Ultrasound-Enhanced Catalytic Ozonation Oxidation of Ammonia in Aqueous Solution

Abstract
:1. Introduction
2. Experimental
2.1. Materials
2.2. Preparation of Supported Catalyst
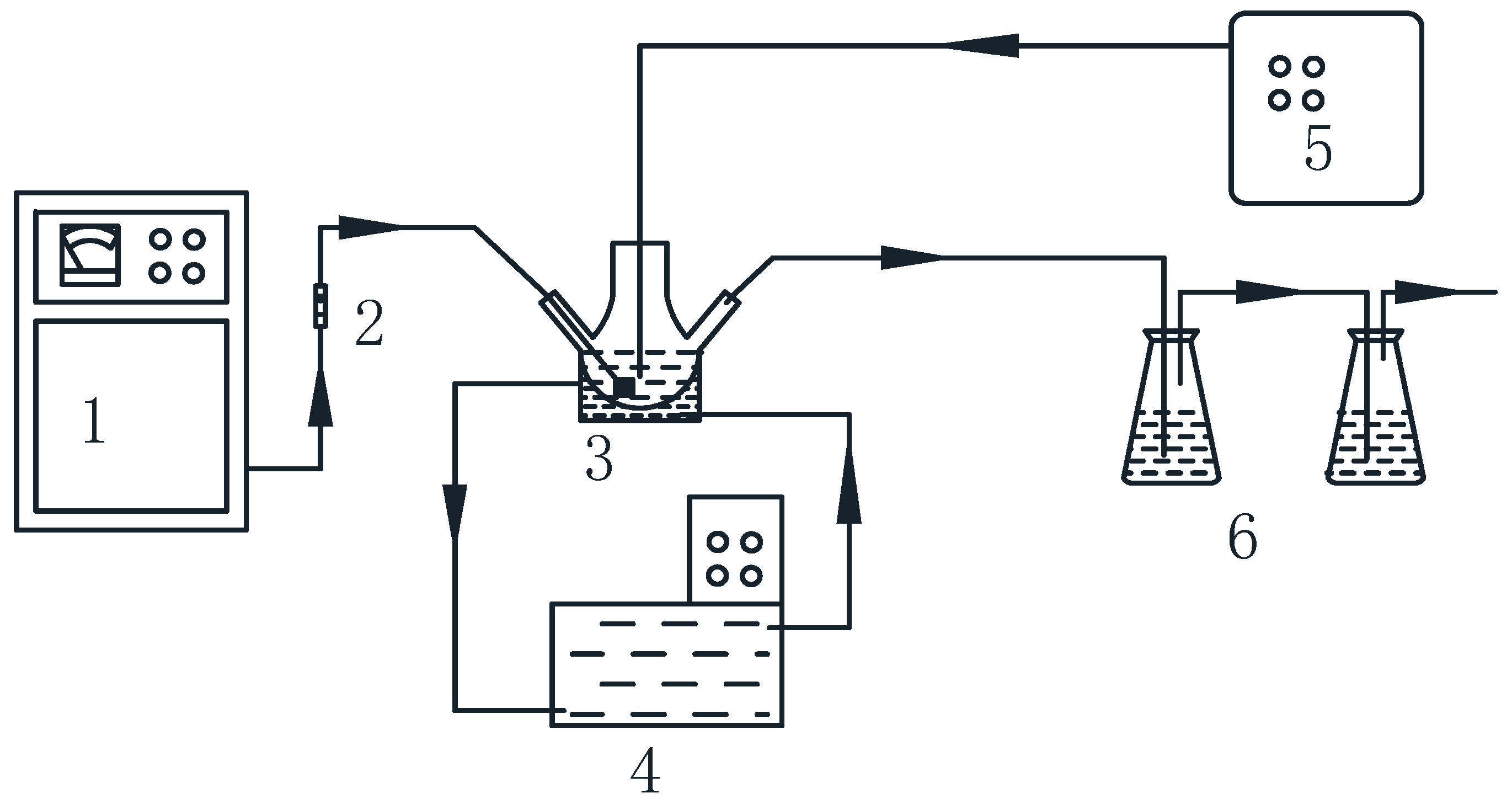
2.3. Procedure
2.4. Analysis
3. Results and Discussions
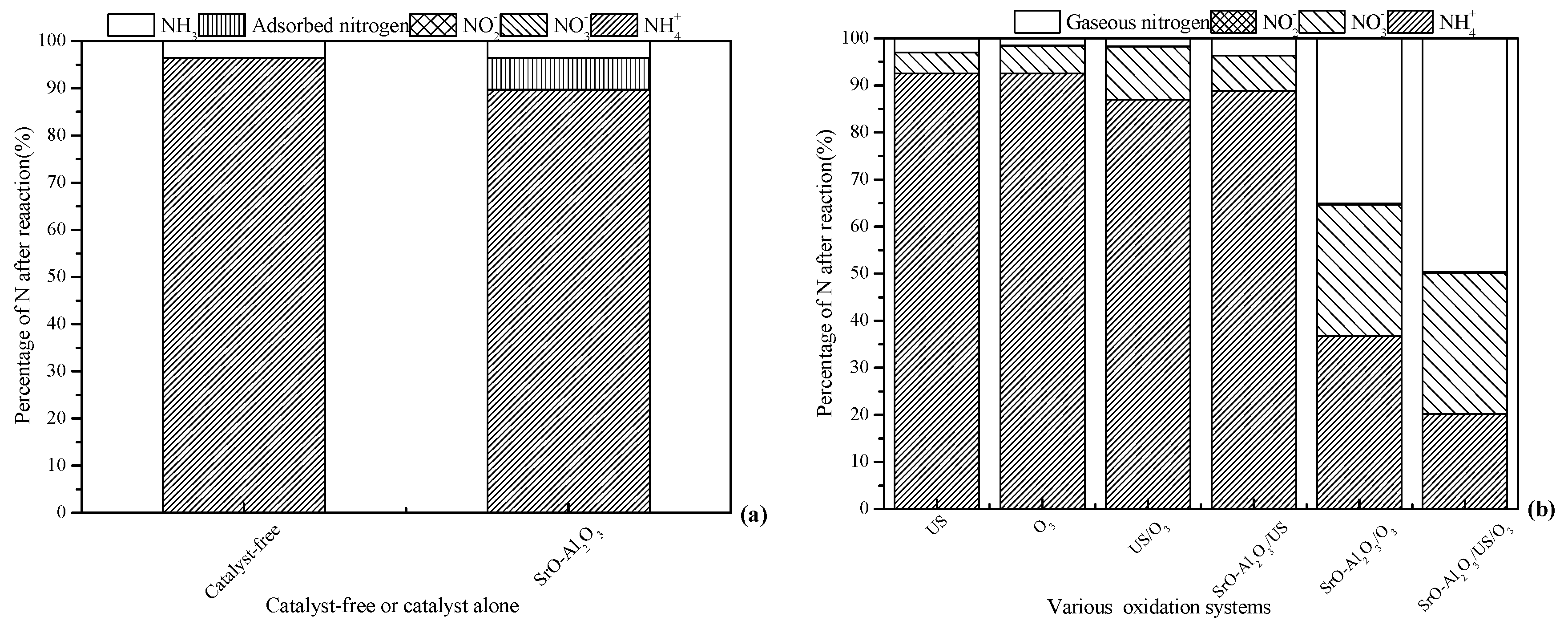
3.1. Performance Comparison of Different Treatment Systems
3.2. Effect of Operating Parameters on Ammonia Conversion
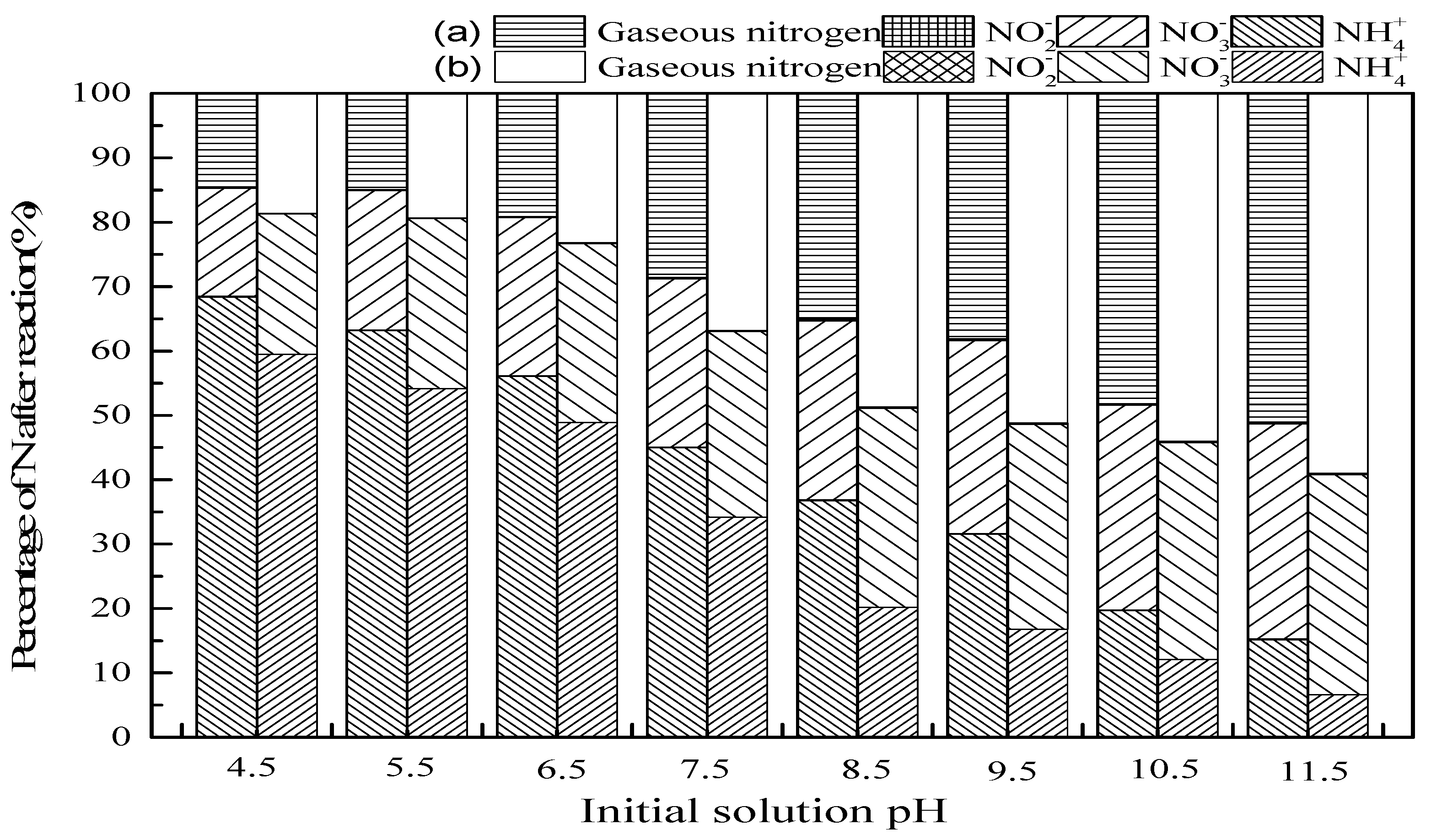
3.2.1. Initial Solution pH
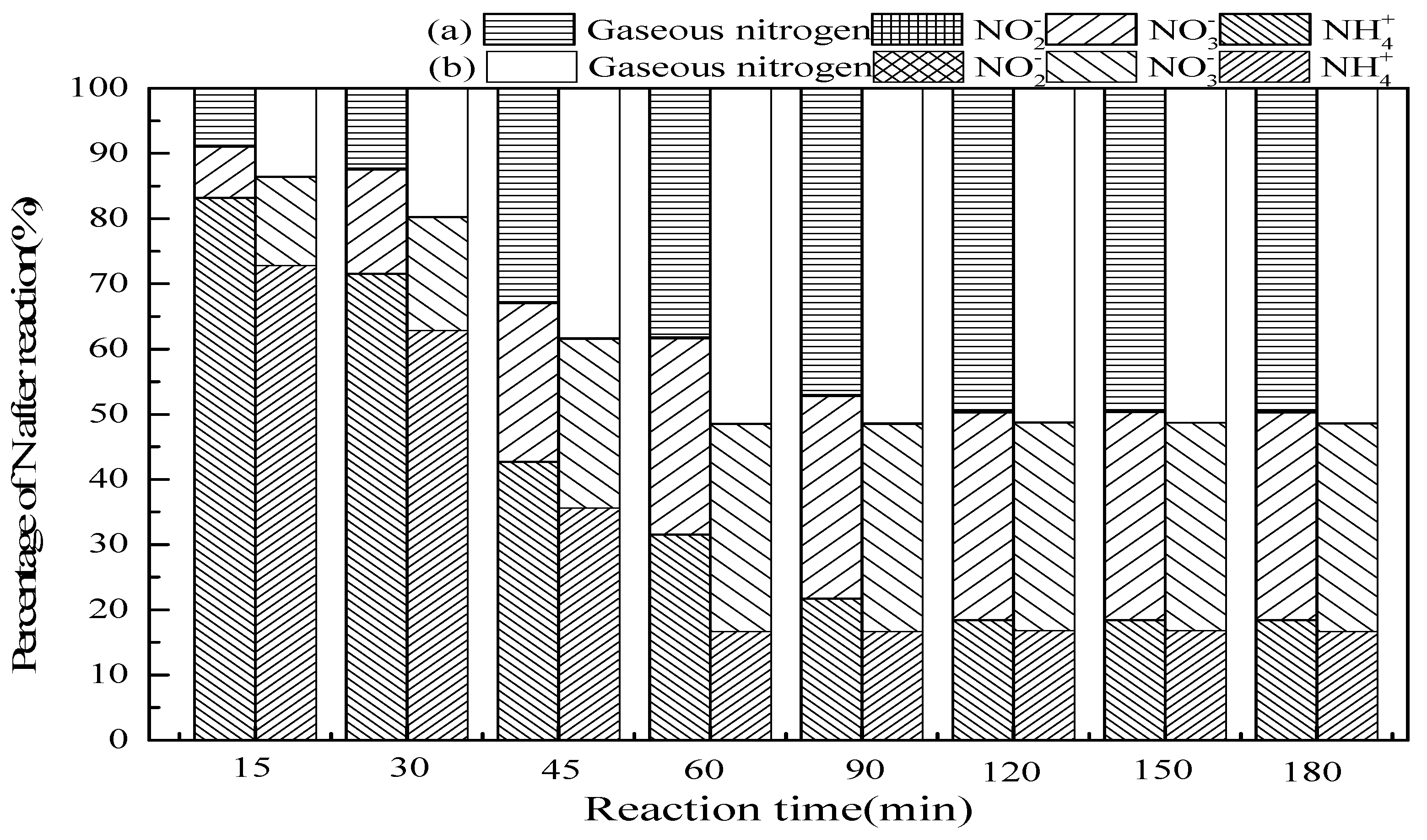
3.2.2. Reaction Time
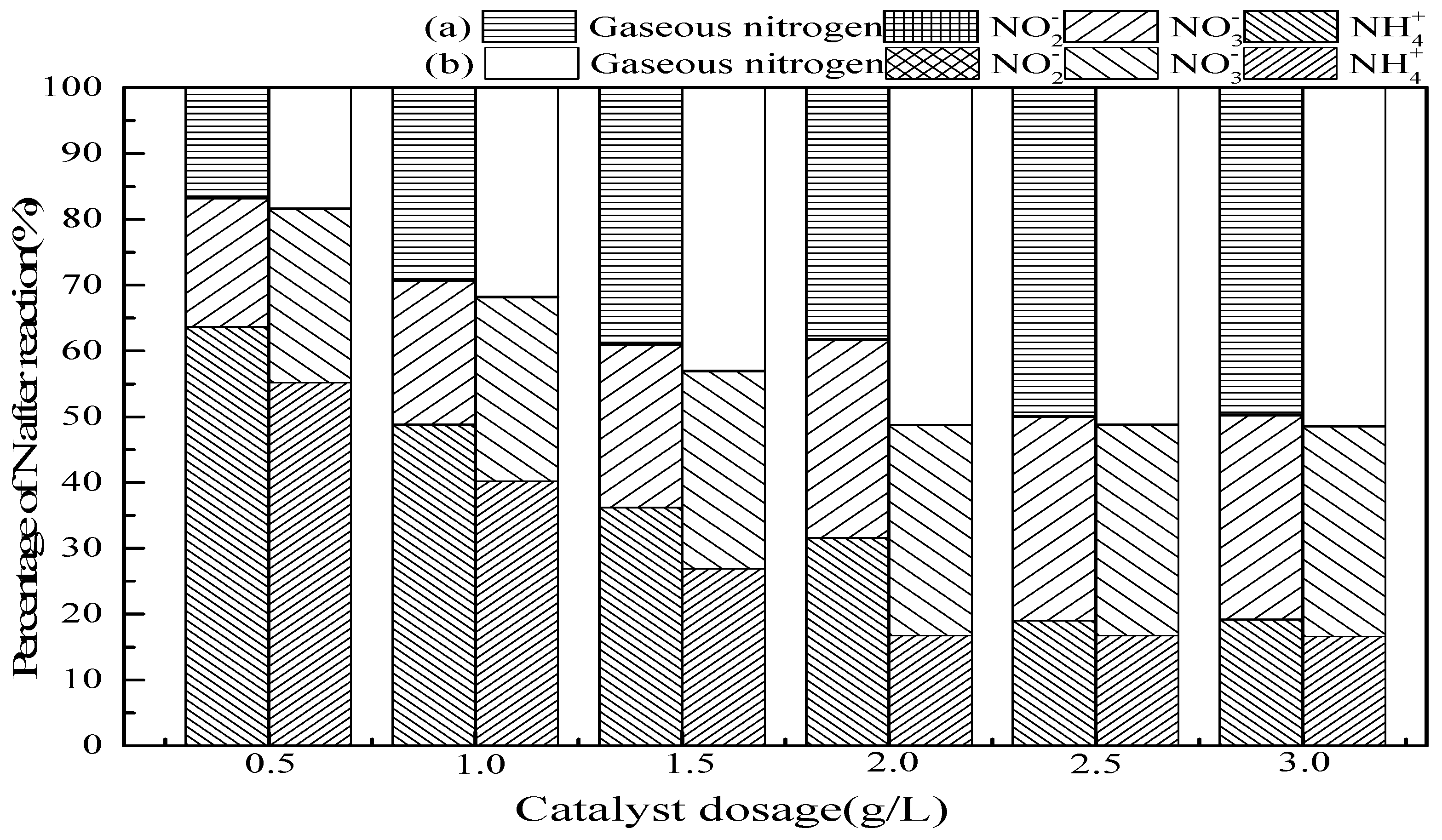
3.2.3. Catalyst Dosage
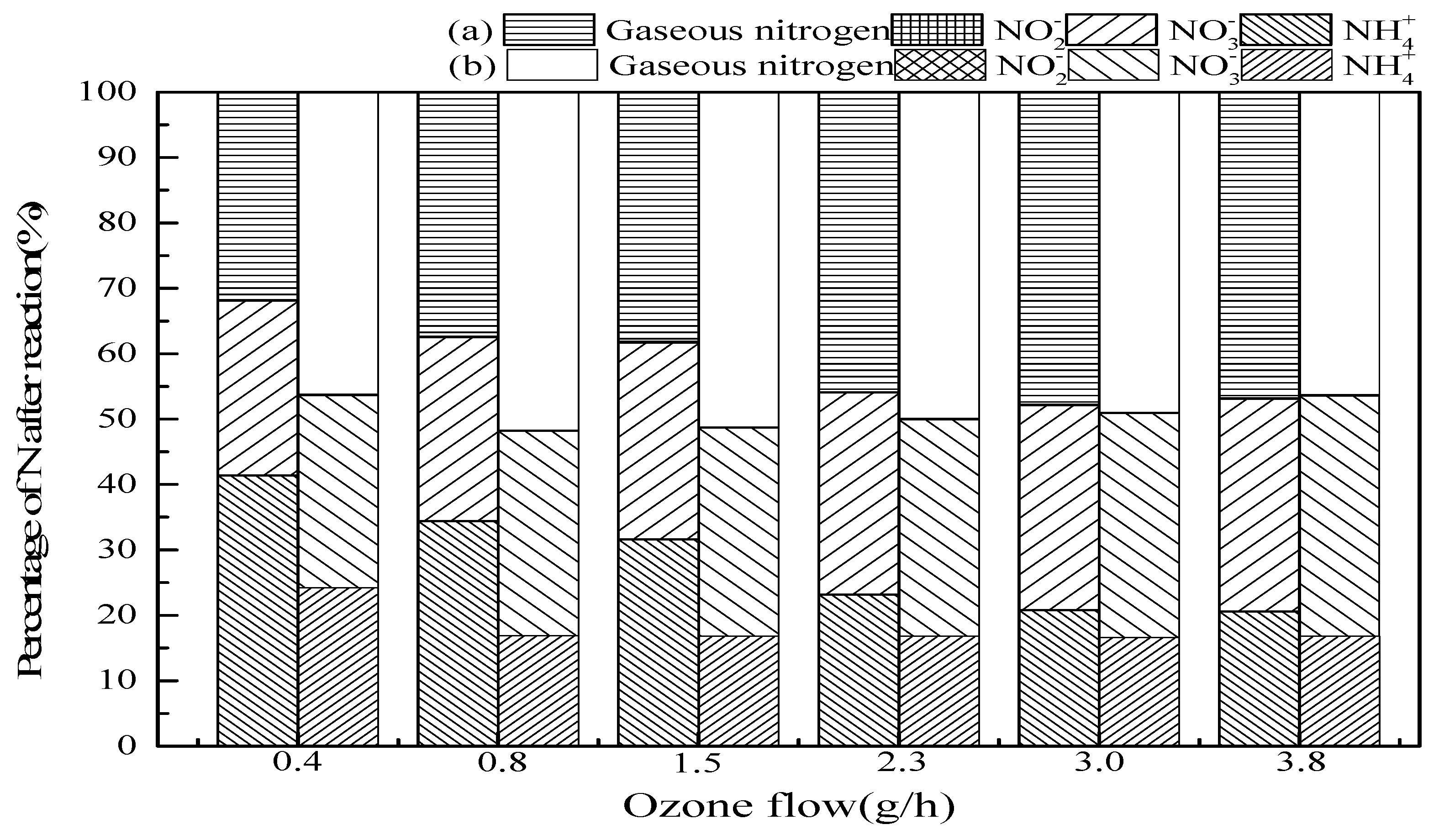
3.2.4. Ozone Flow
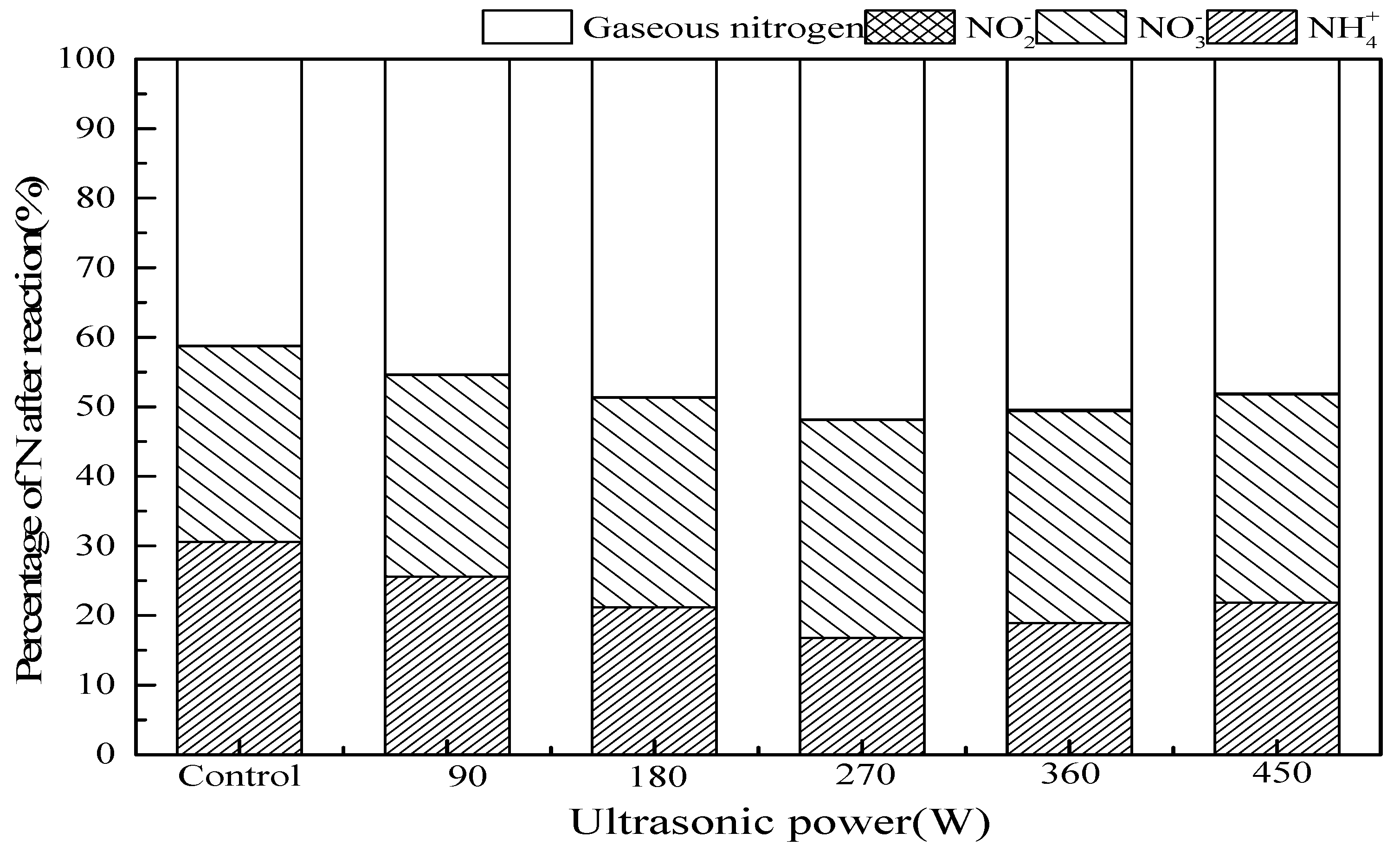
3.2.5. Ultrasonic Power
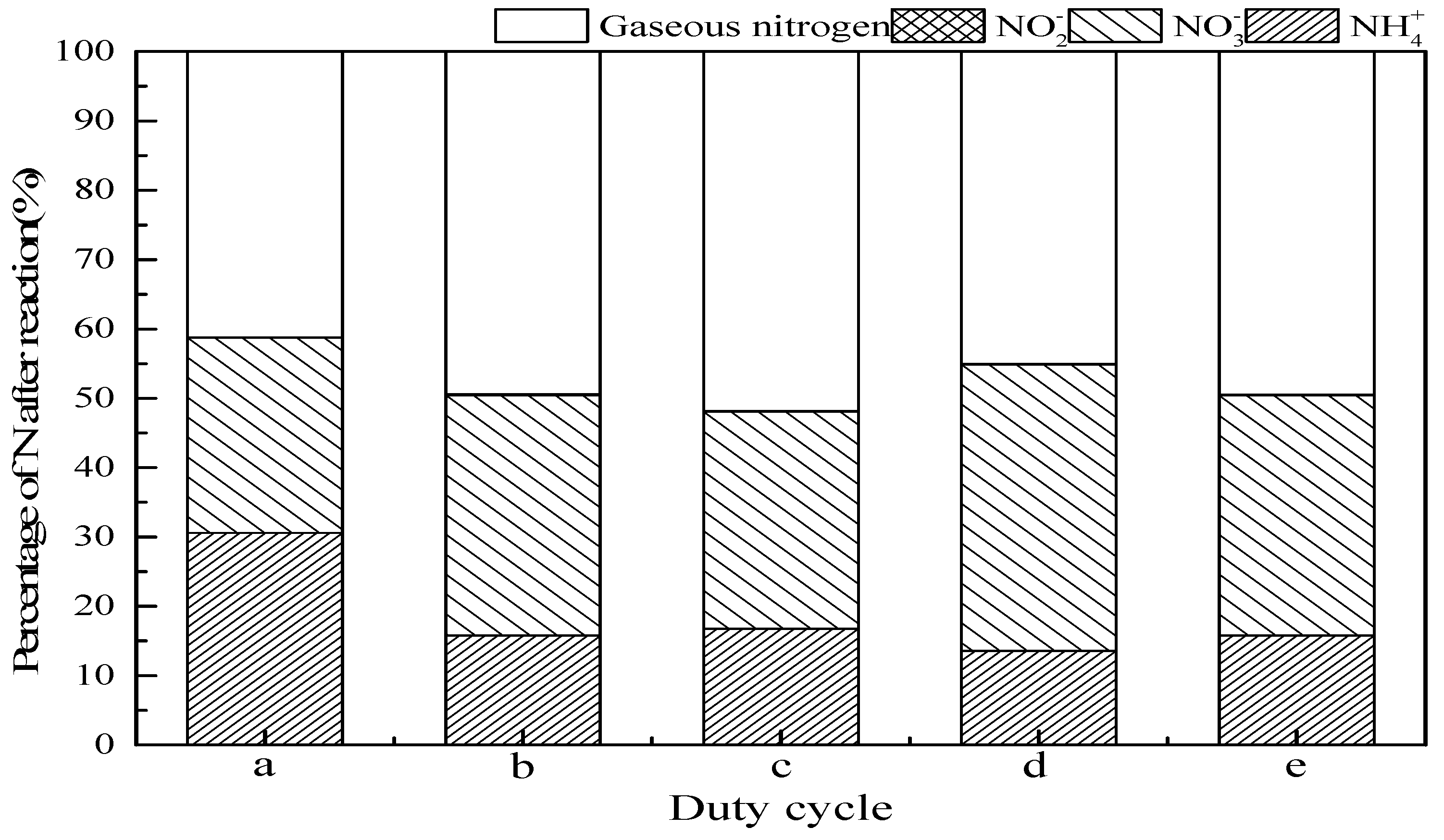
3.2.6. Duty Cycle
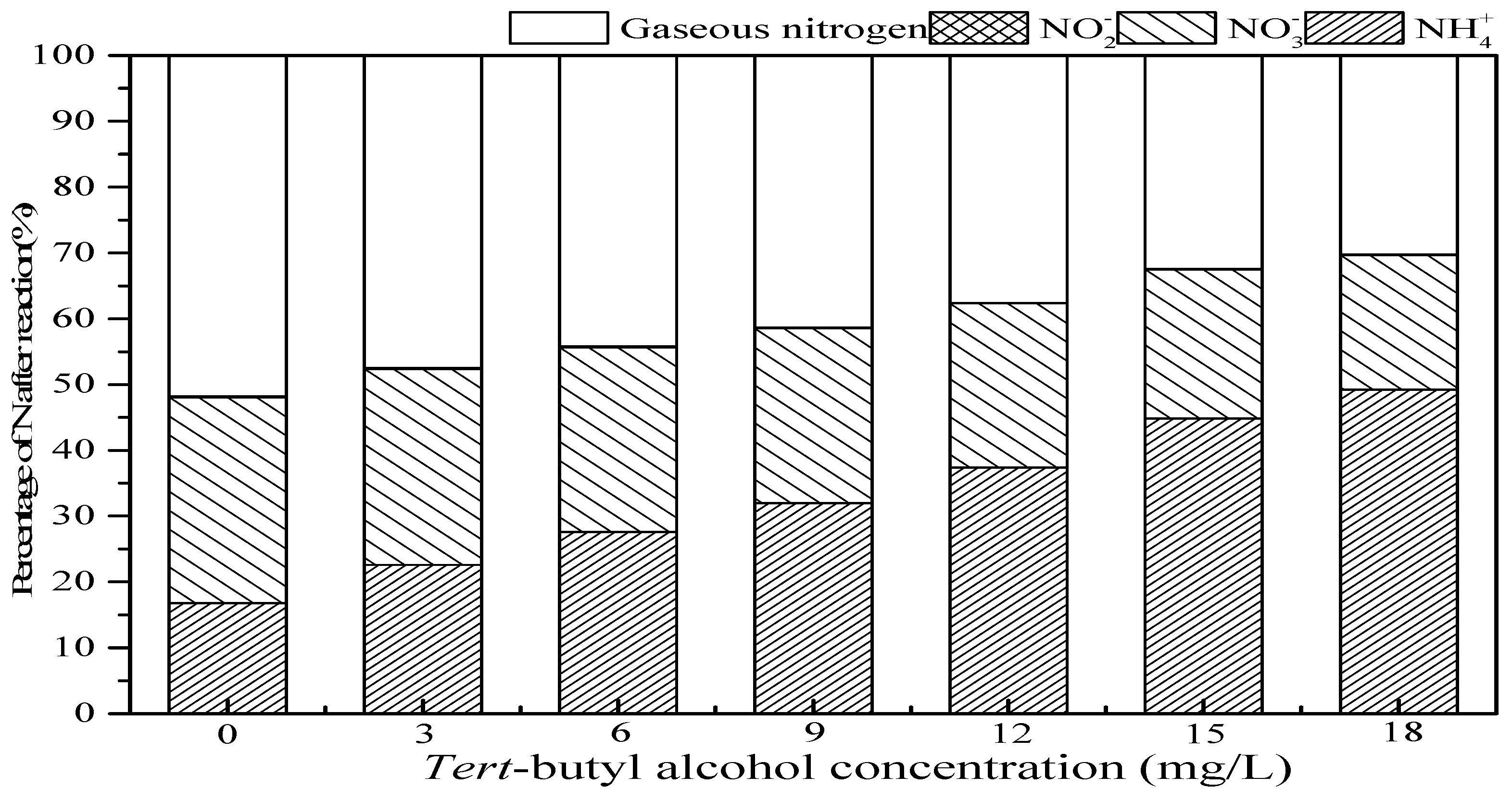
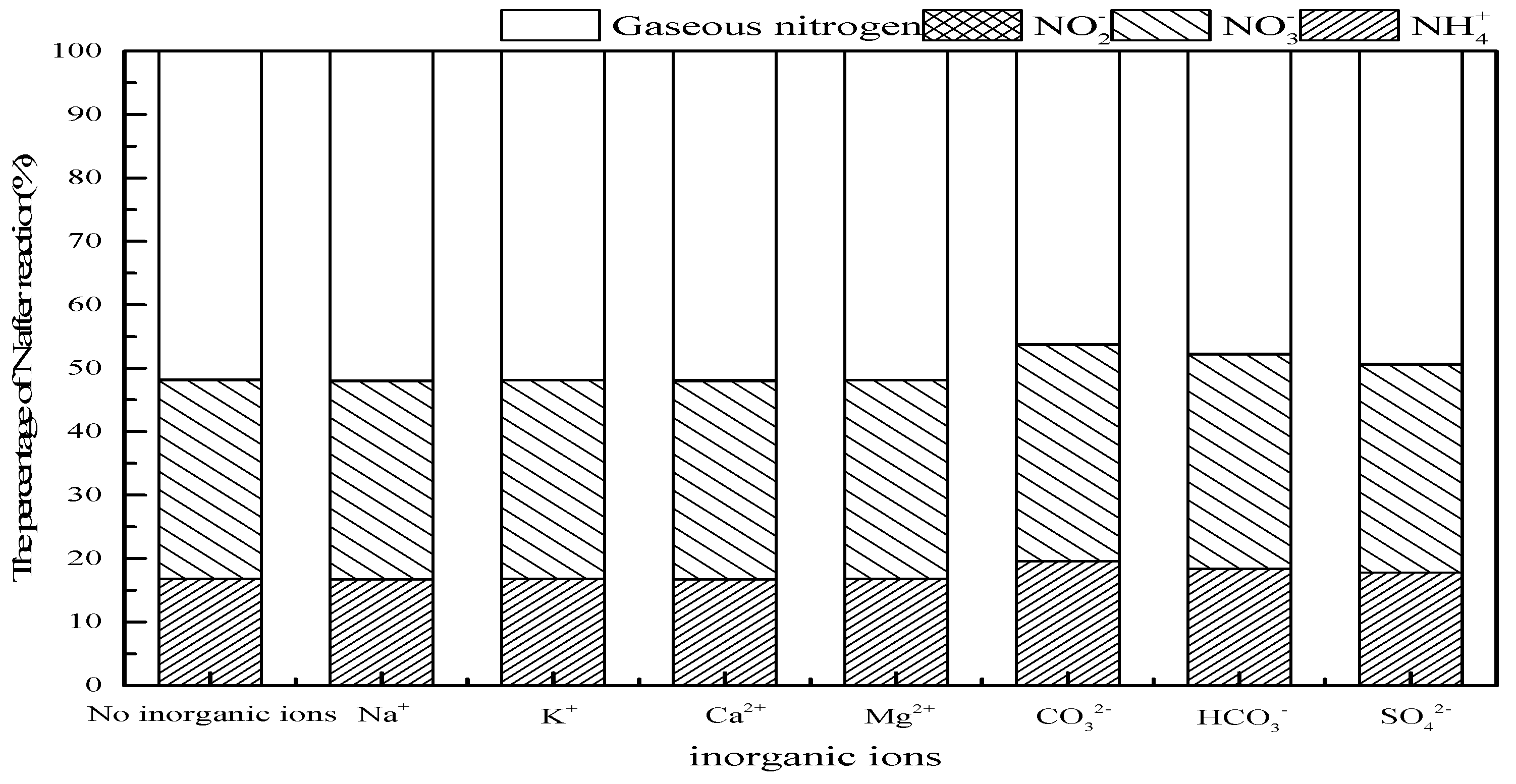
3.3. Effect of Tert-Butanol and Inorganic Ions on Ammonia Conversion
3.3.1. Effect of Tert-Butanol
3.3.2. Effect of Inorganic Ions
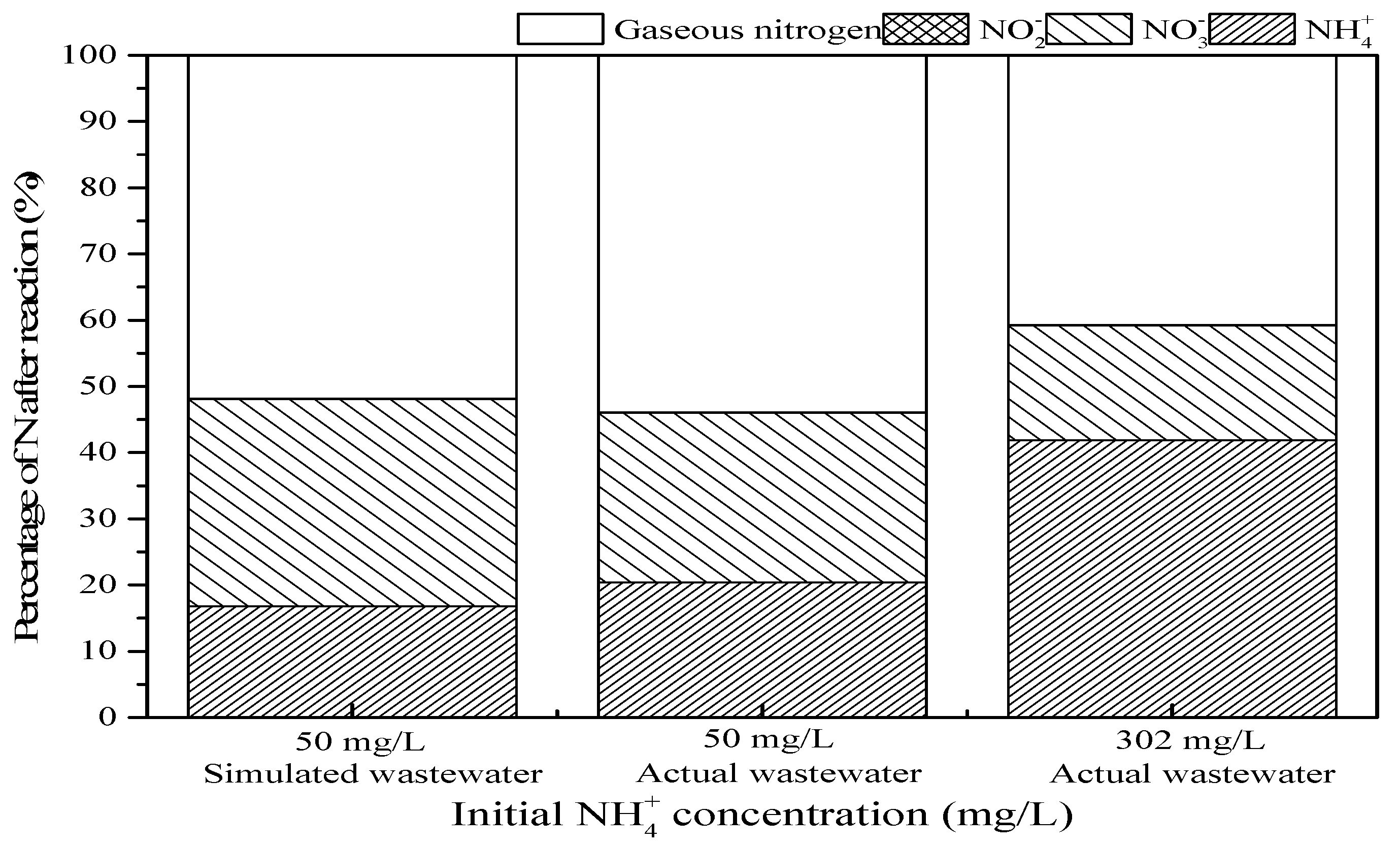
3.4. Actual Ammonium-Containing Wastewater Oxidized in the SrO-Al2O3/US/O3 System
3.5. Morphology Analysis of Catalyst
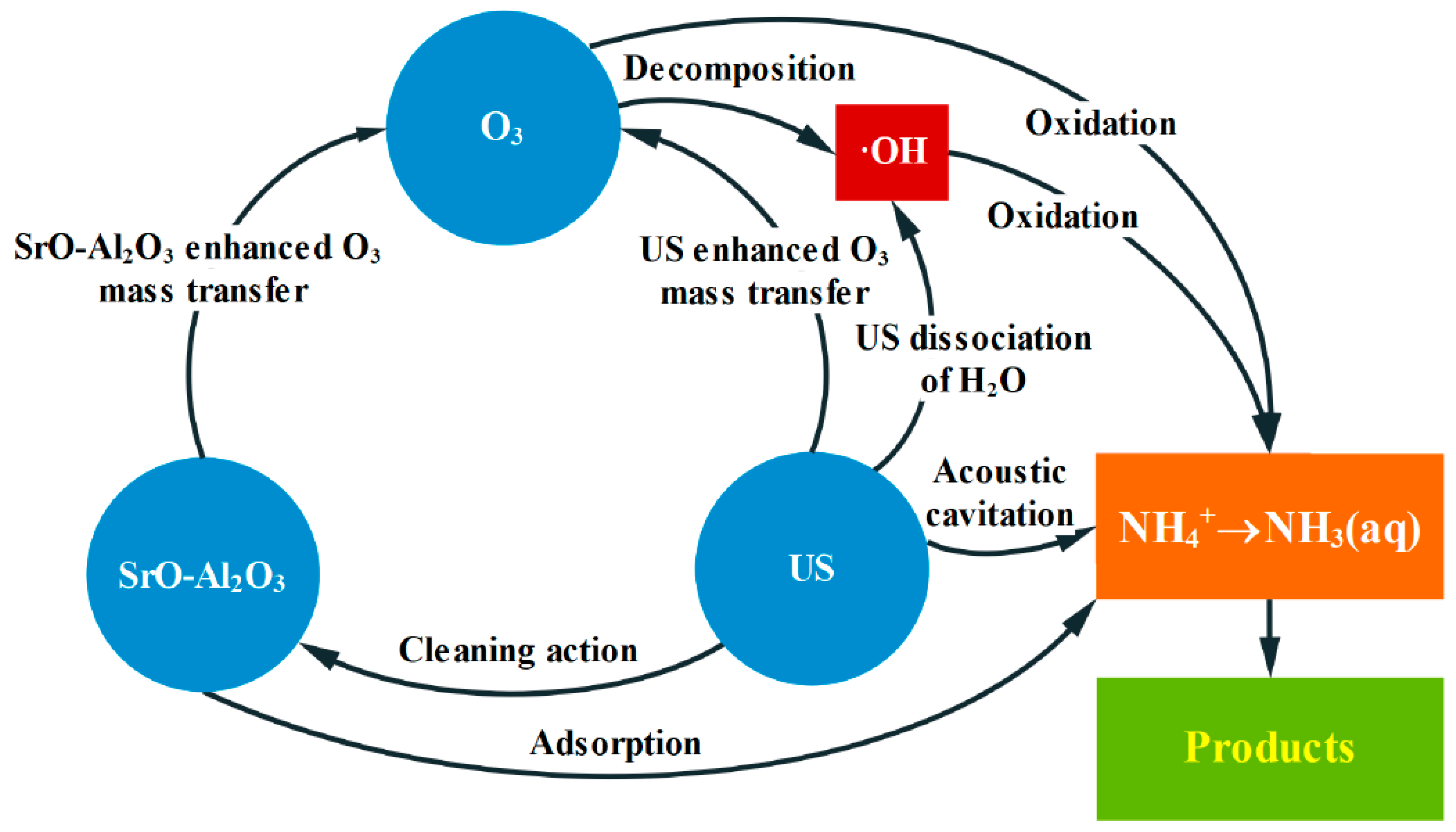
3.6. The Pathway for Ammonia in Water Oxidized in the SrO-Al2O3/US/O3 System
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Luo, X.P.; Yan, Q.; Wang, C.Y.; Luo, C.G.; Zhou, N.N.; Jian, C.S. Treatment of Ammonia Nitrogen Wastewater in Low Concentration by Two-Stage Ozonation. Int. J. Environ. Res. Public Health 2015, 12, 11975–11987. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.M.; Xiao, X.M.; Yan, B. Complex treatment of the ammonium nitrogen wastewater from rare-earth separation plant. Desalin. Water Treat. 2009, 8, 109–117. [Google Scholar] [CrossRef]
- Khuntia, S.; Majumder, S.K.; Ghosh, P. Removal of ammonia from water by ozone microbubbles. Ind. Eng. Chem. Res. 2013, 52, 318–326. [Google Scholar] [CrossRef]
- Evelin, P.N.; Iiho, K.T.; Hiroaki, T.; Chikashim, S. Ozone treatment process for the removal of pharmaceuticals and personal care products in wastewater. Ozone Sci. Eng. 2019, 41, 3–16. [Google Scholar] [CrossRef]
- Gunten, U.V. Ozonation of drinking water: Part I. Oxidation kinetics and product formation. Water Res. 2003, 37, 1443–1467. [Google Scholar] [CrossRef]
- Rakness, K.L. Ozone in Drinking Water Treatment: Process Design, Operation and Optimization; American Water Works Association: Denver, CO, USA, 2011; ISBN 1613000227. [Google Scholar]
- Haag, W.R.; Hoigne, J.; Bader, H. Improved ammonia oxidation by ozone in the presence of bromide ion during water treatment. Water Res. 1984, 18, 1125–1128. [Google Scholar] [CrossRef]
- Mahardiani, L.; Kamiya, Y. Enhancement of catalytic activity of cobalt oxide for catalytic ozonation of ammonium ion in water with repeated use. J. Jpn. Petrol. Inst. 2016, 59, 31–34. [Google Scholar] [CrossRef]
- Kasprzyk-Hordern, B.; Ziolek, M.; Nawrocki, J. Catalytic ozonation and methods of enhancing molecular ozone reactions in water treatment. Appl. Catal. B Environ. 2003, 46, 639–669. [Google Scholar] [CrossRef]
- Beltran, F.J.; Rivas, F.J.; Montero-de-Espinosa, R. Iron type catalysts for the ozonation of oxalic acid in water. Water Res. 2005, 39, 3553–3564. [Google Scholar] [CrossRef]
- Sanchez-Polo, M.; Rivera-Utrilla, J. Ozonation of 1,3,6-naphthalenetrisulfonic acid in presence of heavy metals. J. Chem. Technol. Biotechnol. 2004, 79, 902–909. [Google Scholar] [CrossRef]
- Liu, Y.; He, H.P.; Wu, D.L.; Zhang, Y.L. Heterogeneous catalytic ozonation reaction mechanism. Prog. Chem. 2016, 28, 1374–1379. [Google Scholar] [CrossRef]
- Ichikawa, S.; Mahardiani, L.; Kamiya, Y. Catalytic oxidation of ammonium ion in water with ozone over metal oxide catalysts. Catal. Today 2014, 232, 192–197. [Google Scholar] [CrossRef]
- Chen, Y.N.; Wu, Y.; Liu, C.; Guo, L.; Nie, J.X.; Chen, Y.; Qiu, T.S. Low-temperature conversion of ammonia to nitrogen in water with ozone over composite metal oxide catalyst. J. Environ. Sci. 2018, 66, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Subbarao, C.V.; Rao, K.V.; Sriniva, T.; Sumana, V.S.; Prasad, K.M.M.K. Review on heterogeneous catalysis for biodiesel production. Asian J. Res. Chem. 2011, 4, 524–536. [Google Scholar]
- Zhang, G.; Hattori, H.; Tanabe, K. Aldol addition of acetone catalyzed by solid base catalysts: Magnesium oxide, calcium oxide, strontium oxide, barium oxide, lanthanum (III) oxide, and zirconium oxide. Appl. Catal. 1988, 36, 189–197. [Google Scholar] [CrossRef]
- Kabashima, H.; Tsuji, H.; Shibuya, T.; Hattori, H. Michael addition of nitromethane to α, β-unsaturated carbonyl compounds over solid base catalysts. J. Mol. Catal. A Chem. 2000, 155, 23–29. [Google Scholar] [CrossRef]
- Wei, T.; Wang, M.H.; Wei, W.; Sun, H.Y.; Zhong, B. Solid base catalysts. Chem. Bull. 2002, 65, 594–600. [Google Scholar] [CrossRef]
- Cotman, M.; Erjavec, B.; Djinovic, P.; Pintar, A. Catalyst support materials for prominent mineralization of bisphenol A in catalytic ozonation process. Environ. Sci. Pollut. Res. 2016, 23, 10223–10233. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, W.Z.; Yin, X.S.; Liu, Y. The role of Mn-doping for catalytic ozonation of phenol using Mn/γ-Al2O3 nano catalyst: Performance and mechanism. J. Environ. Chem. Eng. 2016, 4, 3415–3425. [Google Scholar] [CrossRef]
- Einage, H.; Futamura, S. Effect of water vapor on catalytic oxidation of benzene with ozone on alumina-supported manganese oxides. J. Catal. 2016, 243, 446–450. [Google Scholar] [CrossRef]
- Ziylan, A.; Ince, N.H. Catalytic ozonation of ibuprofen with ultrasound and Fe-based catalysts. Catal. Today 2015, 240, 2–8. [Google Scholar] [CrossRef]
- Muruganandham, M.; Wu, J.J. Granular α-FeOOH—A stable and efficient catalyst for the decomposition of dissolved ozone in water. Catal. Commun. 2007, 8, 668–672. [Google Scholar] [CrossRef]
- Ince, N.H. Ultrasound-assisted advanced oxidation processes for water decontamination. Ultrason Sonochem. 2018, 40, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Neppolian, B.; Park, J.S.; Choi, H. Effect of Fenton-like oxidation on enhanced oxidative degradation of para-chlorobenzoic acid by ultrasonic irradiation. Ultrason Sonochem. 2004, 11, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Leighton, T.G. The Acoustic Bubble; Academic Press: Cambridge, MA, USA, 1997; ISBN 0124419216. [Google Scholar]
- Wang, Y.; Zhang, H.; Chen, L. Ultrasound enhanced catalytic ozonation of tetracycline in a rectangular air-lift reactor. Catal. Today 2011, 175, 283–292. [Google Scholar] [CrossRef]
- Ministry of Environment Protection. Determination of Ammonia Nitrogen by Nessler’s Reagent Spectrophotometry. Available online: http://kjs.mee.gov.cn/hjbhbz/bzwb/jcffbz/201001/t20100112_184155.shtml (accessed on 21 May 2019).
- Ministry of Environment Protection. Determination of Nitrite by Spectrophotometry. Available online: http://kjs.mee.gov.cn/hjbhbz/bzwb/jcffbz/198708/t19870801_66628.shtml (accessed on 21 May 2019).
- Weavers, L.K.; Hoffmann, M.R. Sonolytic decomposition of ozone in aqueous solution: Mass transfer effects. Environ. Sci. Technol. 1998, 32, 3941–3947. [Google Scholar] [CrossRef]
- Rayaroth, M.P.; Aravind, U.K. Degradation of pharmaceuticals by ultrasound-based advanced oxidation process. Environ. Chem. Lett. 2016, 14, 259–290. [Google Scholar] [CrossRef]
- Holgne, J.; Bader, H. Ozonation of water: Kinetics of oxidation of ammonia by ozone and hydroxyl radicals. Environ. Sci. Technol. 1978, 12, 79–84. [Google Scholar] [CrossRef]
- Moussavi, G.; Mahdavianpour, M. The selective direct oxidation of ammonium in the contaminated water to nitrogen gas using the chemical-less VUV photochemical continuous-flow reactor. Chem. Eng. J. 2016, 295, 57–63. [Google Scholar] [CrossRef]
- Kuo, C.H.; Yuan, F.; Hill, D.O. Kinetics of oxidation of ammonia in solutions containing ozone with or without hydrogen peroxide. Ind. Eng. Chem. Res. 1997, 36, 4106–4113. [Google Scholar] [CrossRef]
- Huang, Y.X.; Cui, C.C.; Zhang, D.F.; Li, L.; Pan, D. Heterogeneous catalytic ozonation of dibutyl phthalate in aqueous solution in the presence of iron-loaded activated carbon. Chemosphere 2015, 119, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Ghuge, S.P.; Saroha, A.K. Catalytic ozonation for the treatment of synthetic and industrial effluents—Application of mesoporous materials: A review. J. Environ. Manag. 2018, 211, 83–102. [Google Scholar] [CrossRef] [PubMed]
- Hua, I.; Hoffmann, M.R. Optimization of ultrasonic irradiation as an advanced oxidation technology. Environ. Sci. Technol. 1997, 31, 2237–2243. [Google Scholar] [CrossRef]
- Gurol, M.D.; Akata, A. Kinetics of ozone photolysis in aqueous solution. Am. Inst. Chem. Eng. 1996, 42, 3258–3292. [Google Scholar] [CrossRef]
- Chen, Z.L.; Qi, F.; Xu, B.B.; Shen, J.M.; Yue, B.; Ye, M.M. Mechanism discussion of γ-alumina catalyzed ozonation for 2-methylisoborneol removal. Environ. Sci. 2007, 28, 563–568. [Google Scholar]
- Buxtion, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical Review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (OH/O) in Aqueous Solution. J. Phys. Chem. Ref. Data 1988, 2, 513–886. [Google Scholar] [CrossRef]
- Acero, J.L.; Gunten, U.V. Influence of carbonate on the ozone/hydrogen peroxide based advanced oxidation process for drinking water treatment. Ozone Sci. Technol. 2000, 22, 305–328. [Google Scholar] [CrossRef]
- Xiao, Y.J.; Zhang, L.F.; Yue, J.Q.; Webster, R.D.; Lim, T. Kinetic modeling and energy efficiency of UV/H2O2 treatment of iodinated trihalomethanes. Water Res. 2015, 75, 259–269. [Google Scholar] [CrossRef]
- Ludvikova, J.; Jablonska, M.; Jiratova, K.; Chmielarz, L.; Balabanova, J.; Kovanda, F.; Obalova, L. Co-Mn-Al mixed oxides as catalysts for ammonia oxidation to N2O. Res. Chem. Intermed. 2016, 42, 2669–2690. [Google Scholar] [CrossRef]
- Rang, R.Q.; Yang, R.T. Selective catalytic oxidation of ammonia to nitrogen over Fe2O3-TiO2 prepared with a sol-gel method. J. Catal. 2002, 207, 158–165. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | pH | NH4+ | NO3− | NO2− | Mg | Si | Ca | Mn | Rb | Na | Sr | Y |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Concentration (mg/L) | 8.45 | 302 | 70.2 | 0.57 | 1.04 | 1.26 | 27.8 | 3.10 | 1.15 | 0.69 | 0.69 | 0.51 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, C.; Chen, Y.; He, C.; Yin, R.; Liu, J.; Qiu, T. Ultrasound-Enhanced Catalytic Ozonation Oxidation of Ammonia in Aqueous Solution. Int. J. Environ. Res. Public Health 2019, 16, 2139. https://doi.org/10.3390/ijerph16122139
Liu C, Chen Y, He C, Yin R, Liu J, Qiu T. Ultrasound-Enhanced Catalytic Ozonation Oxidation of Ammonia in Aqueous Solution. International Journal of Environmental Research and Public Health. 2019; 16(12):2139. https://doi.org/10.3390/ijerph16122139
Chicago/Turabian StyleLiu, Chen, Yunnen Chen, Caiqing He, Ruoyu Yin, Jun Liu, and Tingsheng Qiu. 2019. "Ultrasound-Enhanced Catalytic Ozonation Oxidation of Ammonia in Aqueous Solution" International Journal of Environmental Research and Public Health 16, no. 12: 2139. https://doi.org/10.3390/ijerph16122139