
Hepatic and Nephric NRF2 Pathway Up-Regulation, an Early Antioxidant Response, in Acute Arsenic-Exposed Mice

Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Chemicals
2.2. Animals and Experimental Procedures
2.3. Calculation of the Liver and Kidney Indexes
2.4. Determination of Tissue Arsenic Levels in Liver and Kidney
2.5. Western Blot Analysis
2.6. Total RNA Isolation and Real-Time PCR Analysis
2.7. Analysis of GSH Levels in the Liver and Kidney
2.8. Analysis of Lipid Peroxidation and T-AOC in the Liver and Kidney
2.9. Statistical Analysis
3. Results
3.1. T-As Concentrations, as Well as the Indexes of Liver and Kidney in Control and Acute Arsenic-Exposed Mice

{kind=link}
{kind=link}
{kind=link}
| NaAsO2 (mg/kg) | T-As in Liver | T-As in Kidney | Liver Index (%) | Kidney Index (%) |
|---|---|---|---|---|
| 0 | < LD | < LD | 5.36 ± 0.39 | 1.21 ± 0.07 |
| 5 | 359.86 ± 28.59 | 1165 ± 303.00 | 5.05 ± 0.34 | 1.17 ± 0.14 |
| 10 | 1033.09 ± 106.66 * | 1816 ± 279.8 * | 4.49 ± 0.20 | 1.14 ± 0.10 |
| 20 | 3075.36 ± 485.11 *,# | 2332 ± 174.60 * | 5.22 ± 0.28 | 1.27 ± 0.10 |
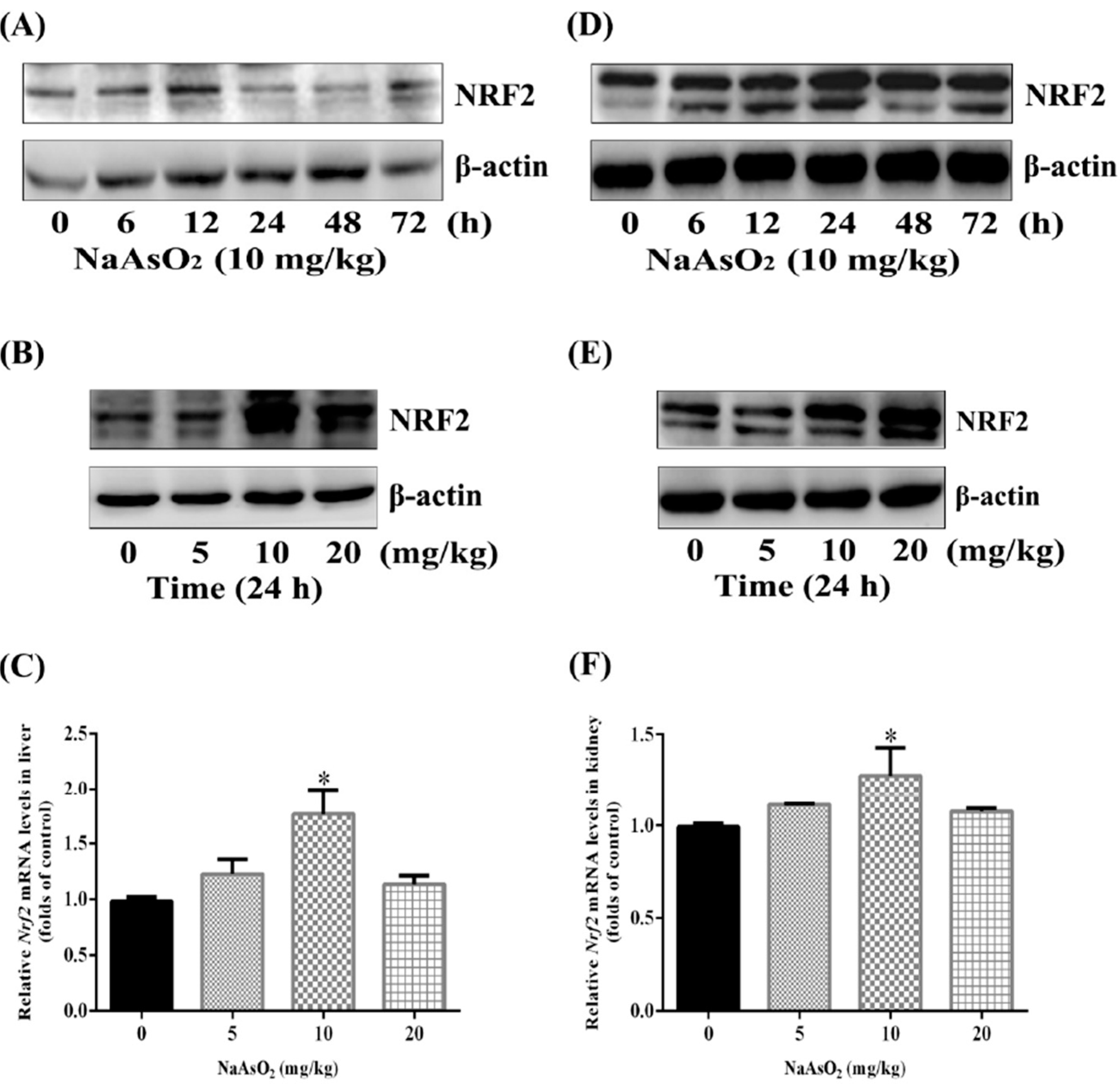
3.2. Up-Regulation of Nuclear Factor NRF2 both in the Liver and Kidney of Control and Acute Arsenic-Exposed Mice

3.3. Up-Regulation of NRF2 Downstream Targets both in the Liver and Kidney of Control and Acute Arsenic-Exposed Mice

3.4. Acute NaAsO2 Exposure Results in Oxidative Stress both in the Liver and Kidney

4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hughes, M.F.; Beck, B.D.; Chen, Y.; Lewis, A.S.; Thomas, D.J. Arsenic exposure and toxicology: A historical perspective. Toxicol. Sci. 2011, 123, 305–332. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.H.; Lingas, E.O.; Rahman, M. Contamination of drinking-water by arsenic in Bangladesh: A public health emergency. Bull. WHO 2000, 78, 1093–1103. [Google Scholar] [PubMed]
- Tchounwou, P.B.; Patlolla, A.K.; Centeno, J.A. Carcinogenic and systemic health effects associated with arsenic exposure-a critical review. Toxicol. Pathol. 2003, 31, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Rossman, T.G. Mechanism of arsenic carcinogenesis: An integrated approach. Mutat. Res. 2003, 533, 37–65. [Google Scholar] [CrossRef] [PubMed]
- IARC. Some drinking-water disinfectants and contaminants, including arsenic. IARC Monogr. Eval. Carcinog. Risks Hum. 2004, 84, 1–477. [Google Scholar]
- Smith, A.H.; Hopenhayn-Rich, C.; Bates, M.N.; Goeden, H.M.; Hertz-Picciotto, I.; Duggan, H.M.; Wood, R.; Kosnett, M.J.; Smith, M.T. Cancer risks from arsenic in drinking water. Environ. Health Perspect. 1992, 97, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Navas-Acien, A.; Silbergeld, E.K.; Streeter, R.A.; Clark, J.M.; Burke, T.A.; Guallar, E. Arsenicexposure and type 2 diabetes: A systematic review of the experimental and epidemiological evidence. Environ. Health Perspect. 2006, 114, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.H. An overview on peripheral vascular disease in blackfoot disease-hyperendemic villages in Taiwan. Angiology 2002, 53, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Ellinsworth, D.C. Arsenic, reactive oxygen, and endothelial dysfunction. J. Pharmacol. Exp. Ther. 2015, 353, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Rehman, K.; Fu, Y.J.; Zhang, Y.F.; Wang, Q.Q.; Wu, B.; Wu, Y.; Zhou, X.Y.; Sun, W.H.; Sun, T.F.; Naranmandura, H. Trivalent methylated arsenic metabolites induce apoptosis in human myeloid leukemic HL-60 cells through generation of reactive oxygen species. Metallomics 2014, 6, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Eblin, K.E.; Bowen, M.E.; Cromey, D.W.; Bredfeldt, T.G.; Mash, E.A.; Lau, S.S.; Gandolfi, A.J. Arsenite and monomethylarsonous acid generate oxidative stress response in human bladder cell culture. Toxicol. Appl. Pharmacol. 2006, 217, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Cooper, K.L.; Liu, K.J.; Hudson, L.G. Enhanced ROS production and redox signaling with combined arsenite and UVA exposure: Contribution of NADPH oxidase. Free Radic. Biol. Med. 2009, 47, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Majhi, C.R.; Khan, S.; Leo, M.D.; Prawez, S.; Kumar, A.; Sankar, P.; Telang, A.G.; Sarkar, S.N. Acetaminophen increases the risk of arsenic-mediated development of hepatic damage in rats by enhancing redox-signaling mechanism. Environ. Toxicol. 2014, 29, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Rizwan, S.; Naqshbandi, A.; Farooqui, Z.; Khan, A.A.; Khan, F. Protective effect of dietary flaxseed oil on arsenic-induced nephrotoxicity and oxidative damage in rat kidney. Food Chem. Toxicol. 2014, 68, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Morris, H.; Cronin, M.T.D. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Misra, V.; Thimmulappa, R.K.; Hannah, L.H.; Ames, S.; Hoque, M.O.; Herman, J.G.; Stephen, B.; Baylin, S.B.; Sidransky, D.; et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006, 3, 1865–1876. [Google Scholar] [CrossRef] [PubMed]
- Aono, J.; Yanagawa, T.; Itoh, K.; Li, B.; Hiroshi, Y.H.; Kumagai, Y.; Yamamoto, M.; Ishii, T. Activation of Nrf2 and accumulation of ubiquitinated A170 by arsenic in osteoblasts. Biochem. Biophys. Res. Commun. 2003, 305, 271–277. [Google Scholar] [CrossRef]
- Zhao, R.; Hou, Y.; Zhang, Q.; Woods, C.G.; Xue, P.; Fu, J.; Yarborough, K.; Guan, D.; Andersen, M.E.; Pi, J. Cross-regulations among NRFs and KEAP1 and effects of their silencing on arsenic-induced antioxidant response and cytotoxicity in human keratinocytes. Environ. Health Perspect. 2012, 120, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Sun, Z.; Chen, W.; Eblin, K.E.; Gandolfi, J.A.; Zhang, D.D. Nrf2 protects human bladder urothelial cells from arsenite and monomethylarsonous acid toxicity. Toxicol. Appl. Pharmacol. 2007, 225, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Massrieh, W.; Derjuga, A.; Blank, V. Induction of endogenous Nrf2/small maf heterodimers by arsenic-mediated stress in placental choriocarcinoma cells. Antioxid. Redox Signal. 2006, 8, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Blank, V.; Sesay, J.S.; Crawford, D.R. Maf genes are involved in multiple stress response in human. Biochem. Biophys. Res. Commun. 2001, 280, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Matulis, S.M.; Morales, A.A.; Yehiayan, L.; Croutch, C.; Gutman, D.; Cai, Y.; Lee, K.P.; Boise, L.H. Darinaparsin induces a unique cellular response and is active in an arsenic trioxide-resistant myeloma cell line. Mol. Cancer Ther. 2009, 8, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Sun, Z.; Chen, W.; Li, Y.; Villeneuve, N.F.; Zhang, D.D. Activation of Nrf2 by arsenite and monomethylarsonous acid is independent of Keap1-C151: Enhanced Keap1-Cul3 interaction. Toxicol. Appl. Pharmacol. 2008, 230, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Waalkes, M.P. Liver is a target of arsenic carcinogenesis. Toxicol. Sci. 2008, 105, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, D.N. Effect of chronic intake of arsenic-contaminated water on liver. Toxicol. Appl. Pharmacol. 2005, 206, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Eom, S.Y.; Lee, Y.C.; Yim, D.H.; Lee, C.H.; Kim, Y.D.; Choi, B.S.; Park, C.H.; Yu, S.D.; Kim, D.S.; Park, J.D.; et al. Effects of low-level arsenic exposure on urinary N-acetyl-β-D-glucosaminidase activity. Hum. Exp. Toxicol. 2011, 30, 1885–1891. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.W.; Chen, H.Y.; Li, W.F.; Liou, S.H.; Chen, C.J.; Wu, J.H.; Wang, S.L. The association between total urinary arsenic concentration and renal dysfunction in a community-based population from central Taiwan. Chemosphere 2011, 84, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Robles-Osorio, M.L.; Sabath-Silva, E.; Sabath, E. Arsenic-mediated nephrotoxicity. Ren. Fail. 2015, 23, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Li, X.; Zhu, B.; Zhang, X.; Wang, Y.; Xu, Y.; Wang, H.; Hou, Y.; Zheng, Q.; Sun, G. Sodium arsenite induced reactive oxygen species generation, nuclear factor (erythroid-2related) factor 2 activation, heme oxygenase-1 expression, and glutathioneElevat ion in Chang human hepatocytes. Environ. Toxicol. 2013, 28, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Huang, Z.; Chan, J.Y.; Zhang, D.D. Nrf2 protects against As(III)-induced damage in mouse liver and bladder. Toxicol. Appl. Pharmacol. 2009, 240, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Sosa, M.; García-Montalvo, E.A.; del Razo, L.M.; Vega, L. Effect of Selenomethionine supplementation in food on the excretion and toxicity of arsenic exposure in female mice. Biol. Trace Elem. Res. 2013, 156, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Anwar-Mohamed, A.; Elshenawy, O.H.; El-Sherbeni, A.A.; Abdelrady, M.; El-Kadi, A.O. Acute arsenic treatment alters arachidonic acid and its associated metabolite levels in the brain of C57BL/6 mice. Can. J. Physiol. Pharmacol. 2014, 92, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Xie, R.J.; Geng, X.X.; Luo, X.H.; Han, B.; Cheng, M.L. Effect of Danshao Huaxian capsule on expression of matrix metalloproteinase-1 and tissue inhibitor of metalloproteinase-1 in fibrotic liver of rats. World J. Gastroenterol. 2005, 11, 4953–4956. [Google Scholar] [PubMed]
- Sánchez-Rodas, D.; Gómez-Ariza, J.L.; Oliveira, V. Development of a rapid extraction procedure for speciation of arsenic in chicken meat. Anal. Bioanal. Chem. 2006, 385, 1172–1177. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical biochemistry 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Sedlak, J.; Lindsay, R.H. Estimation of total, protein-bound, and nonprotein sulfhydryl groups in tissue with Ellman’s reagent. Anal. Biochem. 1968, 25, 192–205. [Google Scholar] [CrossRef]
- Ohkawa, H.; Ohishi, N.; Yagi, K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 1979, 95, 351–358. [Google Scholar] [CrossRef]
- Opara, E.C.; Abdel-Rahman, E.; Soliman, S.; Kamel, W.A.; Souka, S.; Lowe, J.E.; Abdel-Aleem, S. Depletion of total antioxidant capacity in type 2 diabetes. Metabolism 1999, 48, 1414–1417. [Google Scholar] [CrossRef]
- Ercal, N.; Gurer-Orhan, H.; Aykin-Burns, N. Toxic metals and oxidative stress part I: Mechanisms involved in metal-induced oxidative damage. Curr. Top. Med. Chem. 2001, 1, 529–539. [Google Scholar]
- Hellou, J.; Ross, N.W.; Moon, T.W. Glutathione, glutathione S-transferase, and glutathione conjugates, complementary markers of oxidative stress in aquatic biota. Environ. Sci. Pollut. Res. Int. 2012, 19, 2007–2023. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ding, Y.; Niu, Q.; Xu, S.; Pang, L.; Ma, R.; Jing, M.; Feng, G.; Tang, J.X.; Zhang, Q.; et al. Lutein has a protective effect on hepatotoxicity induced by arsenic via Nrf2 signaling. Biomed. Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.W.; Jiang, Y.; Zhang, D.Y.; Wang, M.; Chen, W.S.; Su, H.; Wang, Y.T.; Wan, J.B. Protective effects of Penthorum chinense Pursh against chronic ethanol-induced liver injury in mice. J. Ethnopharmacol. 2015, 161, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Barchowsky, A.; Klei, L.R.; Dudek, E.J.; Swartz, H.M.; James, P.E. Stimulation of reactive oxygen, but not reactive nitrogen species, in vascular endothelial cells exposed to low levels of arsenite. Free Radic. Biol. Med. 1999, 27, 1405–1412. [Google Scholar] [CrossRef]
- Sarath, T.S.; Waghe, P.; Gupta, P.; Choudhury, S.; Kannan, K.; Pillai, A.H.; Harikumar, S.K.; Mishra, S.K.; Sarkar, S.N. Atorvastatin ameliorates arsenic-induced hypertension and enhancement of vascular redox signaling in rats. Toxicol. Appl. Pharmacol. 2014, 280, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Engel, R.H.; Evens, A.M. Oxidative stress and apoptosis: A new treatment paradigm in cancer. Front. Biosci. 2006, 11, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Mitsuishi, Y.; Motohashi, H.; Yamamoto, M. The Keap1-Nrf2 system in cancers: Stress response and anabolic metabolism. Front. Oncol. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Whitman, S.A.; Jaramillo, M.C.; Zhang, D.D. Arsenic-mediated activation of the Nrf2-Keap1 antioxidant pathway. J. Biochem. Mol. Toxicol. 2013, 27, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Yamamoto, M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid. Redox Signal. 2005, 7, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Su, Y.; Zhang, M.; Sun, Z. Opposite effects of arsenic trioxide on the Nrf2 pathway in oral squamous cell carcinoma in vitro and in vivo. Cancer Lett. 2012, 318, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Saw, C.L.; Kong, A.N. Nuclear factor-erythroid 2-related factor 2 as a chemopreventive target in colorectal cancer. Expert Opin. Ther. Targets 2011, 15, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Abiko, Y.; Shinkai, Y.; Sumi, D.; Kumagai, Y. Reduction of arsenic-induced cytotoxicity through Nrf2/HO-1 signaling in HepG2 cells. J. Toxicol. Sci. 2010, 35, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Duan, X.; Dong, D.; Bai, C.; Li, X.; Sun, G.; Li, B. Activation of the Nrf2 pathway by inorganic arsenic in human hepatocytes and the role of transcriptional repressor Bach1. Oxid. Med. Cell. Longev. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Pi, J.; Qu, W.; Reece, J.M.; Kumagai, Y.; Waalkes, M.P. Transcription factor Nrf2 activation by inorganic arsenic in cultured keratinocytes: Involvement of hydrogen peroxide. Exp. Cell Res. 2003, 290, 234–245. [Google Scholar] [CrossRef]
- Sampsonas, F.; Archontidou, M.A.; Salla, E.; Karkoulias, K.; Tsoukalas, G.; Spiropoulos, K. Genetic alterations of glutathione S-transferases in asthma: Do they modulate lung growth and response to environmental stimuli. Allergy Asthma Proc. 2007, 28, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Li.; Chcn, W.P.; Yang, T.Y.; Chen, Y.H.; Lo, W.C.; Wang, Y.H.; Liao, Y.T.; Hsueh, Y.M.; Chiou, H.Y.; Wu, M.M.; et al. Genetic polymorphisms in glutathione S-transferase (GST) superfamily and risk of arsenic-induced urothelial carcinoma in residents of southwestern Taiwan. J. Biomed. Sci. 2011, 18. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shertzer, H.G.; Schneider, S.N.; Nebert, D.W.; Dalton, T.P. Glutamate cysteine ligase catalysis: Dependence on ATP and modifier subunit for regulation of tissue glutathione levels. J Biol. Chem. 2005, 280, 33766–33774. [Google Scholar] [CrossRef] [PubMed]
- Griffith, O.W.; Mulcahy, R.T. The enzymes of glutathione synthesis: Gamma-glutamylcysteine synthetase. Adv. Enzymol. Relat. Areas Mol. Biol. 1999, 73, 209–267. [Google Scholar] [PubMed]
- Mattson, M.P.; Cheng, A. Neurohormetic phytochemicals: Low-dose toxins that induce adaptive neuronal stress responses. Trends Neurosci. 2006, 29, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Chen, C.; Liu, Y.; Zhang, P.; Zhang, Z. Critical role of cellular glutathione homeostasis for trivalent inorganic arsenite-induced oxidative damage in human bronchial epithelial cells. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2014, 770, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, Y.; Wang, H.; Xue, P.; Li, X.; Li, B.; Zheng, Q.; Sun, G. Arsenic induces mitochondria-dependent apoptosis by reactive oxygen species generation rather than glutathione depletion in Chang human hepatocytes. Arch. Toxicol. 2009, 83, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Li, S.G.; Ding, Y.S.; Niu, Q.; Xu, S.Z.; Pang, L.J.; Ma, R.L.; Jing, M.X.; Feng, G.L.; Liu, J.M.; Guo, S.X. Grape seed proanthocyanidin extract alleviates arsenic-induced oxidative reproductive toxicity in male mice. Biomed. Environ. Sci. 2015, 28, 272–280. [Google Scholar] [PubMed]
- Gao, S.; Duan, X.; Wang, X.; Dong, D.; Liu, D.; Li, X.; Sun, G.; Li, B. Curcumin attenuates arsenic-induced hepatic injuries and oxidative stress in experimental mice through activation of Nrf2 pathway, promotion of arsenic methylation and urinary excretion. Food Chem. Toxicol. 2013, 59, 739–747. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Duan, X.; Dong, D.; Zhang, Y.; Li, W.; Zhao, L.; Nie, H.; Sun, G.; Li, B. Hepatic and Nephric NRF2 Pathway Up-Regulation, an Early Antioxidant Response, in Acute Arsenic-Exposed Mice. Int. J. Environ. Res. Public Health 2015, 12, 12628-12642. https://doi.org/10.3390/ijerph121012628
Li J, Duan X, Dong D, Zhang Y, Li W, Zhao L, Nie H, Sun G, Li B. Hepatic and Nephric NRF2 Pathway Up-Regulation, an Early Antioxidant Response, in Acute Arsenic-Exposed Mice. International Journal of Environmental Research and Public Health. 2015; 12(10):12628-12642. https://doi.org/10.3390/ijerph121012628
Chicago/Turabian StyleLi, Jinlong, Xiaoxu Duan, Dandan Dong, Yang Zhang, Wei Li, Lu Zhao, Huifang Nie, Guifan Sun, and Bing Li. 2015. "Hepatic and Nephric NRF2 Pathway Up-Regulation, an Early Antioxidant Response, in Acute Arsenic-Exposed Mice" International Journal of Environmental Research and Public Health 12, no. 10: 12628-12642. https://doi.org/10.3390/ijerph121012628