandey, M.K.; Aggarwal, B.B. Nuclear factor-κB links carcinogenic and chemopreventive agents. Front. Biosci. 2009, 1, 45–60. [Google Scholar] [CrossRef]
- Gupta, S.C.; Sundaram, C.; Reuter, S.; Aggarwal, B.B. Inhibiting NF-κB activation by small molecules as a therapeutic strategy. Biochim. Biophys. Acta 2010, 1799, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Zhong, S.; Shen, Z. Targeting the inflammatory pathways to enhance chemotherapy of cancer. Cancer Biol. Ther. 2011, 12, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Sovak, M.A.; Zanieski, G.; Nonet, G.; Romieu-Mourez, R.; Lau, A.W.; Hafer, L.J.; Yaswen, P.; Stampfer, M.; Rogers, A.E.; et al. Activation of NF-κB/Rel occurs early during neoplastic transformation of mammary cells. Carcinogenesis 2000, 21, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Currier, N.; Solomon, S.E.; Demicco, E.G.; Chang, D.L.; Farago, M.; Ying, H.; Dominguez, I.; Sonenshein, G.E.; Cardiff, R.D.; Xiao, Z.X.; et al. Oncogenic signaling pathways activated in DMBA-induced mouse mammary tumors. Toxicol. Pathol. 2005, 33, 726–737. [Google Scholar] [CrossRef] [PubMed]
- Vinothini, G.; Manikandan, P.; Anandan, R.; Nagini, S. Chemoprevention of rat mammary carcinogenesis by Azadirachta indica leaf fractions: Modulation of hormone status, xenobiotic-metabolizing enzymes, oxidative stress, cell proliferation and apoptosis. Food Chem. Toxicol. 2009, 47, 1852–1863. [Google Scholar] [CrossRef] [PubMed]
- Baird, L.; Dinkova-Kostova, A.T. The cytoprotective role of the Keap1-Nrf2 pathway. Arch. Toxicol. 2011, 85, 241–272. [Google Scholar] [CrossRef] [PubMed]
- Khor, T.O.; Yu, S.; Kong, A.N. Dietary cancer chemopreventive agents—Targeting inflammation and Nrf2 signaling pathway. Planta Med. 2008, 74, 1540–1547. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Saw, C.L.; Yu, R.; Kong, A.N. Regulation of NF-E2-related factor 2 signaling for cancer chemoprevention: Antioxidant coupled with antiinflammatory. Antioxid. Redox Signal. 2010, 13, 1679–1698. [Google Scholar] [CrossRef] [PubMed]
- Kundu, J.K.; Surh, Y.J. Nrf2-Keap1 signaling as a potential target for chemoprevention of inflammation-associated carcinogenesis. Pharm. Res. 2010, 27, 999–1013. [Google Scholar] [CrossRef] [PubMed]
- Slocum, S.L.; Kensler, T.W. Nrf2: Control of sensitivity to carcinogens. Arch. Toxicol. 2011, 85, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Cardozo, L.F.; Pedruzzi, L.M.; Stenvinkel, P.; Stockler-Pinto, M.B.; Daleprane, J.B.; Leite, M., Jr.; Mafra, D. Nutritional strategies to modulate inflammation and oxidative stress pathways via activation of the master antioxidant switch Nrf2. Biochimie 2013, 95, 1525–1533. [Google Scholar] [CrossRef] [PubMed]
- Carayol, N.; Chen, J.; Yang, F.; Jin, T.; Jin, L.; States, D.; Wang, C.Y. A dominant function of IKK/NF-κB signaling in global lipopolysaccharide-induced gene expression. J. Biol. Chem. 2006, 281, 31142–31151. [Google Scholar] [CrossRef] [PubMed]
- Khor, T.O.; Huang, M.T.; Kwon, K.H.; Chan, J.Y.; Reddy, B.S.; Kong, A.N. Nrf2-deficient mice have an increased susceptibility to dextran sulfate sodium-induced colitis. Cancer Res. 2006, 66, 11580–11584. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Khor, T.O.; Xu, C.; Shen, G.; Jeong, W.S.; Yu, S.; Kong, A.N. Activation of Nrf2-antioxidant signaling attenuates NFκB-inflammatory response and elicits apoptosis. Biochem. Pharmacol. 2008, 76, 1485–1489. [Google Scholar] [CrossRef] [PubMed]
- Becks, L.; Prince, M.; Burson, H.; Christophe, C.; Broadway, M.; Itoh, K.; Yamamoto, M.; Mathis, M.; Orchard, E.; Shi, R.; et al. Aggressive mammary carcinoma progression in Nrf2 knockout mice treated with 7,12-dimethylbenz[a]anthracene. BMC Cancer 2010, 10. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Bhat, N.K.; Bhat, H.K. Induction of NAD(P)H-quinone oxidoreductase 1 by antioxidants in female ACI rats is associated with decrease in oxidative DNA damage and inhibition of estrogen-induced breast cancer. Carcinogenesis 2012, 33, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Bhat, H.K. Superoxide dismutase 3 is induced by antioxidants, inhibits oxidative DNA damage and is associated with inhibition of estrogen-induced breast cancer. Carcinogenesis 2012, 33, 2601–2610. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Chatterjee, A.; Ronghe, A.M.; Bhat, N.K.; Bhat, H.K. Antioxidant-mediated up-regulation of OGG1 via NRF2 induction is associated with inhibition of oxidative DNA damage in estrogen-induced breast cancer. BMC Cancer 2013, 13. [Google Scholar] [CrossRef]
- Yao, Y.; Brodie, A.M.; Davidson, N.E.; Kensler, T.W.; Zhou, Q. Inhibition of estrogen signaling activates the NRF2 pathway in breast cancer. Breast Cancer Res. Treat. 2010, 124, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Manzanares, M.A.; Solanas, M.; Moral, R.; Escrich, R.; Vela, E.; Costa, I.; Escrich, E. Dietary extra-virgin olive oil and corn oil differentially modulate the mRNA expression of xenobiotic-metabolizing enzymes in the liver and in the mammary gland in a rat chemically induced breast cancer model. Eur. J. Cancer Prev. 2014, in press. [Google Scholar]
- Chen, B.; Zhang, Y.; Wang, Y.; Rao, J.; Jiang, X.; Xu, Z. Curcumin inhibits proliferation of breast cancer cells through Nrf2-mediated down-regulation of Fen1 expression. J. Steroid Biochem. Mol. Biol. 2014, 143, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Na, H.K.; Surh, Y.J. Oncogenic potential of Nrf2 and its principal target protein heme oxygenase-1. Free Radic. Biol. Med. 2014, 67, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.H. Potential synergy of phytochemicals in cancer prevention: Mechanism of action. J. Nutr. 2004, 134, 3479S–3485S. [Google Scholar] [PubMed]
- De Kok, T.M.; van Breda, S.G.; Manson, M.M. Mechanisms of combined action of different chemopreventive dietary compounds: A review. Eur. J. Nutr. 2008, 47, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Bode, A.M.; Dong, Z. Epigallocatechin 3-gallate and green tea catechins: United they work, divided they fail. Cancer Prev. Res. 2009, 2, 514–517. [Google Scholar] [CrossRef]
- Bishayee, A.; Mandal, A.; Thoppil, R.J.; Darvesh, A.S.; Bhatia, D. Chemopreventive effect of a novel oleanane triterpenoid in a chemically induced rodent model of breast cancer. Int. J. Cancer 2013, 133, 1054–1064. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.; Bhatia, D.; Bishayee, A. Suppression of inflammatory cascade is implicated in methyl amooranin-mediated inhibition of experimental mammary carcinogenesis. Mol. Carcinogenes. 2014, 53, 999–1010. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mandal, A.; Bishayee, A. Trianthema portulacastrum Linn. Displays Anti-Inflammatory Responses during Chemically Induced Rat Mammary Tumorigenesis through Simultaneous and Differential Regulation of NF-κB and Nrf2 Signaling Pathways. Int. J. Mol. Sci. 2015, 16, 2426-2445. https://doi.org/10.3390/ijms16022426
Mandal A, Bishayee A. Trianthema portulacastrum Linn. Displays Anti-Inflammatory Responses during Chemically Induced Rat Mammary Tumorigenesis through Simultaneous and Differential Regulation of NF-κB and Nrf2 Signaling Pathways. International Journal of Molecular Sciences. 2015; 16(2):2426-2445. https://doi.org/10.3390/ijms16022426
Chicago/Turabian StyleMandal, Animesh, and Anupam Bishayee. 2015. "Trianthema portulacastrum Linn. Displays Anti-Inflammatory Responses during Chemically Induced Rat Mammary Tumorigenesis through Simultaneous and Differential Regulation of NF-κB and Nrf2 Signaling Pathways" International Journal of Molecular Sciences 16, no. 2: 2426-2445. https://doi.org/10.3390/ijms16022426