
Designing of a Recombinant Multi-Epitopes Based Vaccine against Enterococcus mundtii Using Bioinformatics and Immunoinformatics Approaches

 ,
,  ,
,  ,
, 
Abstract
:1. Introduction
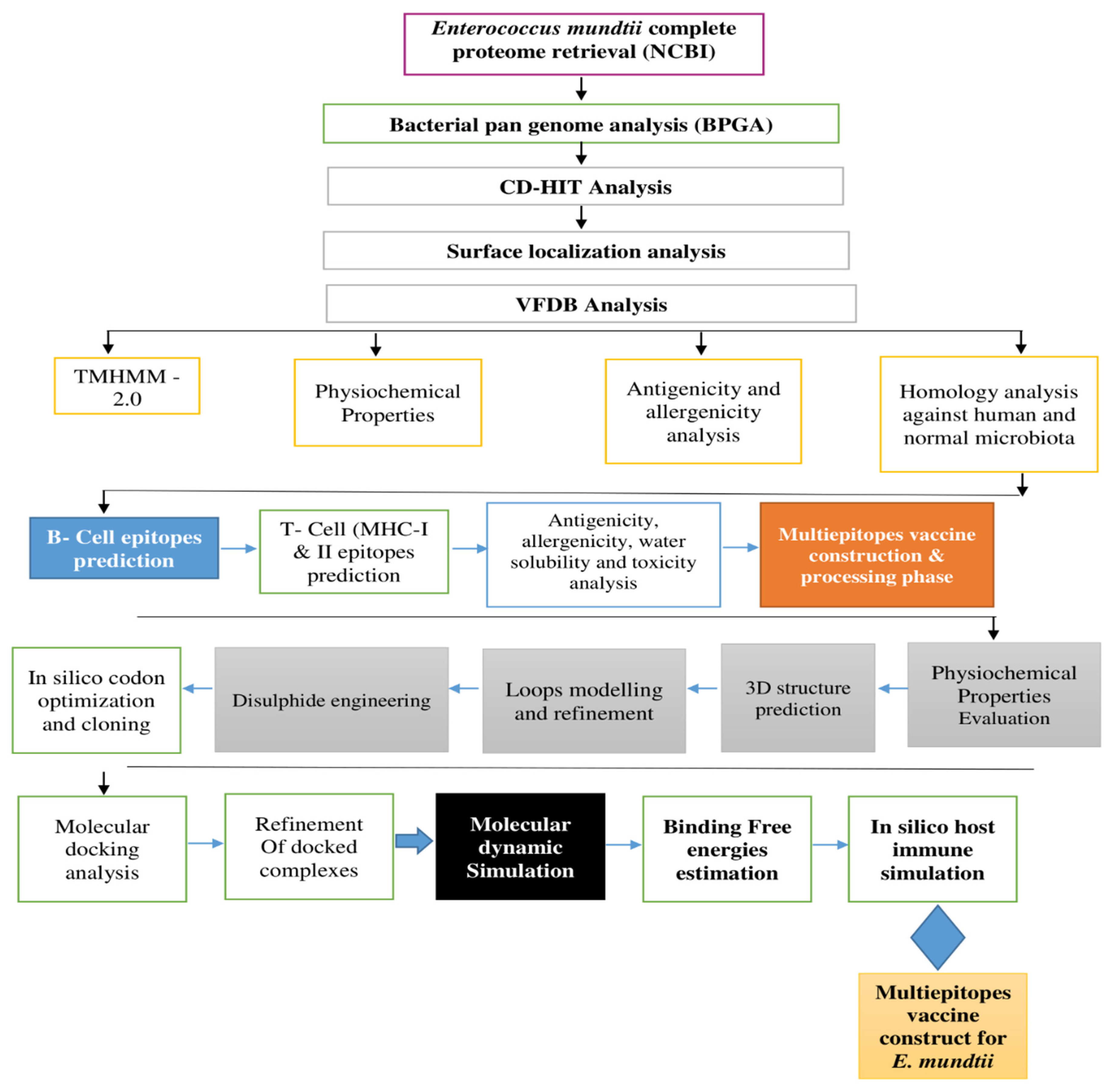
2. Research Methodology
2.1. E. mundtii Complete Proteome Extraction and Analysis
2.2. Epitope Prediction and Processing
2.3. Multi-Epitope Vaccine Construction and Processing Phase
2.4. Structure Prediction, Loops Modelling, Refinement, Codon Optimization, and Cloning
2.5. Molecular Docking Interaction Analysis
2.6. Molecular Dynamic Simulation
2.7. Free Binding Energies Calculation
2.8. Host Immune Simulation
3. Results
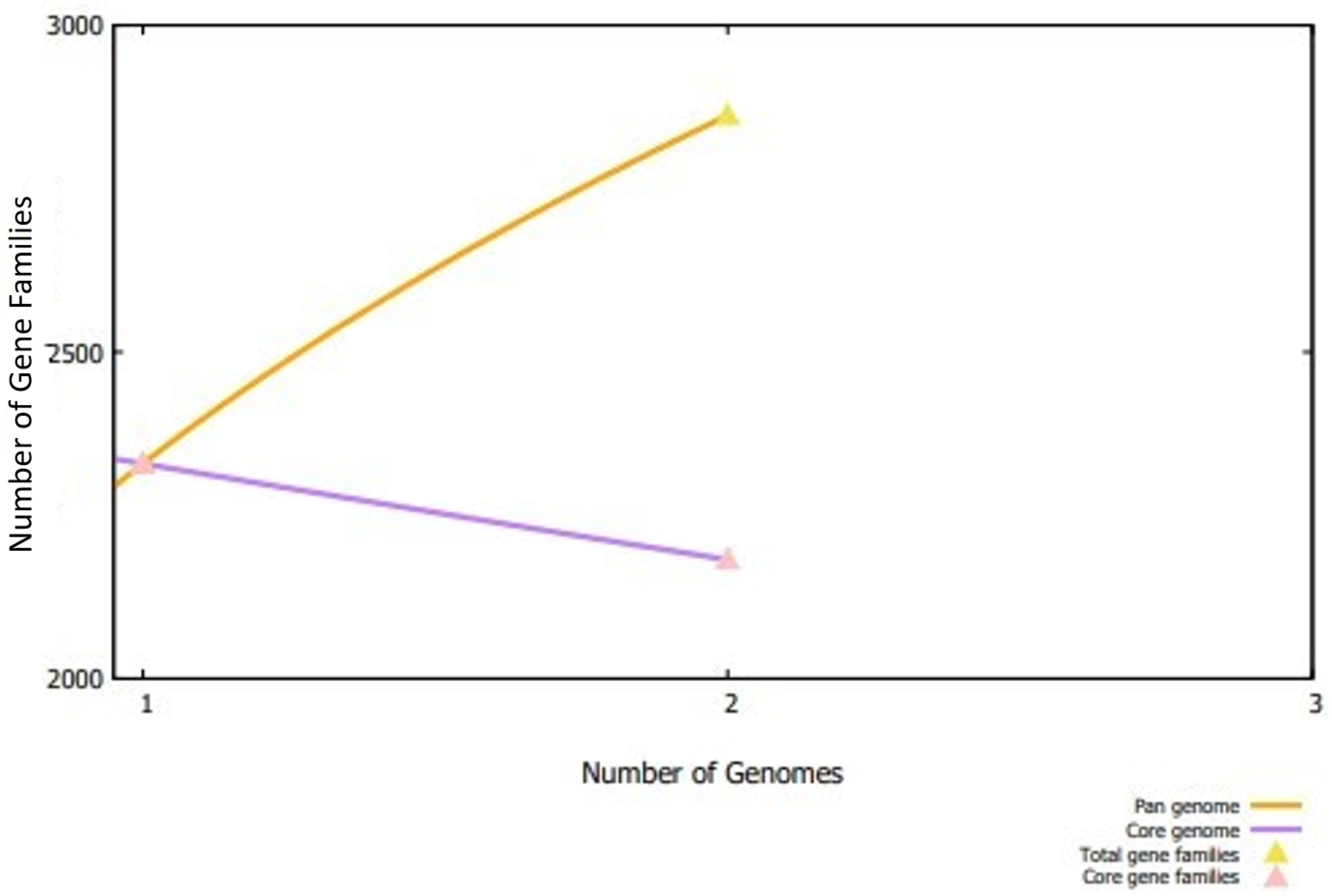
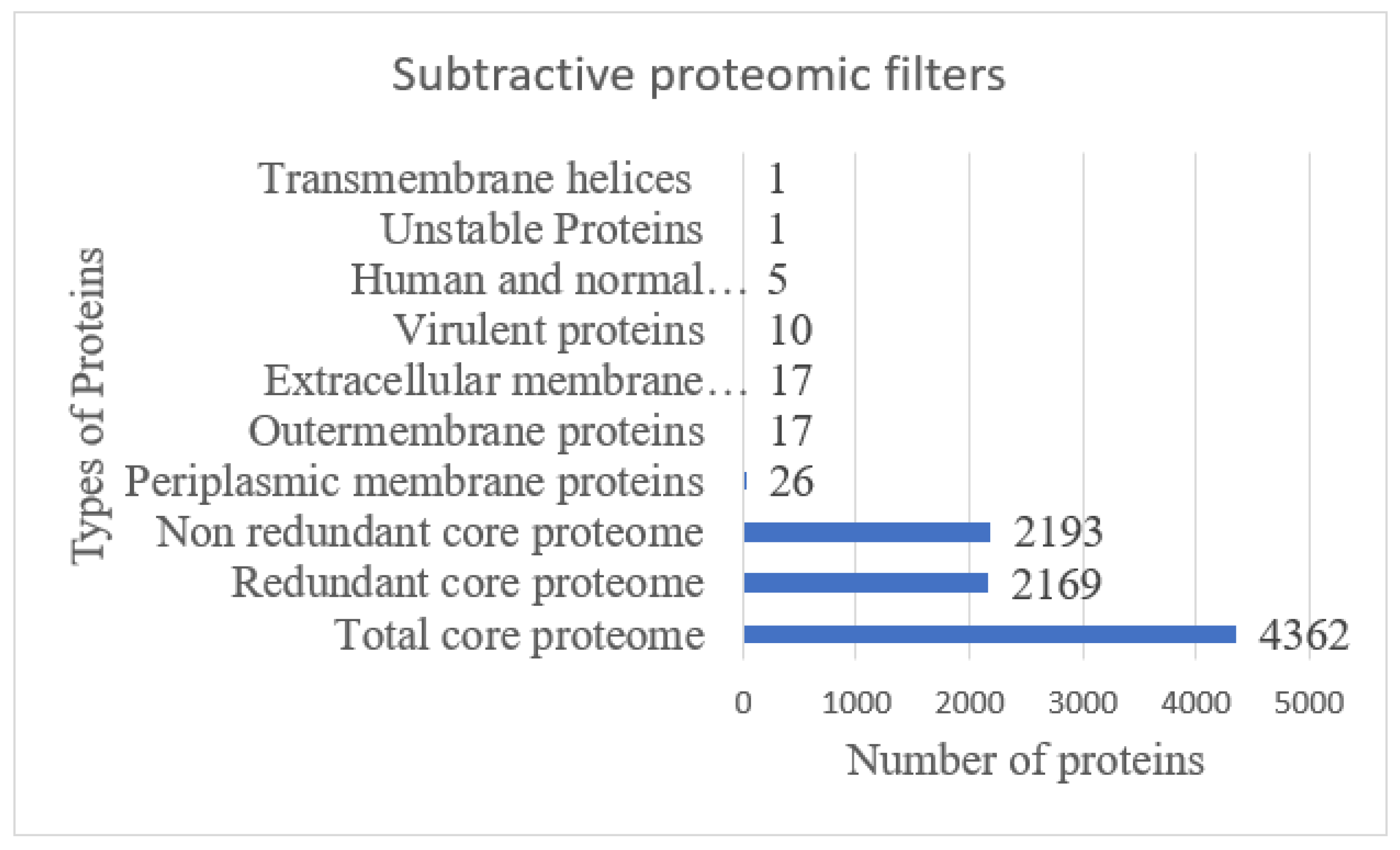
3.1. Subtractive Proteomics Analysis
3.2. CD-HIT, Surface Localization, VFDB, Antigenicity, Allergenicity, Homology, Transmembrane Helices, and Physiochemical Properties Analysis
3.3. B-Cell Derived T-Cell Epitope Prediction
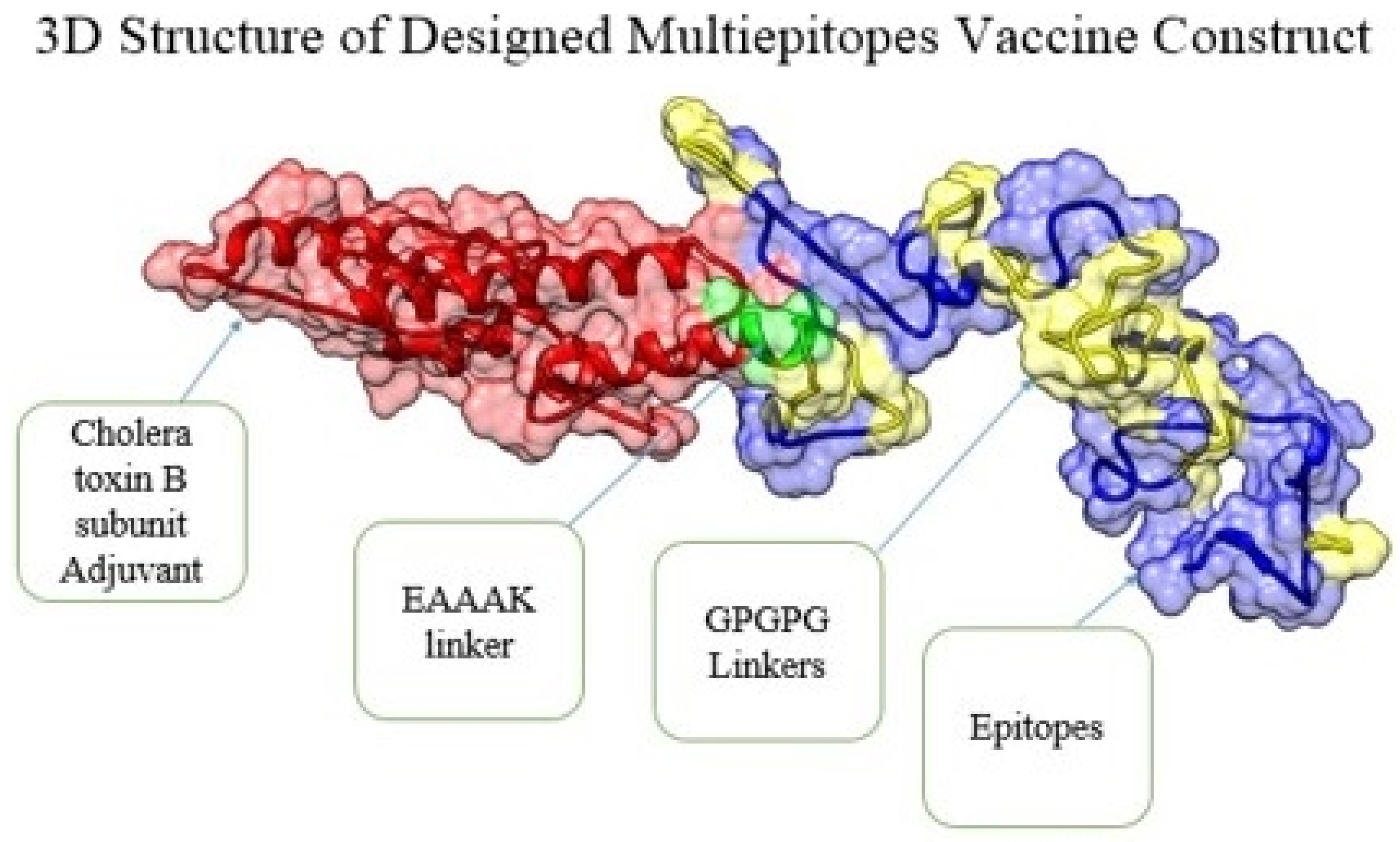
3.4. Multi-Epitope Vaccine Construction and Processing Phase
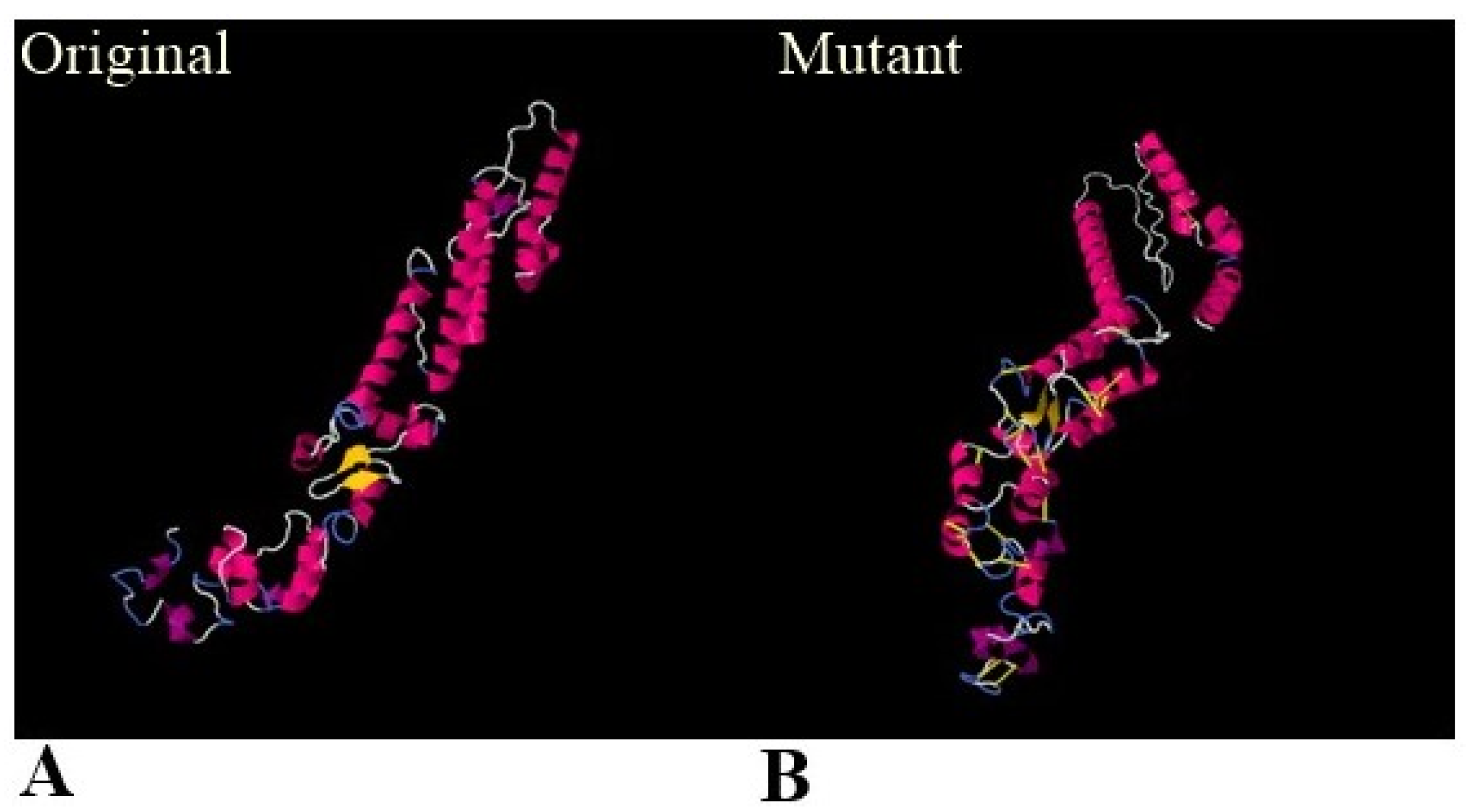
3.5. Physiochemical Properties, 3D Structure, Loops Modeling and Refinement, and Disulphide Engineering
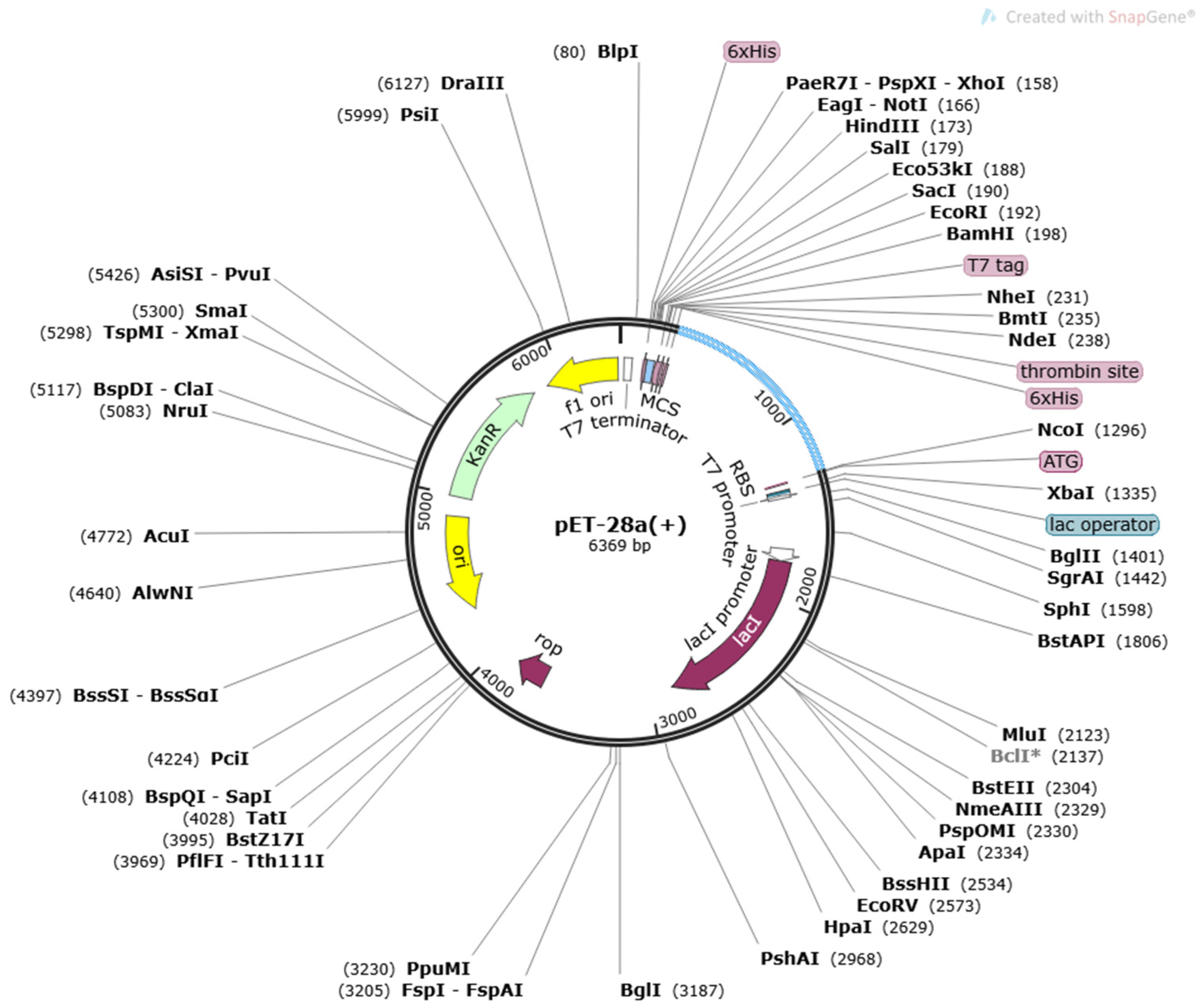
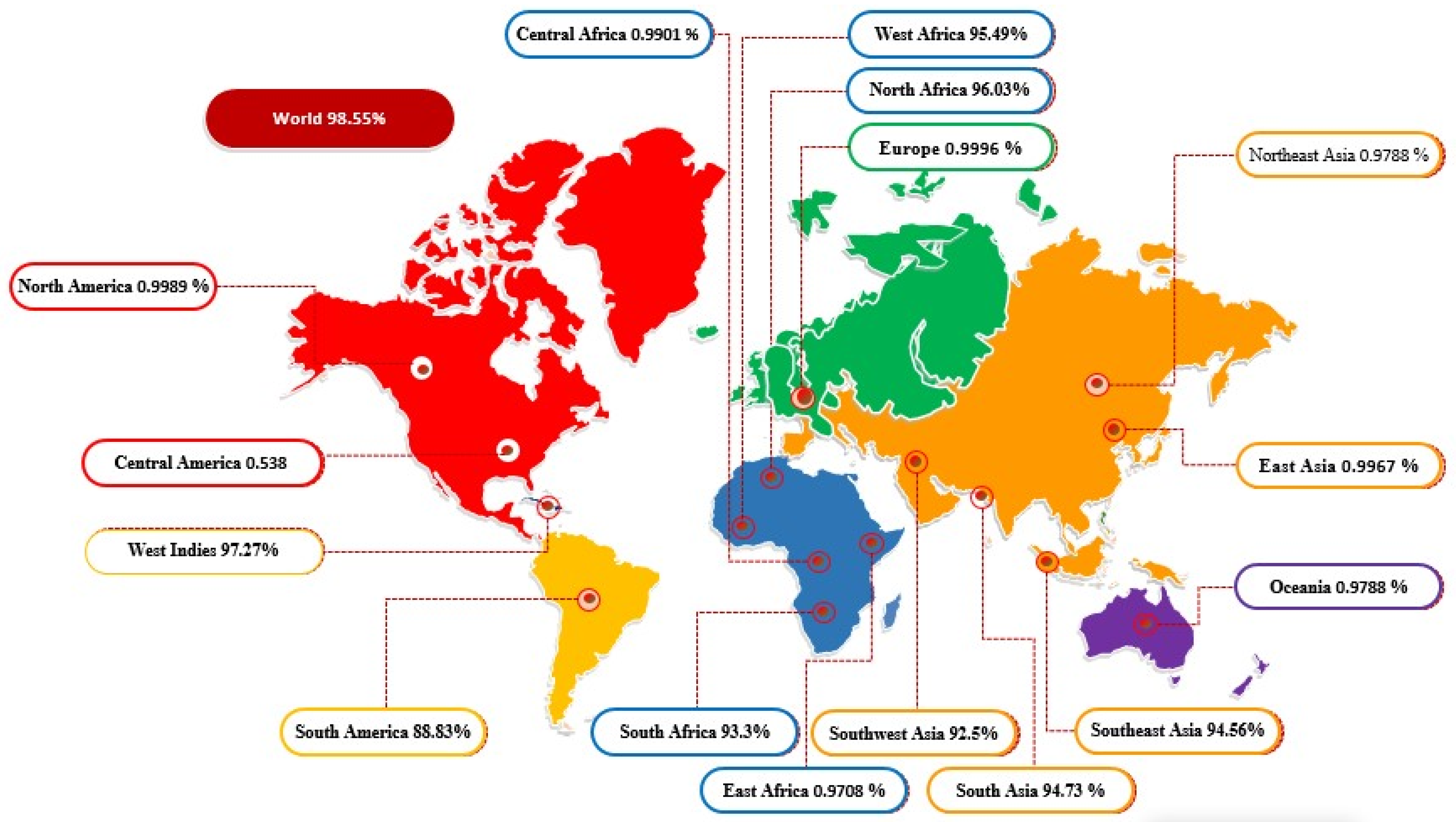
3.6. In Codon Optimization Cloning and Population Coverage Analysis
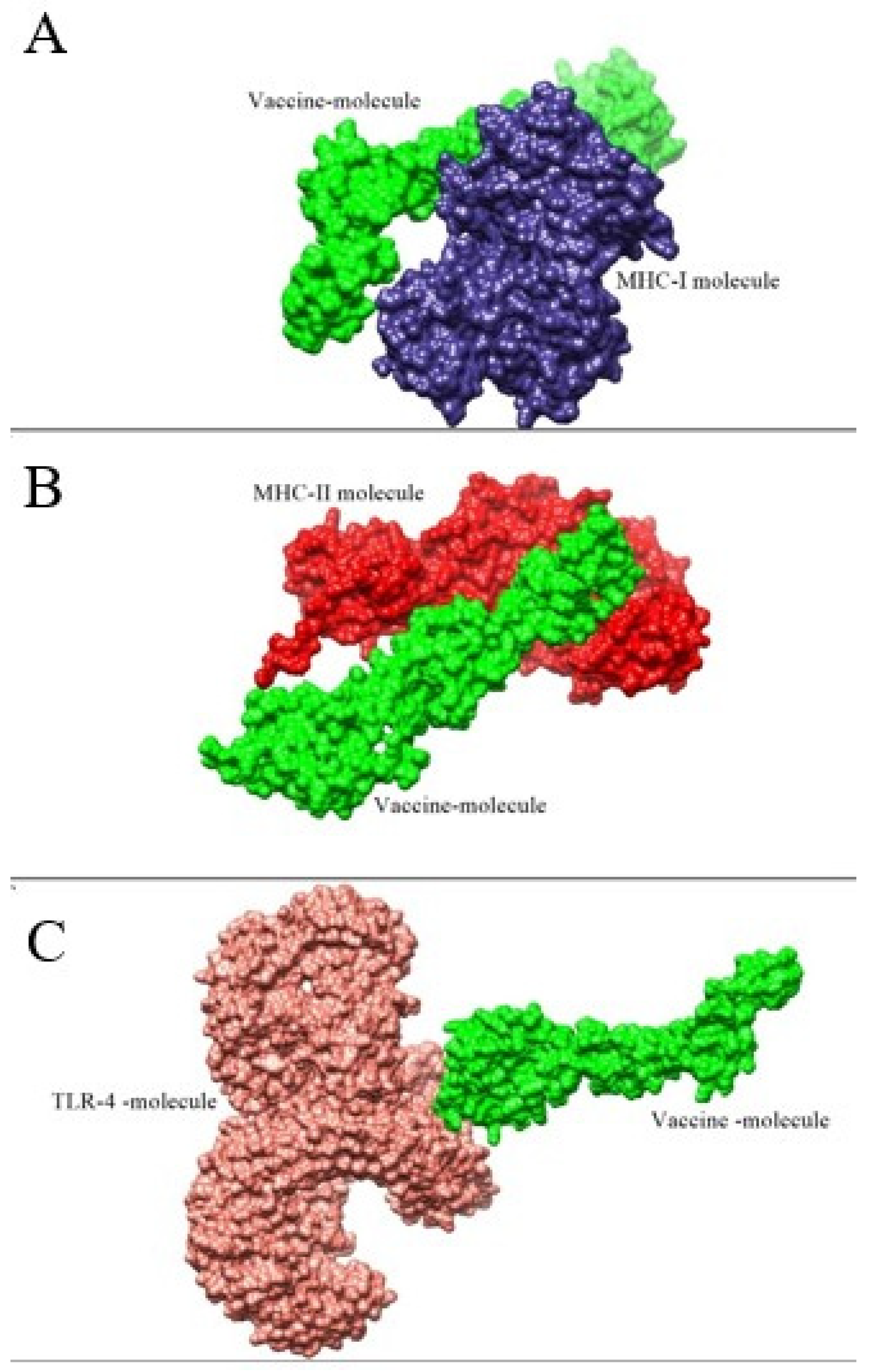
3.7. Molecular Docking and Refinement Studies
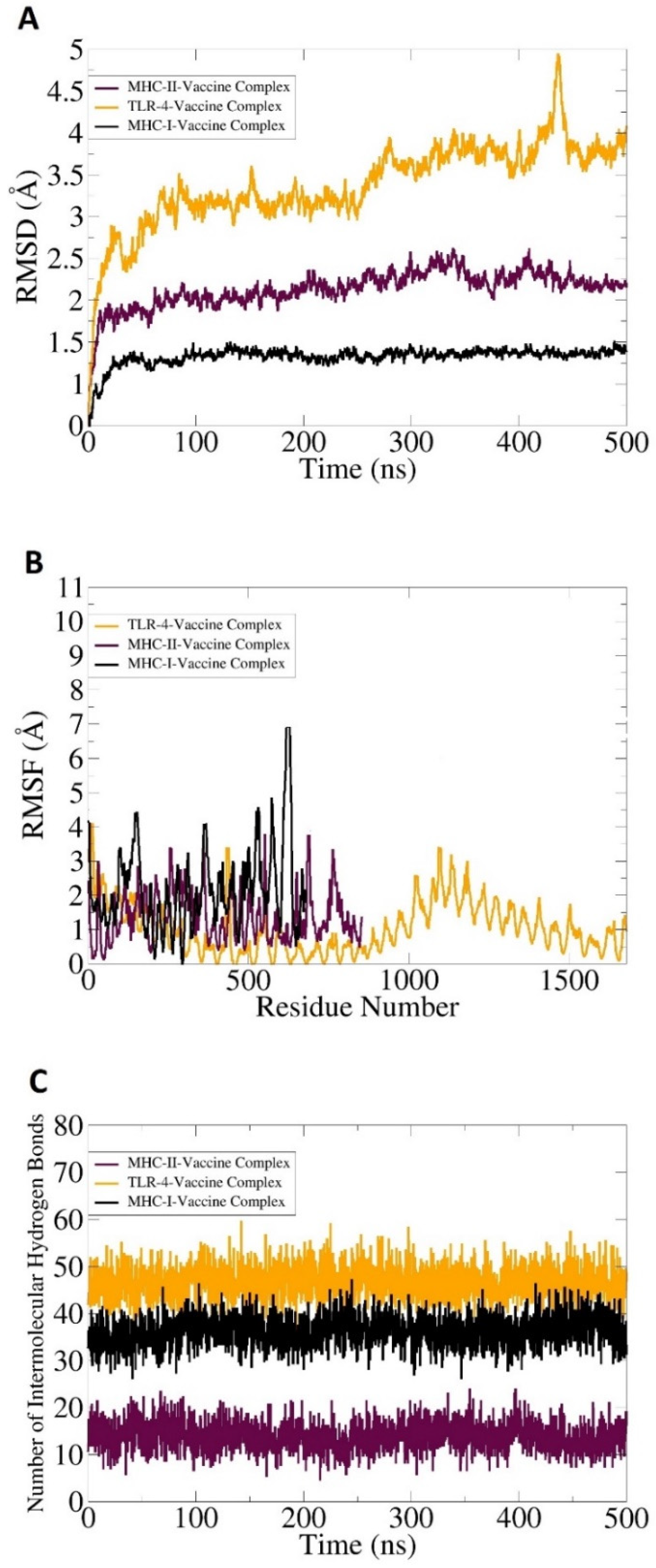
3.8. Molecular Dynamic Simulation, Hydrogen Bonding, and Free Binding Energies Calculation
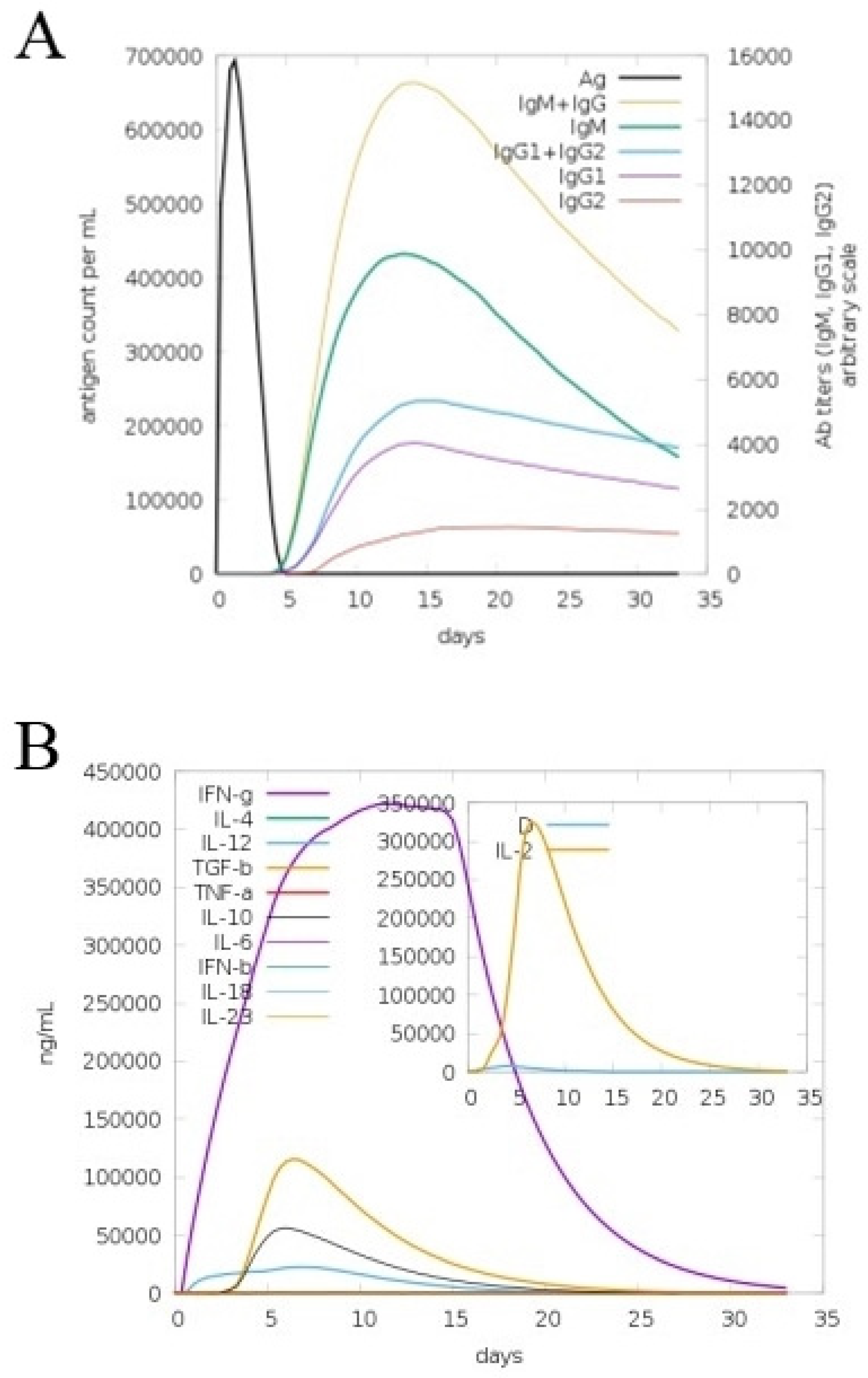
3.9. In Silico Immune Simulation
4. Discussion
5. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ramsey, A.M.; Zilberberg, M.D. Secular trends of hospitalization with vancomycin-resistant enterococcus infection in the United States, 2000–2006. Infect. Control Hosp. Epidemiol. 2009, 30, 184–186. [Google Scholar] [CrossRef] [PubMed]
- Higuita, N.I.A.; Huycke, M.M. Enterococcal disease, epidemiology, and implications for treatment. In Enterococci: From Commensals to Leading Causes of Drug Resistant Infection [Internet]; Massachusetts Eye and Ear Infirmary: Boston, MA, USA, 2014. [Google Scholar]
- Heath, C.H.; Blackmore, T.K.; Gordon, D.L. Emerging resistance in Enterococcus spp. Med. J. Aust. 1996, 164, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Bekele, B.; Ashenafi, M. Distribution of drug resistance among enterococci and Salmonella from poultry and cattle in Ethiopia. Trop. Anim. Health Prod. 2010, 42, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Werner, G.; Coque, T.M.; Franz, C.M.A.P.; Grohmann, E.; Hegstad, K.; Jensen, L.; van Schaik, W.; Weaver, K. Antibiotic resistant enterococci—Tales of a drug resistance gene trafficker. Int. J. Med. Microbiol. 2013, 303, 360–379. [Google Scholar] [CrossRef]
- Výrostková, J.; Regecová, I.; Dudriková, E.; Marcinčák, S.; Vargová, M.; Kováčová, M.; Maľová, J. Antimicrobial Resistance of Enterococcus sp. Isolated from Sheep and Goat Cheeses. Foods 2021, 10, 1844. [Google Scholar] [CrossRef]
- Igbinosa, E.O.; Beshiru, A. Antimicrobial resistance, virulence determinants, and biofilm formation of Enterococcus species from ready-to-eat seafood. Front. Microbiol. 2019, 10, 728. [Google Scholar] [CrossRef]
- Olawale, K.O.; Fadiora, S.O.; Taiwo, S.S. Prevalence of hospital acquired enterococci infections in two primary-care hospitals in Osogbo, Southwestern Nigeria. Afr. J. Infect. Dis. 2011, 5, 40–46. [Google Scholar] [CrossRef]
- Müller, T.; Ulrich, A.; Ott, E.; Müller, M. Identification of plant-associated enterococci. J. Appl. Microbiol. 2001, 91, 268–278. [Google Scholar] [CrossRef]
- Higashide, T.; Takahashi, M.; Kobayashi, A.; Ohkubo, S.; Sakurai, M.; Shirao, Y.; Tamura, T.; Sugiyama, K. Endophthalmitis caused by Enterococcus mundtii. J. Clin. Microbiol. 2005, 43, 1475–1476. [Google Scholar] [CrossRef] [Green Version]
- Fisher, K.; Phillips, C. The ecology, epidemiology and virulence of Enterococcus. Microbiology 2009, 155, 1749–1757. [Google Scholar] [CrossRef] [Green Version]
- Dhanda, S.K.; Usmani, S.S.; Agrawal, P.; Nagpal, G.; Gautam, A.; Raghava, G.P.S. Novel in silico tools for designing peptide-based subunit vaccines and immunotherapeutics. Brief. Bioinform. 2017, 18, 467–478. [Google Scholar] [PubMed]
- Bibi, N.; Zaidi, N.-S.S.; Tahir, M.; Babar, M.M. Vaccinomics driven proteome-wide screening of Haemophilus influenzae for the prediction of common putative vaccine candidates. Can. J. Microbiol. 2021, 67, 799–812. [Google Scholar] [CrossRef] [PubMed]
- Pulendran, B.; Ahmed, R. Immunological mechanisms of vaccination. Nat. Immunol. 2011, 12, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Rappuoli, R. Reverse vaccinologay. Curr. Opin. Microbiol. 2000, 3, 445–450. [Google Scholar] [CrossRef]
- Ullah, A.; Ahmad, S.; Ismail, S.; Afsheen, Z.; Khurram, M.; Tahir ul Qamar, M.; AlSuhaymi, N.; Alsugoor, M.H.; Allemailem, K.S. Towards a Novel Multi-Epitopes Chimeric Vaccine for Simulating Strong Immune Responses and Protection against Morganella morganii. Int. J. Environ. Res. Public Health 2021, 18, 10961. [Google Scholar] [CrossRef]
- Albutti, A. An integrated computational framework to design a multi-epitopes vaccine against Mycobacterium tuberculosis. Sci. Rep. 2021, 11, 21929. [Google Scholar] [CrossRef]
- Gul, S.; Ahmad, S.; Ullah, A.; Ismail, S.; Khurram, M.; Tahir ul Qamar, M.; Hakami, A.R.; Alkhathami, A.G.; Alrumaihi, F.; Allemailem, K.S. Designing a Recombinant Vaccine against Providencia rettgeri Using Immunoinformatics Approach. Vaccines 2022, 10, 189. [Google Scholar] [CrossRef]
- Mahapatra, S.R.; Dey, J.; Kaur, T.; Sarangi, R.; Bajoria, A.A.; Kushwaha, G.S.; Misra, N.; Suar, M. Immunoinformatics and molecular docking studies reveal a novel Multi-Epitope peptide vaccine against pneumonia infection. Vaccine 2021, 39, 6221–6237. [Google Scholar] [CrossRef]
- Sette, A.; Rappuoli, R. Reverse vaccinology: Developing vaccines in the era of genomics. Immunity 2010, 33, 530–541. [Google Scholar] [CrossRef] [Green Version]
- Hajissa, K.; Zakaria, R.; Suppian, R.; Mohamed, Z. Epitope-based vaccine as a universal vaccination strategy against Toxoplasma gondii infection: A mini-review. J. Adv. Vet. Anim. Res. 2019, 6, 174. [Google Scholar] [CrossRef]
- Coordinators, N.R. Nucleic Acids Res. Database Resour. Natl. Cent. Biotechnol. Inf. 2017, 45, D12–D17. [Google Scholar]
- Tahir ul Qamar, M.; Zhu, X.; Khan, M.S.; Xing, F.; Chen, L.L. Pan-genome: A promising resource for noncoding RNA discovery in plants. Plant Genome 2020, 13, e20046. [Google Scholar] [CrossRef] [PubMed]
- Tahir Ul Qamar, M.; Zhu, X.; Xing, F.; Chen, L.-L. ppsPCP: A plant presence/absence variants scanner and pan-genome construction pipeline. Bioinformatics 2019, 35, 4156–4158. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, N.M.; Gupta, V.K.; Dutta, C. BPGA-an ultra-fast pan-genome analysis pipeline. Sci. Rep. 2016, 6, 24373. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef]
- Yu, N.Y.; Wagner, J.R.; Laird, M.R.; Melli, G.; Rey, S.; Lo, R.; Dao, P.; Sahinalp, S.C.; Ester, M.; Foster, L.J. PSORTb 3.0: Improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 2010, 26, 1608–1615. [Google Scholar] [CrossRef]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Yu, P.; Luo, J.; Jiang, Y. Secreted protein prediction system combining CJ-SPHMM, TMHMM, and PSORT. Mamm. Genome 2003, 14, 859–865. [Google Scholar] [CrossRef]
- ProtParam, E. ExPASy-ProtParam Tool. 2017. Available online: https://web.expasy.org/protparam/ (accessed on 12 February 2022).
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Dimitrov, I.; Flower, D.R.; Doytchinova, I. AllerTOP-a server for in silico prediction of allergens. BMC Bioinform. 2013, 14, S4. [Google Scholar] [CrossRef] [Green Version]
- Blast, N. Basic local alignment search tool. Natl. Libr. Med. Natl. Cent. Biotechnol. Inf. 2015, 14, 1–9. [Google Scholar]
- Ahmadi, F.; Dorosti, H.; Ghasemi, Y.; Nezafat, N. In silico design of epitope-based allergy vaccine against bellatella germanica cockroach allergens. Int. J. Pept. Res. Ther. 2020, 26, 1739–1749. [Google Scholar] [CrossRef]
- Vita, R.; Overton, J.A.; Greenbaum, J.A.; Ponomarenko, J.; Clark, J.D.; Cantrell, J.R.; Wheeler, D.K.; Gabbard, J.L.; Hix, D.; Sette, A. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2015, 43, D405–D412. [Google Scholar] [CrossRef] [PubMed]
- Ismail, S.; Ahmad, S.; Azam, S.S. Vaccinomics to design a novel single chimeric subunit vaccine for broad-spectrum immunological applications targeting nosocomial Enterobacteriaceae pathogens. Eur. J. Pharm. Sci. 2020, 146, 105258. [Google Scholar] [CrossRef]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Consortium, O.S.D.D.; Raghava, G.P.S. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misra, N.; Panda, P.K.; Shah, K.; Sukla, L.B.; Chaubey, P. Population coverage analysis of T-Cell epitopes of Neisseria meningitidis serogroup B from Iron acquisition proteins for vaccine design. Bioinformation 2011, 6, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A protein structure and structural feature prediction server. Nucleic Acids Res. 2005, 33, W72–W76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef] [Green Version]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef]
- Craig, D.B.; Dombkowski, A.A. Disulfide by Design 2.0: A web-based tool for disulfide engineering in proteins. BMC Bioinform. 2013, 14, 346. [Google Scholar] [CrossRef] [Green Version]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: Fast interaction refinement in molecular docking. Proteins Struct. Funct. Bioinform. 2007, 69, 139–159. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.-S.; Allen, B.K.; Giese, T.J.; Guo, Z.; Li, P.; Lin, C.; McGee, T.D., Jr.; Pearlman, D.A.; Radak, B.K.; Tao, Y. Alchemical binding free energy calculations in AMBER20: Advances and best practices for drug discovery. J. Chem. Inf. Model. 2020, 60, 5595–5623. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Navid, A.; Farid, R.; Abbas, G.; Ahmad, F.; Zaman, N.; Parvaiz, N.; Azam, S.S. Design of a novel multi epitope-based vaccine for pandemic coronavirus disease (COVID-19) by vaccinomics and probable prevention strategy against avenging zoonotics. Eur. J. Pharm. Sci. 2020, 151, 105387. [Google Scholar] [CrossRef]
- Ahmad, S.; Navid, A.; Akhtar, A.S.; Azam, S.S.; Wadood, A.; Pérez-Sánchez, H. Subtractive genomics, molecular docking and molecular dynamics simulation revealed LpxC as a potential drug target against multi-drug resistant Klebsiella pneumoniae. Interdiscip. Sci. Comput. Life Sci. 2019, 11, 508–526. [Google Scholar] [CrossRef]
- Kanduc, D.; Lucchese, A.; Mittelman, A. Non-redundant peptidomes from DAPs: Towards “the vaccine”? Autoimmun. Rev. 2007, 6, 290–294. [Google Scholar] [CrossRef]
- Peabody, M.A.; Laird, M.R.; Vlasschaert, C.; Lo, R.; Brinkman, F.S.L. PSORTdb: Expanding the bacteria and archaea protein subcellular localization database to better reflect diversity in cell envelope structures. Nucleic Acids Res. 2016, 44, D663–D668. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Xiao, D.; Pang, B.; Zhang, Q.; Zhou, H.; Zhang, L.; Zhang, J.; Kan, B. The core proteome and pan proteome of Salmonella Paratyphi A epidemic strains. PLoS ONE 2014, 9, e89197. [Google Scholar] [CrossRef] [Green Version]
- Ferrieri, P. Surface-localized protein antigens of group B streptococci. Rev. Infect. Dis. 1988, S363–S366. [Google Scholar] [CrossRef]
- Segal, Y.; Shoenfeld, Y. Vaccine-induced autoimmunity: The role of molecular mimicry and immune crossreaction. Cell. Mol. Immunol. 2018, 15, 586–594. [Google Scholar] [CrossRef]
- Jensen, P.E. Recent advances in antigen processing and presentation. Nat. Immunol. 2007, 8, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Ismail, S.; Shahid, F.; Khan, A.; Bhatti, S.; Ahmad, S.; Naz, A.; Almatroudi, A.; ul Qamar, M.T. Pan-Vaccinomics Approach Towards a Universal Vaccine Candidate Against WHO Priority Pathogens to Address Growing Global Antibiotic Resistance. Comput. Biol. Med. 2021, 104705. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.S.; Hossan, M.I.; Mizan, S.; Moin, A.T.; Yasmin, F.; Akash, A.-S.; Powshi, S.N.; Hasan, A.K.R.; Chowdhury, A.S. Immunoinformatics approach to designing a multi-epitope vaccine against Saint Louis Encephalitis Virus. Inform. Med. Unlocked 2021, 22, 100500. [Google Scholar] [CrossRef]
- Gupta, N.; Kumar, A. Designing an efficient multi-epitope vaccine against Campylobacter jejuni using immunoinformatics and reverse vaccinology approach. Microb. Pathog. 2020, 147, 104398. [Google Scholar] [CrossRef]
- Bibi, S.; Ullah, I.; Zhu, B.; Adnan, M.; Liaqat, R.; Kong, W.B.; Niu, S. In silico analysis of epitope-based vaccine candidate against tuberculosis using reverse vaccinology. Sci. Rep. 2021, 11, 1249. [Google Scholar] [CrossRef]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham III, T.E.; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. The FF14SB force field. Amber 2014, 14, 29–31. [Google Scholar]
- Nestor, G.; Ruda, A.; Anderson, T.; Oscarson, S.; Widmalm, G.; Gronenborn, A.M. A detailed picture of a protein–carbohydrate hydrogen-bonding network revealed by NMR and MD simulations. Glycobiology 2021, 31, 508–518. [Google Scholar] [CrossRef]
- Tuccinardi, T. What is the current value of MM/PBSA and MM/GBSA methods in drug discovery? Expert Opin. Drug Discov. 2021, 16, 1233–1237. [Google Scholar] [CrossRef]
- Bianconi, I.; Alcalá-Franco, B.; Scarselli, M.; Dalsass, M.; Buccato, S.; Colaprico, A.; Marchi, S.; Masignani, V.; Bragonzi, A. Genome-based approach delivers vaccine candidates against Pseudomonas aeruginosa. Front. Immunol. 2019, 9, 3021. [Google Scholar] [CrossRef] [Green Version]
- Shi, F.; Ogawa, Y.; Sano, A.; Harada, T.; Hirota, J.; Eguchi, M.; Oishi, E.; Shimoji, Y. Characterization and identification of a novel candidate vaccine protein through systematic analysis of extracellular proteins of Erysipelothrix rhusiopathiae. Infect. Immun. 2013, 81, 4333–4340. [Google Scholar] [CrossRef] [Green Version]
- Tarang, S.; Kesherwani, V.; LaTendresse, B.; Lindgren, L.; Rocha-Sanchez, S.M.; Weston, M.D. In silico design of a multivalent vaccine against Candida albicans. Sci. Rep. 2020, 10, 1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dar, H.A.; Zaheer, T.; Shehroz, M.; Ullah, N.; Naz, K.; Muhammad, S.A.; Zhang, T.; Ali, A. Immunoinformatics-aided design and evaluation of a potential multi-epitope vaccine against Klebsiella pneumoniae. Vaccines 2019, 7, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dar, H.A.; Ismail, S.; Waheed, Y.; Ahmad, S.; Jamil, Z.; Aziz, H.; Hetta, H.F.; Muhammad, K. Designing a multi-epitope vaccine against Mycobacteroides abscessus by pangenome-reverse vaccinology. Sci. Rep. 2021, 11, 11197. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Coveney, P.V.; Flower, D.R. Peptide recognition by the T cell receptor: Comparison of binding free energies from thermodynamic integration, Poisson–Boltzmann and linear interaction energy approximations. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2005, 363, 2037–2053. [Google Scholar] [CrossRef]
- Dorosti, H.; Eslami, M.; Negahdaripour, M.; Ghoshoon, M.B.; Gholami, A.; Heidari, R.; Dehshahri, A.; Erfani, N.; Nezafat, N.; Ghasemi, Y. Vaccinomics approach for developing multi-epitope peptide pneumococcal vaccine. J. Biomol. Struct. Dyn. 2019, 37, 3524–3535. [Google Scholar] [CrossRef]
- Singh, R.; Garg, N.; Shukla, G.; Capalash, N.; Sharma, P. Immunoprotective efficacy of Acinetobacter baumannii outer membrane protein, FilF, predicted in silico as a potential vaccine candidate. Front. Microbiol. 2016, 7, 158. [Google Scholar] [CrossRef]
- Todorov, S.D.; Wachsman, M.B.; Knoetze, H.; Meincken, M.; Dicks, L.M.T. An antibacterial and antiviral peptide produced by Enterococcus mundtii ST4V isolated from soya beans. Int. J. Antimicrob. Agents 2005, 25, 508–513. [Google Scholar] [CrossRef]
- Rehman, A.; Ahmad, S.; Shahid, F.; Albutti, A.; Alwashmi, A.S.S.; Aljasir, M.A.; Alhumeed, N.; Qasim, M.; Ashfaq, U.A.; Tahir ul Qamar, M. Integrated Core Proteomics, Subtractive Proteomics, and Immunoinformatics Investigation to Unveil a Potential Multi-Epitope Vaccine against Schistosomiasis. Vaccines 2021, 9, 658. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteins Accession Number/Name | Predicted B-Cell Peptides |
|---|---|
| >core/490/2/Org2_Gene1203 (lytic polysaccharide monooxygenase) | GSLDRNVNHNAALAKYGPVIYEPQSLEALKGFPQAGPADGRIASANGAVGNNFNLDRQTSTMWTKQDLNTG |
| ADWNPNDQLDRSDFELLTTINHGGAQASTN | |
| VDDTAMAFYQVIDVNLKGDSAIPVAPTAPRNVRTTNVTSS | |
| LLGNTASPDFSDQNLTAE | |
| QTGLVSERTALSVTTLSETTEEKPTAPSHL | |
| DRDENGGGDENGGDGGNGGGEVVTGRQWTVGSFFSPVS | |
| ITWQSHLNYGDTNWAPGIAHSL | |
| >core/1058/1/Org1_Gene1225 (siderophore ABC transporter substrate-binding protein) | NEAQTRETTASSTIATD |
| NLPAYLEKYQEVESAGGIKEPDLEKINEM | |
| KQADDRIEASTHGQSVSYEYVL | |
| TQAIGGDTSNDN | |
| >core/1868/1/Org1_Gene902(lytic polysaccharide monooxygenase) | GNLNQNVGRAQWEPQSIEAPKNTFIDGKIASAGVSGFEPLDEQTASRWHKSVINSGA |
| PGWNQNQPLKFSDFELITKIDDKATIPP |
| Structure Information | ||||||
|---|---|---|---|---|---|---|
| Model | RMSD | MolProbity | Clash Score | Poor Rotamers | Rama Favored | GALAXY Energy |
| Initial | 0 | 2.99 | 39.1 | 3 | 89.7 | 13,957.59 |
| MODEL 1 | 2.01 | 1.199 | 0.5 | 0 | 90.5 | −4775.14 |
| MODEL 2 | 2.156 | 1.483 | 1.9 | 0.5 | 90.5 | −4771.87 |
| MODEL 3 | 2.724 | 1.214 | 0.7 | 0.5 | 91.9 | −4765.4 |
| MODEL 4 | 2.272 | 1.352 | 1.2 | 0 | 90.8 | −4764.62 |
| MODEL 5 | 2.675 | 1.329 | 1.2 | 0 | 91.6 | −4754.89 |
| MODEL 6 | 2.227 | 1.25 | 0.7 | 0 | 90.8 | −4751.34 |
| MODEL 7 | 2.208 | 1.214 | 0.7 | 0 | 91.9 | −4750.35 |
| MODEL 8 | 2.093 | 1.315 | 0.9 | 0 | 90.5 | −4748.02 |
| MODEL 9 | 2.478 | 1.341 | 1.2 | 0 | 91.2 | −4747.58 |
| MODEL 10 | 2.095 | 1.326 | 0.9 | 0.5 | 90.1 | −4746.33 |
| A.A Residues Pairs | Chi3 Value | Energy | Sum B-Factors |
|---|---|---|---|
| GLU32-ASN35 | 126.99 | 4.04 | 0 |
| MET 89-PRO114 | −116 | 5.52 | 0 |
| GLU 100-GLY161 | 103.67 | 2.53 | 0 |
| LYS 129-GLY134 | 99.26 | 3.2 | 0 |
| ALA 137-ARG140 | 79.22 | 4.58 | 0 |
| ASN152-GLY181 | 114.66 | 2.76 | 0 |
| GLY155-ASP180 | 74.31 | 4.37 | 0 |
| THR 173-GLY 217 | 121.34 | 3.34 | 0 |
| GLY 174-GLY193 | −57.19 | 4.85 | 0 |
| GLY179-ASN183 | 100.56 | 2.89 | 0 |
| GLY189-GLY217 | −100 | 4.12 | 0 |
| PRO190-GLY219 | −94.95 | 4 | 0 |
| LYS196-ASP199 | 125.12 | 5.84 | 0 |
| LYS202-LYS208 | 85.73 | 3.3 | 0 |
| PRO204-GLY231 | −85.8 | 4.06 | 0 |
| GLY205-LYS208 | 74.84 | 2.52 | 0 |
| ALA210-GLU236 | 93.44 | 0.73 | 0 |
| GLN222-GLY226 | 68.24 | 6.37 | 0 |
| PRO237-THR242 | 94.65 | 2.39 | 0 |
| SER257-GLY263 | 107.81 | 5.71 | 0 |
| GLY261-GLN266 | 107.38 | 0.93 | 0 |
| PRO262-PHE270 | 113.73 | 2.28 | 0 |
| Energy Parameter | TLR-4-Vaccine Complex | MHC-I-Vaccine Complex | MHC-II-Vaccine Complex |
|---|---|---|---|
| MM-GBSA | |||
| van der Waals | −397.98 | −306.66 | −407.08 |
| electrostatic | −115.66 | −112.37 | −128.39 |
| polar | 81.97 | 62.09 | 39.14 |
| non-polar | −25.00 | −30.23 | −19.66 |
| gas phase | −513.64 | −419.03 | −535.47 |
| solvation | 56.97 | 31.86 | 19.48 |
| net | −456.67 | −387.17 | −515.99 |
| MM-PBSA | |||
| van der Waals | −397.98 | −306.66 | −407.08 |
| electrostatic | −115.66 | −112.37 | −128.39 |
| polar | 73.12 | 71.00 | 58.51 |
| non-polar | −29.04 | −24.67 | −17.15 |
| gas phase | −513.64 | −419.03 | −535.47 |
| solvation | 44.08 | 46.33 | 41.36 |
| net | −469.56 | −372.7 | −494.11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alharbi, M.; Alshammari, A.; Alasmari, A.F.; Alharbi, S.M.; Tahir ul Qamar, M.; Ullah, A.; Ahmad, S.; Irfan, M.; Khalil, A.A.K. Designing of a Recombinant Multi-Epitopes Based Vaccine against Enterococcus mundtii Using Bioinformatics and Immunoinformatics Approaches. Int. J. Environ. Res. Public Health 2022, 19, 3729. https://doi.org/10.3390/ijerph19063729
Alharbi M, Alshammari A, Alasmari AF, Alharbi SM, Tahir ul Qamar M, Ullah A, Ahmad S, Irfan M, Khalil AAK. Designing of a Recombinant Multi-Epitopes Based Vaccine against Enterococcus mundtii Using Bioinformatics and Immunoinformatics Approaches. International Journal of Environmental Research and Public Health. 2022; 19(6):3729. https://doi.org/10.3390/ijerph19063729
Chicago/Turabian StyleAlharbi, Metab, Abdulrahman Alshammari, Abdullah F. Alasmari, Salman Mansour Alharbi, Muhammad Tahir ul Qamar, Asad Ullah, Sajjad Ahmad, Muhammad Irfan, and Atif Ali Khan Khalil. 2022. "Designing of a Recombinant Multi-Epitopes Based Vaccine against Enterococcus mundtii Using Bioinformatics and Immunoinformatics Approaches" International Journal of Environmental Research and Public Health 19, no. 6: 3729. https://doi.org/10.3390/ijerph19063729