
The First Complete Mitochondrial Genome of the Genus Litostrophus: Insights into the Rearrangement and Evolution of Mitochondrial Genomes in Diplopoda

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Specimen Collection and DNA Extraction
2.2. Sequence Analysis
2.3. Phylogenetic Analysis
3. Results and Discussion
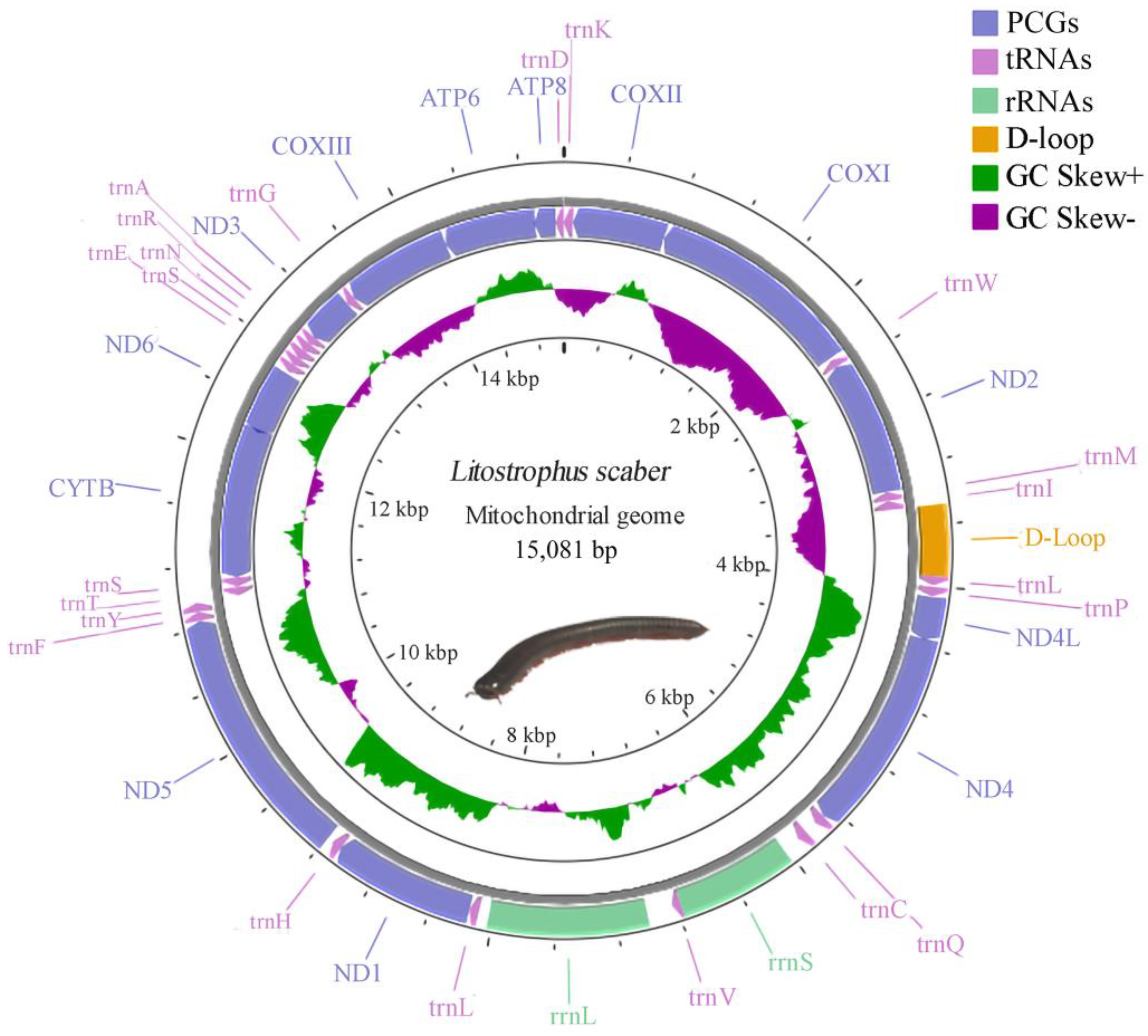
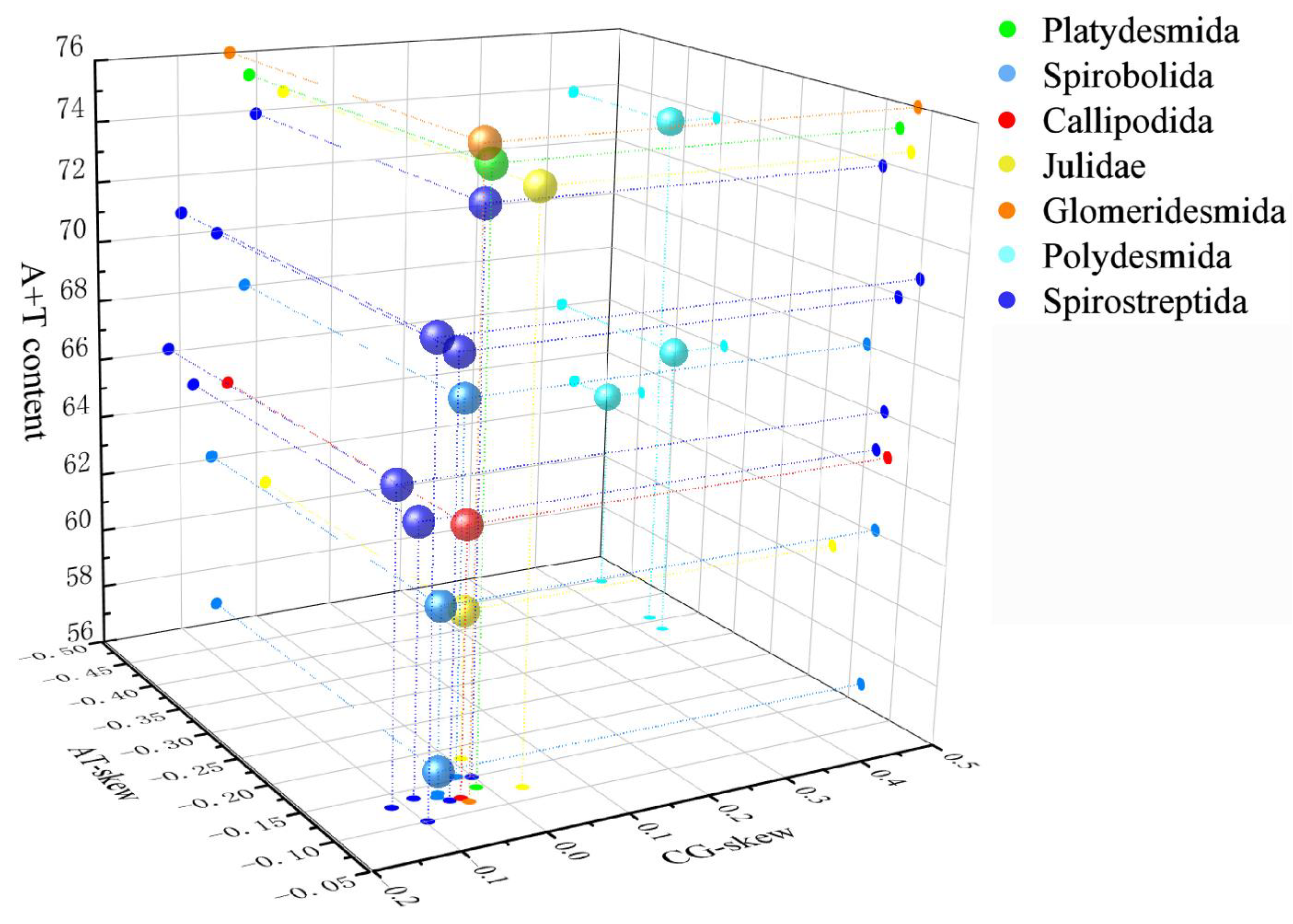
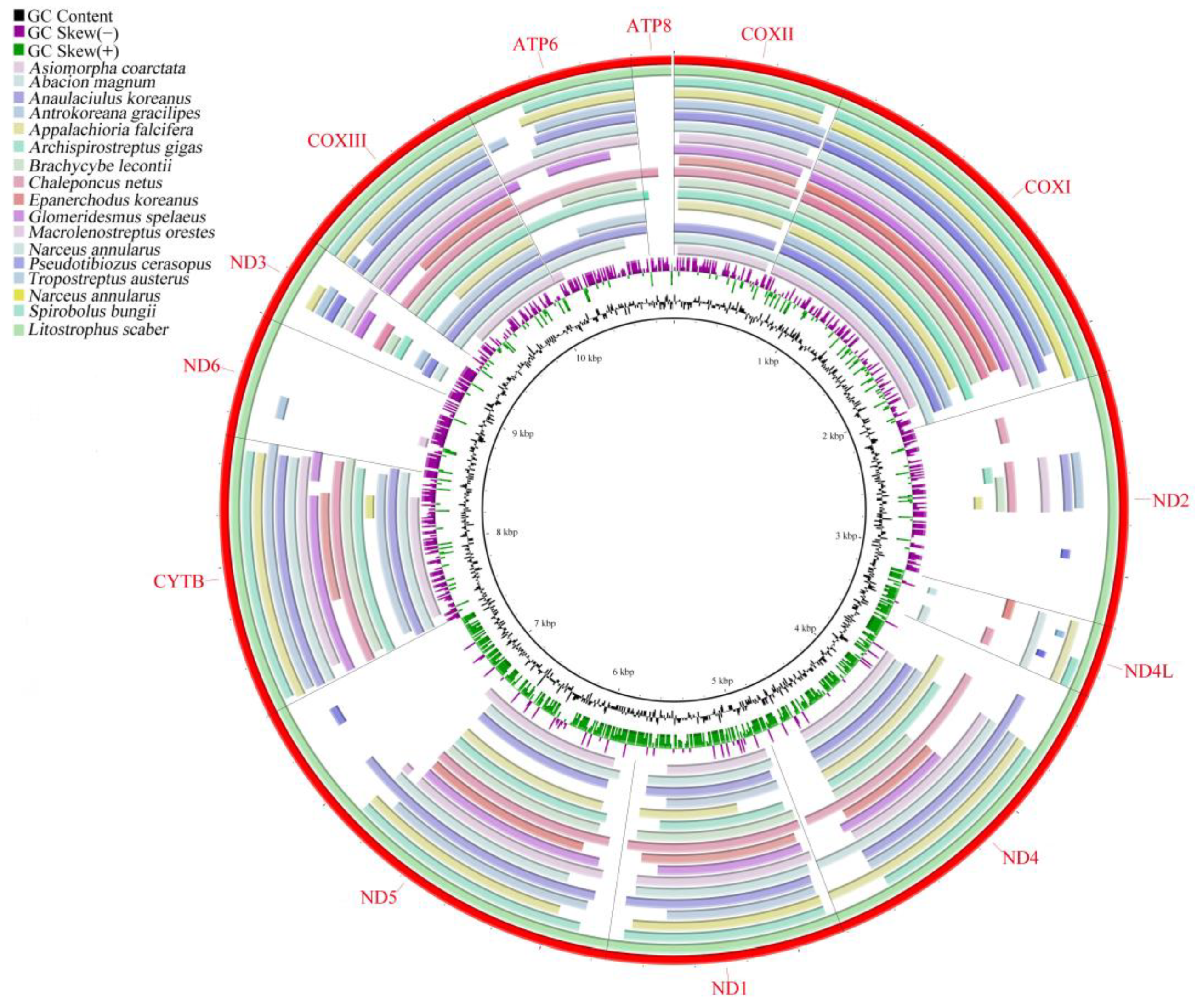
3.1. Mitogenomic Structure and Comparison
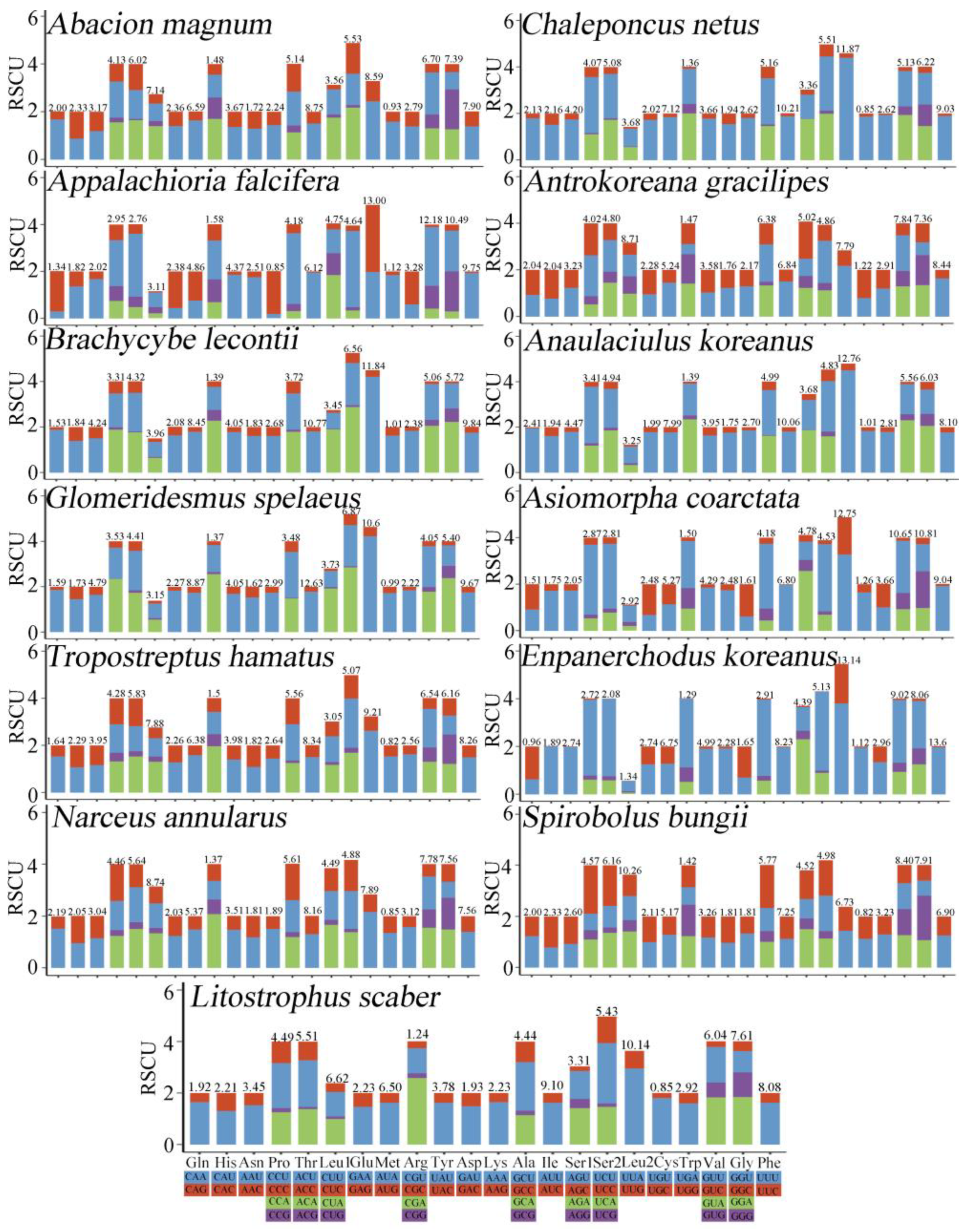
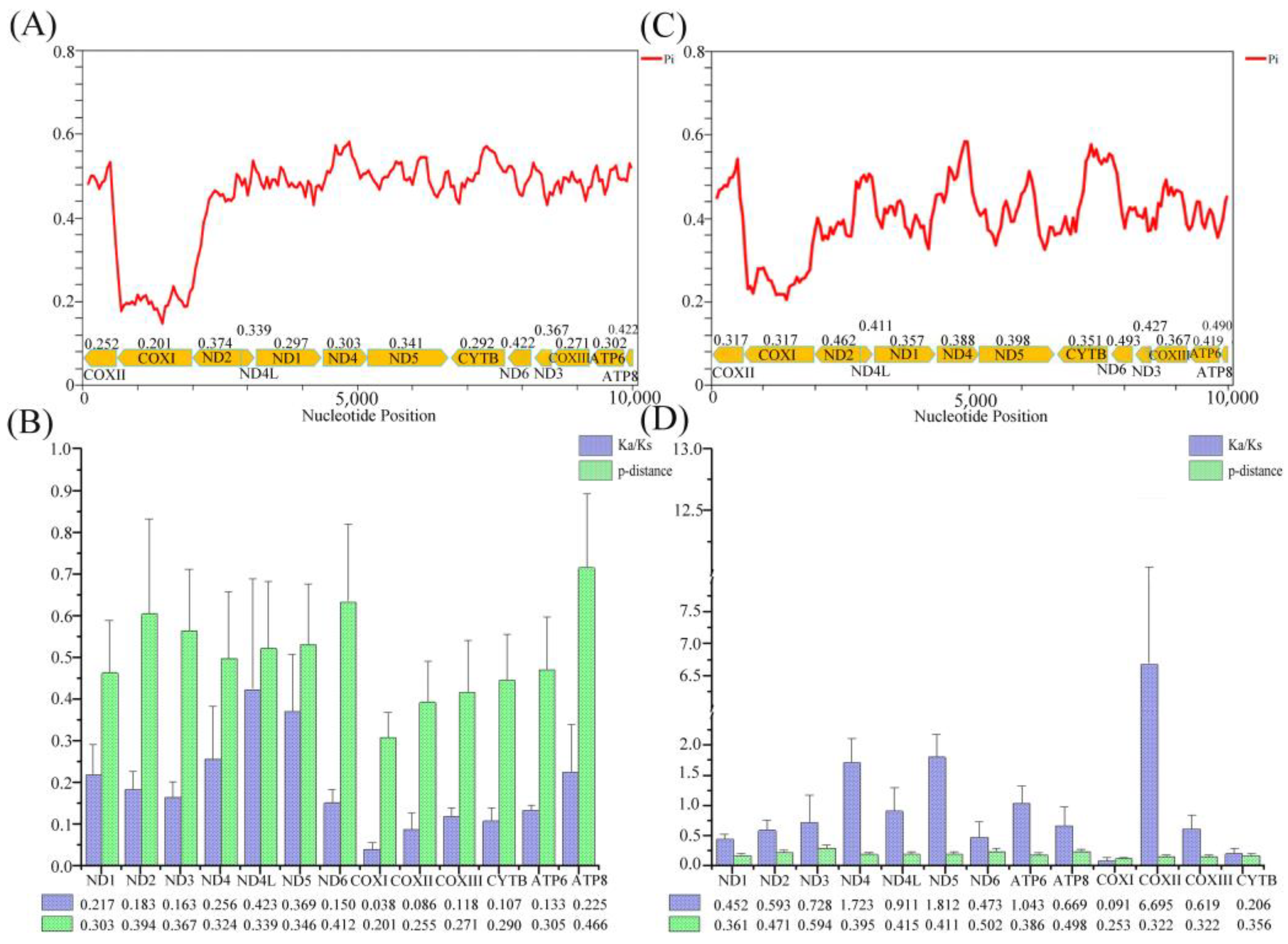
3.2. PCGs
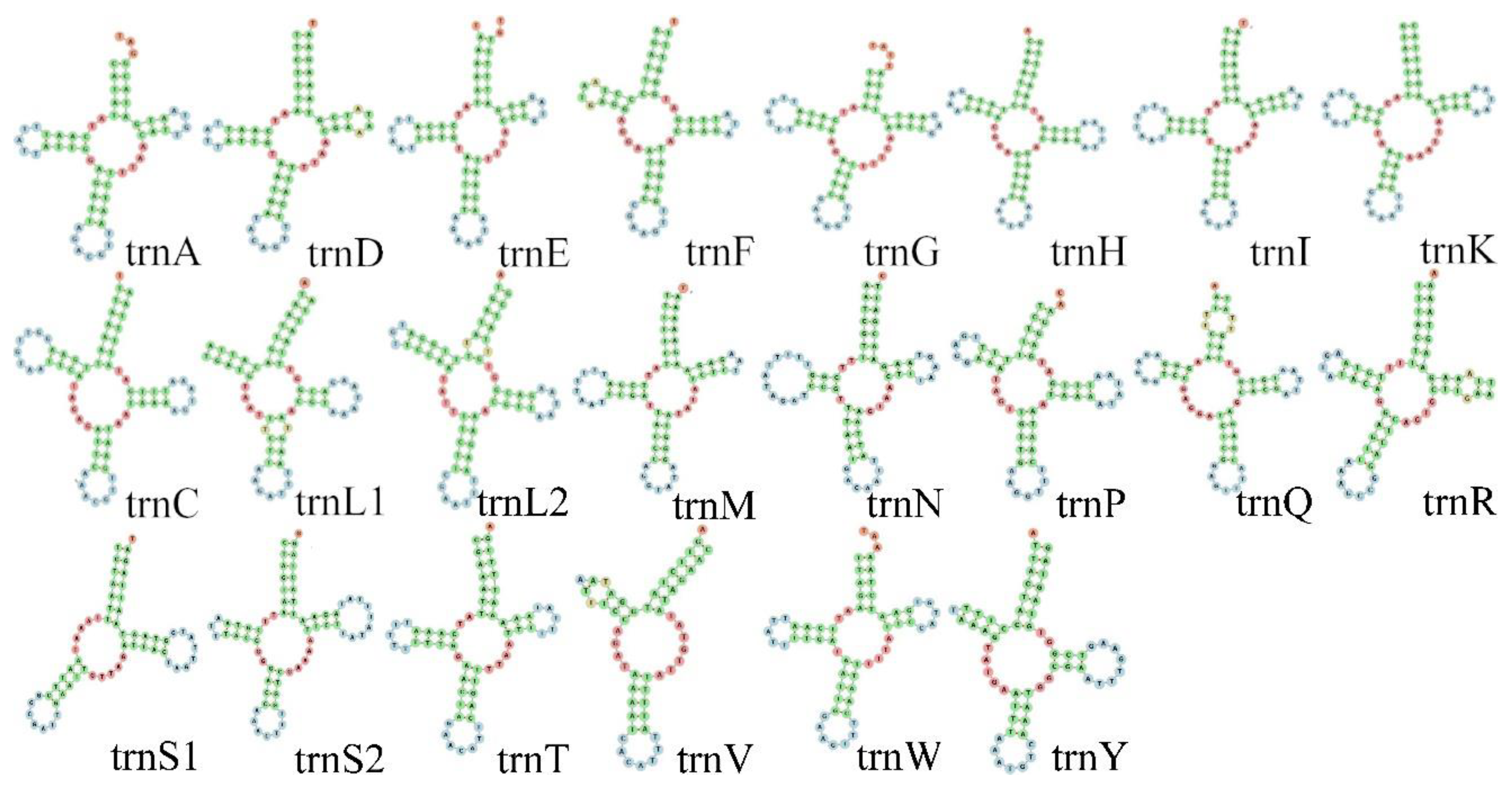
3.3. RNA Genes and Non-Coding Regions
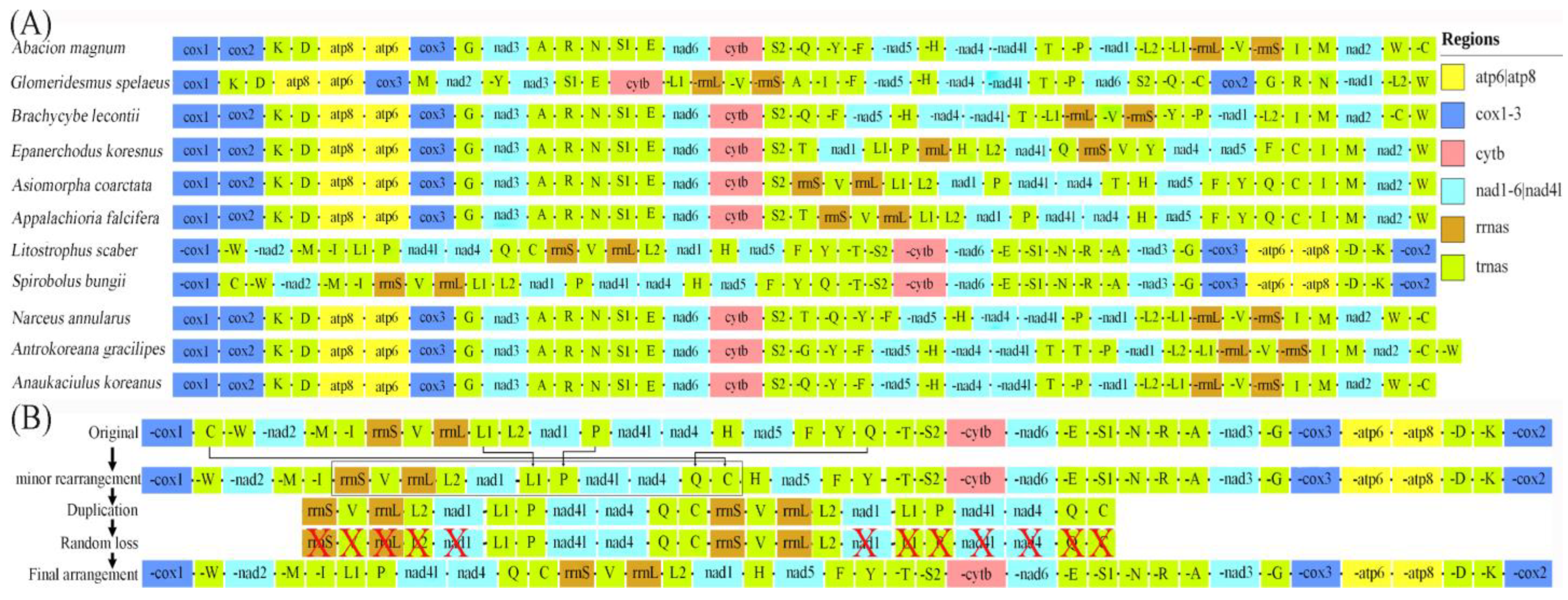
3.4. Gene Rearrangement
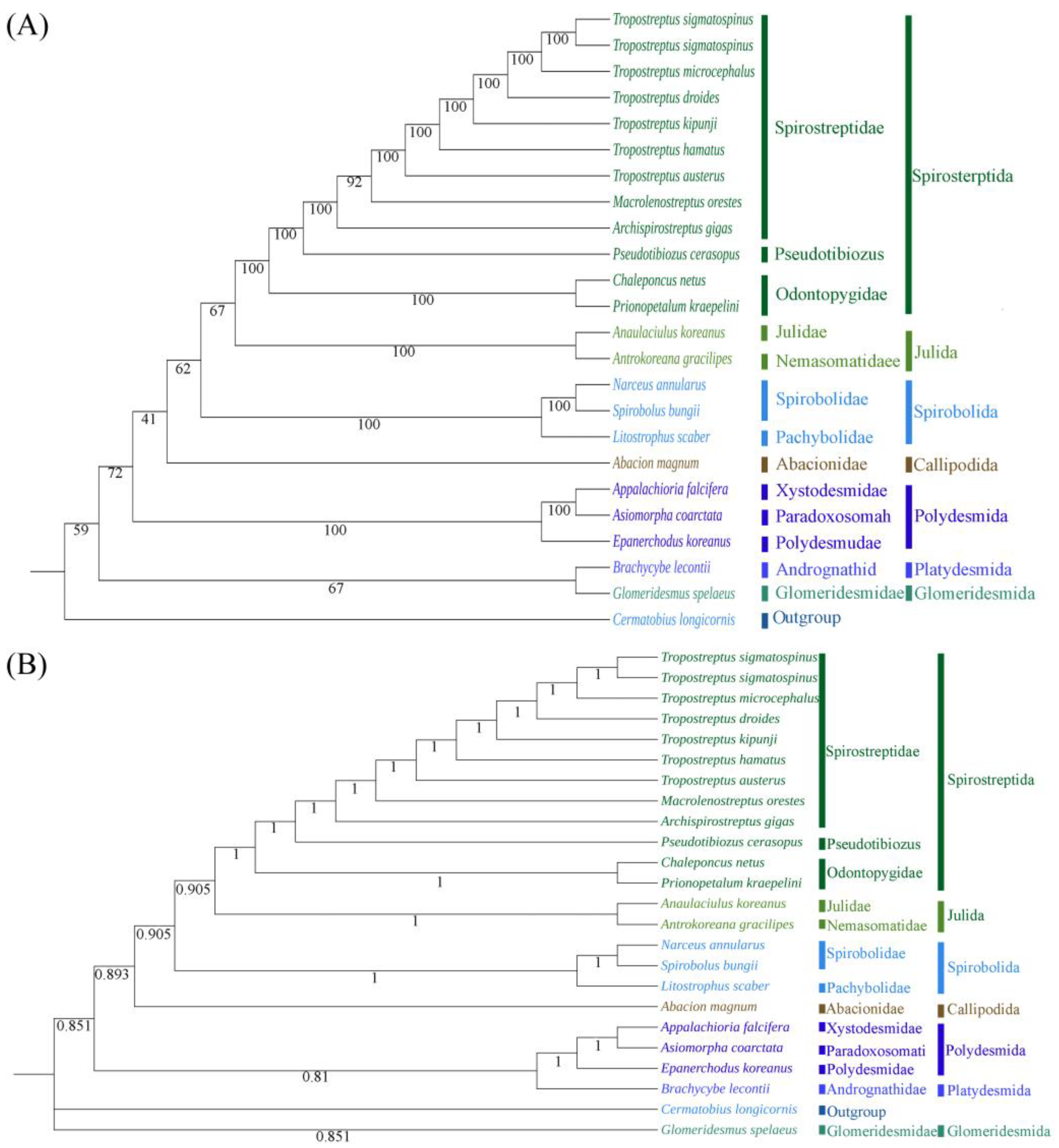
3.5. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sierwald, P.; Bond, J.E. Current status of the myriapod class diplopoda (millipedes): Taxonomic diversity and phylogeny. Annu. Rev. Entomol. 2007, 52, 401–420. [Google Scholar] [CrossRef]
- Pimvichai, P.; Enghoff, H.; Panha, S.; Backeljau, T. Morphological and mitochondrial DNA data reshuffle the taxonomy of the genera Atopochetus Attems, Litostrophus Chamberlin and Tonkinbolus Verhoeff (Diplopoda: Spirobolida: Pachybolidae), with descriptions of nine new species. Invertebr. Syst. 2018, 32, 159–195. [Google Scholar] [CrossRef]
- Snyder, B.A.; Hendrix, P. Current and potential roles of soil macroinvertebrates (earthworms, millipedes, and isopods) in ecological restoration. Restor. Ecol. 2008, 16, 629–636. [Google Scholar] [CrossRef]
- Li, Y.Y.; Liao, J.H.; Chen, H.Y.H.; Zou, X.M.; Delgado-Baquerizo, M.; Ni, J.P.; Ren, T.T.; Xu, H.M.; Ruan, H.H. Soil fauna alter the responses of greenhouse gas emissions to changes in water and nitrogen availability. Soil Biol. Biochem. 2023, 179, 108990. [Google Scholar] [CrossRef]
- Sustr, V.; Simek, M.; Faktorova, L.; Mackova, J.; Tajovsky, K. Release of greenhouse gases from millipedes as related to food, body size, and other factors. Soil Biol. Biochem. 2020, 144, 107765. [Google Scholar] [CrossRef]
- David, J.F.; Handa, I.T. The ecology of saprophagous macroarthropods (millipedes, woodlice) in the context of global change. Biol. Rev. 2010, 85, 881–895. [Google Scholar] [CrossRef]
- Braschler, B.; Gilgado, J.D.; Zwahlen, V.; Rusterholz, H.P.; Buchholz, S.; Baur, B. Ground-dwelling invertebrate diversity in domestic gardens along a rural-urban gradient: Landscape characteristics are more important than garden characteristics. PLoS ONE 2020, 15, e0240061. [Google Scholar] [CrossRef]
- Brewer, M.S.; Sierwald, P.; Bond, J.E. Millipede taxonomy after 250 years: Classification and taxonomic practices in a mega-diverse yet understudied arthropod group. PLoS ONE 2012, 7, e37240. [Google Scholar] [CrossRef]
- Garcia, A.; Krummel, G.; Priya, S. Fundamental understanding of millipede morphology and locomotion dynamics. Bioinspiration Biomim. 2020, 16, 026003. [Google Scholar] [CrossRef]
- Gerlach, J.; Samways, M.; Pryke, J. Terrestrial invertebrates as bioindicators: An overview of available taxonomic groups. J. Insect Conserv. 2013, 17, 831–850. [Google Scholar] [CrossRef]
- Reip, H.S.; Wesener, T. Intraspecific variation and phylogeography of the millipede model organism, the black pill millipede Glomeris marginata (Villers, 1789) (Diplopoda, Glomerida, Glomeridae). Zookyes 2018, 741, 93–131. [Google Scholar] [CrossRef] [PubMed]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef] [PubMed]
- Cameron, L.S. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef]
- Boyce, T.M.; Zwick, M.E.; Aquadro, C.F. Mitochondrial DNA in the bark weevils: Size, structure and heteroplasmy. Genetics 1989, 123, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.F.; Kirkness, E.F.; Barker, S.C. The single mitochondrial chromosome typical of animals has evolved into 18 minichromosomes in the human body louse, Pediculus humanus. Genome Res. 2009, 19, 904–912. [Google Scholar] [CrossRef]
- Liu, H.R.; Li, H.; Song, F.; Gu, W.Y.; Feng, J.N.; Cai, W.Z.; Shao, R.F. Novel insights into mitochondrial gene rearrangement in thrips (Insecta: Thysanoptera) from the grass thrips, Anaphothrips obscurus. Sci. Rep. 2017, 7, 4284. [Google Scholar] [CrossRef]
- Ma, C.; Yang, P.C.; Jiang, F.; Chapuis, M.P.; Shali, Y.; Sword, G.A.; Kang, L. Mitochondrial genomes reveal the global phylogeography and dispersal routes of the migratory locust. Mol. Ecol. 2012, 21, 4344–4358. [Google Scholar] [CrossRef]
- Xu, R.F.; Chen, J.; Pan, Y.; Wang, J.C.; Chen, L.; Ruan, H.H.; Wu, Y.B.; Xu, H.M.; Wang, G.B.; Liu, H.Y. Genetic diversity and population structure of Spirobolus bungii as revealed by mitochondrial DNA sequences. Insects 2022, 13, 729. [Google Scholar] [CrossRef]
- Nelson, L.A.; Lambkin, C.L.; Batterham, P.; Wallman, J.F.; Dowton, M.; Whiting, M.F.; Yeates, D.K.; Cameron, S.L. Beyond barcoding: A mitochondrial genomics approach to molecular phylogenetics and diagnostics of blowflies (Diptera: Calliphoridae). Gene 2012, 511, 131–142. [Google Scholar] [CrossRef]
- Lavrov, D.V.; Boore, J.L.; Brown, W.M. Complete mtDNA sequences of two millipedes suggest a new model for mitochondrial gene rearrangements: Duplication and nonrandom loss. Mol. Biol. Evol. 2002, 19, 163–169. [Google Scholar] [CrossRef]
- Salvato, P.; Simonato, M.; Battisti, A.; Negrisolo, E. The complete mitochondrial genome of the bag-shelter moth Ochrogaster lunifer (Lepidoptera, Notodontidae). BMC Genom. 2008, 9, 331. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, K.; Chakraborty, R.; Cameron, S.L.; Sweet, A.D.; Chandra, K.; Kumar, V. Rearrangement and evolution of mitochondrial genomes in Thysanoptera (Insecta). Sci. Rep. 2020, 10, 695. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Lei, Z.M.; Li, W.J.; Zhang, W.; Zhou, C.F. Comparative mitogenomic analysis of Heptageniid Mayflies (Insecta: Ephemeroptera): Conserved intergenic spacer and tRNA gene duplication. Insects 2021, 12, 170. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Havird, J.C.; Santos, S.R. Performance of single and concatenated sets of mitochondrial genes at inferring metazoan relationships relative to full mitogenome data. PLoS ONE 2014, 9, e84080. [Google Scholar] [CrossRef]
- Feng, J.T.; Guo, Y.H.; Yan, C.R.; Ye, Y.Y.; Yan, X.J.; Li, J.J.; Xu, K.D.; Guo, B.Y.; Lü, Z.M. Novel gene rearrangement in the mitochondrial genome of Siliqua minima (Bivalvia, Adapedonta) and phylogenetic implications for Imparidentia. PLoS ONE 2021, 16, e0249446. [Google Scholar] [CrossRef]
- Boore, J.L.; Collins, T.M.; Stanton, D.; Daehler, L.L.; Brown, W.M. Deducing the pattern of arthropod phylogeny from mitochondrial DNA rearrangements. Nature 1995, 376, 163–165. [Google Scholar] [CrossRef]
- Zuo, Q.; Zhang, Z.S.; Shen, Y.J. Novel mitochondrial gene rearrangements pattern in the millipede Polydesmus sp. GZCS-2019 and phylogenetic analysis of the Myriapoda. Ecol. Evol. 2022, 12, e8764. [Google Scholar] [CrossRef]
- Brewer, M.S.; Swafford, L.; Spruill, C.L.; Bond, J.E. Arthropod phylogenetics in light of three novel millipede (Myriapoda: Diplopoda) mitochondrial genomes with comments on the appropriateness of mitochondrial genome sequence data for inferring deep level relationships. PLoS ONE 2013, 8, e68005. [Google Scholar] [CrossRef]
- Xu, H.M.; Fang, Y.; Cao, G.H.; Shen, C.Q.; Liu, H.Y.; Ruan, H.H. The complete mitochondrial genome of Spirobolus bungii (Diplopoda, Spirobolidae): The first sequence for the genus Spirobolus. Genes 2022, 13, 1587. [Google Scholar] [CrossRef]
- Qu, Z.; Nong, W.Y.; So, W.L.; Barton-Owen, T.; Li, Y.Q.; Leung, T.C.N.; Li, C.D.; Baril, T.; Wong, A.Y.P.; Swale, T.; et al. Millipede genomes reveal unique adaptations during myriapod evolution. PLoS Biol. 2020, 18, e3000636. [Google Scholar] [CrossRef] [PubMed]
- Caravas, J.; Friedrich, M. Of mites and millipedes: Recent progress in resolving the base of the arthropod tree. Bioessays 2010, 32, 488–495. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Juhling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Putz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenetics Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Alikhan, N.F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST ring image generator (BRIG): Simple prokaryote genome comparisons. BMC Genom. 2011, 12, 402. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Kerpedjiev, P.; Hammer, S.; Hofacker, I.L. Forna (force-directed RNA): Simple and effective online RNA secondary structure diagrams. Bioinformatics 2015, 31, 3374–3376. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.L.; Jakovlic, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Rozewicki, J.; Li, S.L.; Amada, K.M.; Standley, D.M.; Katoh, K. MAFFT-DASH: Integrated protein sequence and structural alignment. Nucleic Acids Res. 2019, 47, W5–W10. [Google Scholar] [CrossRef]
- Ranwez, V.; Douzery, E.J.P.; Cambon, C.; Chantret, N.; Delsuc, F. MACSE v2: Toolkit for the alignment of coding sequences accounting for frameshifts and stop codons. Mol. Biol. Evol. 2018, 35, 2582–2584. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.J.; Lee, Y.S.; Park, S.J.; Lim, J.T.; Jang, K.H.; Choi, E.H.; Choi, Y.G.; Hwang, U.W. Complete mitochondrial genome of a troglobite millipede Antrokoreana gracilipes (Diplopoda, Juliformia, Julida), and juliformian phylogeny. Mol. Cells 2007, 23, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.J.; Nguyen, A.D.; Jang, K.H.; Choi, E.H.; Ryu, S.H.; Hwang, U.W. The complete mitochondrial genome of the Korean endemic millipede Anaulaciulus koreanus (Verhoeff, 1937), with notes on the gene arrangement of millipede orders. Zootaxa 2017, 4329, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Bai, Y.; Dong, Y. A rearrangement of the mitochondrial genes of Centipedes (Arthropoda, Myriapoda) with a phylogenetic analysis. Genes 2022, 13, 1787. [Google Scholar] [CrossRef] [PubMed]
- Roehrdanz, R.L.; Degrugillier, M.E.; Black, W.C. Novel rearrangements of arthropod mitochondrial DNA detected with Long-PCR: Applications to arthropod phylogeny and evolution. Mol. Biol. Evol. 2002, 19, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Ding, J.Y.; Lan, H.; Xu, W.; Chen, Y.N.; Wu, H.; Jiang, H.M.; Wang, J.C.; Wu, Y.B.; Liu, H.Y. Two complete mitochondrial genomes in Scolopendra and a comparative analysis of tRNA rearrangements in centipedes. Mol. Biol. Rep. 2022, 49, 6173–6180. [Google Scholar] [CrossRef] [PubMed]
- Nunes, G.L.; Oliveira, R.R.M.; Pires, E.S.; Pietrobon, T.; Prous, X.; Oliveira, G.; Vasconcelos, S. Complete mitochondrial genome of Glomeridesmus spelaeus (Diplopoda, Glomeridesmida), a troglobitic species from iron-ore caves in Eastern Amazon. Mitochondrial DNA Part B 2020, 5, 3290–3291. [Google Scholar] [CrossRef]
- van Knippenberg, P.H.; Formenoy, L.J.; Heus, H.A. Is there a special function for U.G basepairs in ribosomal RNA? Biochim. Biophys. Acta 1990, 1050, 14–17. [Google Scholar] [CrossRef]
- Yuan, M.L.; Zhang, Q.L.; Guo, Z.L.; Wang, J.; Shen, Y.Y. The complete mitochondrial genome of Corizus tetraspilus (Hemiptera: Rhopalidae) and phylogenetic analysis of Pentatomomorpha. PLoS ONE 2017, 10, e0129003. [Google Scholar] [CrossRef]
- Cook, C.E. The complete mitochondrial genome of the stomatopod crustacean Squilla mantis. BMC Genom. 2005, 6, 105. [Google Scholar] [CrossRef]
- Roberti, M.; Polosa, P.L.; Bruni, F.; Musicco, C.; Gadaleta, M.N.; Cantatore, P. DmTTF, a novel mitochondrial transcription termination factor that recognises two sequences of Drosophila melanogaster mitochondrial DNA. Nucleic Acids Res. 2003, 31, 1597–1604. [Google Scholar] [CrossRef]
- Boore, J.L.; Brown, W.M. Big trees from little genomes: Mitochondrial gene order as a phylogenetic tool. Curr. Opin. Genet. Dev. 1998, 8, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Zhu, L.X.; Bai, Y.; Ou, Y.Y.; Wang, C.B. Complete mitochondrial genomes of two flat-backed millipedes by next-generation sequencing (Diplopoda, Polydesmida). Zookeys 2016, 637, 1–20. [Google Scholar] [CrossRef]
- Feng, Z.B.; Wu, Y.F.; Yang, C.; Gu, X.H.; Wilson, J.J.; Li, H.; Cai, W.Z.; Yang, H.L.; Song, F. Evolution of tRNA gene rearrangement in the mitochondrial genome of ichneumonoid wasps (Hymenoptera: Ichneumonoidea). Int. J. Biol. Macromol. 2020, 164, 540–547. [Google Scholar] [CrossRef]
- Blanke, A.; Wesener, T. Revival of forgotten characters and modern imaging techniques help to produce a robust phylogeny of the Diplopoda (Arthropoda, Myriapoda). Arthropod Struct. Dev. 2014, 43, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Dowton, M.; Castro, R.L.; Austin, D.A. Mitochondrial gene rearrangements as phylogenetic characters in the invertebrates: The examination of genome ‘morphology’. Invertebr. Syst. 2002, 16, 345–356. [Google Scholar] [CrossRef]
- Enghoff, H.; Dohle, W.; Blower, J.G. Anamorphosis in millipedes (Diplopoda)—The present state of knowledge with some developmental and phylogenetic considerations. Zool. J. Linn. Soc. 1993, 109, 103–234. [Google Scholar] [CrossRef]
- Joo, S.; Lee, J.; Lee, D.Y.; Xi, H.; Park, J. The complete mitochondrial genome of the millipede Epanerchodus koreanus Verhoeff, 1937 collected in limestone cave of Korea (Polydesmidae: Polydesmida). Mitochondrial DNA Part B 2020, 5, 3845–3847. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.K.; Shear, W.A.; Ye, L.P.; Chen, H.M.; Xie, Z.C. Recovery of the family status of Pericambalidae silvestri, 1909, stat. nov. (Diplopoda: Spirostreptida: Cambalidea), with a revision of the genera and species from China. Invertebr. Syst. 2023, 37, 78–100. [Google Scholar] [CrossRef]
- Mwabvu, T.; Lamb, J.; Slotow, R.; Hamer, M.; Barraclough, D. Is millipede taxonomy based on gonopod morphology too inclusive? Observations on genetic variation and cryptic speciation in Bicoxidens flavicollis (Diplopoda: Spirostreptida: Spirostreptidae). Afr. Invertebr. 2013, 54, 349–356. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Order | Family | Genus | Species | Accession | Length (bp) |
|---|---|---|---|---|---|---|
| Diplopoda | Callipodida | Callipodidae | Abacion | Abacion magnum | JX437062.1 | 15,160 |
| Glomeridesmida | Glomeridesmidae | Glomeridesmus | Glomeridesmus spelaeus | MH590615.1 | 14,863 | |
| Julida | Julidae | Anaulaciulus | Anaulaciulus koreanus | KX096886.1 | 14,916 | |
| Nemasomatidae | Antrokoreana | Antrokoreana gracilipes | DQ344025.1 | 14,747 | ||
| Playtdesmida | Andrognathidae | Brachycybe | Brachycybe lecontii | JX437064.1 | 15,644 | |
| Polydesmida | Paradoxosomatidae | Asiomorpha | Asiomorpha coarctata | KU721885.1 | 15,644 | |
| Polydesmidae | Epanerchodus | Epanerchodus koreanus | MT898420.1 | 15,581 | ||
| Xystodesmidae | Appalachioria | Appalachioria falcifera | JX437063.1 | 15,828 | ||
| Spirobolida | Spirobolidae | Narceus | Narceus annularus | AY055727.1 | 14,868 | |
| Spirobolus | Spirobolus bungii | MT767838.1 | 14,879 | |||
| Pachybolidae | Litostrophus | Litostrophus scaber | OR139892.1 | 15,081 | ||
| Spirostreptida | Odontopygidae | Chaleponcus | Chaleponcus netus | MT394513.1 | 15,093 | |
| Spirostreptidae | Archispirostreptus | Archispirostreptus gigas | MT394525.1 | 15,117 | ||
| Macrolenostreptus | Macrolenostreptus orestes | MT394512.1 | 15,367 | |||
| Pseudotibiozus | Pseudotibiozus cerasopus | MT394506.1 | 15,121 | |||
| Prionopetalum | Prionopetalum kraepelini | MT394524.1 | 15,114 | |||
| Tropostreptus | Tropostreptus austerus | MT394523.1 | 15,261 | |||
| Tropostreptus droides | MT394522.1 | 15,172 | ||||
| Tropostreptus hamatus | MT394521.1 | 15,156 | ||||
| Tropostreptus kipunji | MT394511.1 | 15,170 | ||||
| Tropostreptus microcephalus | MT394516.1 | 15,169 | ||||
| Tropostreptus severus | MT394517.1 | 15,209 | ||||
| Tropostreptus sigmatospinus | MT394526.1 | 15,176 | ||||
| Chilopoda | Lithobiomorpha | Henicopidae | Cermatobius | Cermatobius longicornis | KC155628.1 | 16,833 |
| Gene Name | Location | Length (bp) | Intergenic | Codon | Stand | ||
|---|---|---|---|---|---|---|---|
| From | To | Start | Stop | ||||
| trnK | 1 | 56 | 56 | J | |||
| COX2 | 66 | 744 | 679 | 9 | ATG | T | J |
| COX1 | 745 | 2269 | 1525 | ATA | T | J | |
| trnW | 2287 | 2347 | 61 | 17 | J | ||
| ND2 | 2348 | 3340 | 993 | ATC | TAA | J | |
| trnM | 3341 | 3406 | 66 | J | |||
| trnI | 3407 | 3474 | 68 | J | |||
| D-loop | 3475 | 3933 | 459 | / | |||
| trnL1 | 3934 | 3995 | 62 | N | |||
| trnP | 4007 | 4067 | 61 | 11 | N | ||
| ND4L | 4068 | 4352 | 285 | TGA | TAG | N | |
| ND4 | 4346 | 5683 | 1338 | −6 | ATG | TAA | N |
| trnQ | 5686 | 5751 | 66 | 2 | N | ||
| trnC | 5834 | 5902 | 69 | 82 | N | ||
| rrnS | 6020 | 6776 | 757 | 117 | N | ||
| trnV | 6777 | 6830 | 54 | N | |||
| rrnL | 7000 | 8030 | 1031 | 169 | N | ||
| trnL2 | 8097 | 8161 | 65 | 66 | N | ||
| ND1 | 8162 | 9087 | 926 | ATT | TA | N | |
| trnH | 9088 | 9152 | 65 | N | |||
| ND5 | 9192 | 10,854 | 1663 | 39 | ATA | T | N |
| trnF | 10,855 | 10,912 | 58 | N | |||
| trnY | 10,913 | 10,974 | 62 | N | |||
| trnT | 10,983 | 11,045 | 63 | 8 | J | ||
| trnS2 | 11,048 | 11,114 | 67 | 2 | J | ||
| CYTB | 11,118 | 12,219 | 1102 | 3 | ATT | T | J |
| ND6 | 12,238 | 12,676 | 439 | 18 | ATA | T | J |
| trnE | 12,695 | 12,757 | 63 | 18 | J | ||
| trnS1 | 12,758 | 12,817 | 60 | J | |||
| trnN | 12,818 | 12,884 | 67 | J | |||
| trnR | 12,886 | 12,948 | 63 | 1 | J | ||
| trnA | 12,949 | 13,007 | 59 | J | |||
| ND3 | 13,008 | 13,355 | 348 | ATT | TAG | J | |
| trnG | 13,356 | 13,417 | 62 | J | |||
| COX3 | 13,418 | 14,198 | 781 | ATG | T | J | |
| ATP6 | 14,199 | 14,871 | 673 | ATG | T | J | |
| ATP8 | 14,865 | 15,020 | 156 | −6 | ATT | TAA | J |
| trnD | 15,021 | 15,081 | 61 | J | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, G.; Gao, M.; Chen, Y.; Wang, Y.; Gan, T.; Zhu, F.; Liu, H. The First Complete Mitochondrial Genome of the Genus Litostrophus: Insights into the Rearrangement and Evolution of Mitochondrial Genomes in Diplopoda. Genes 2024, 15, 254. https://doi.org/10.3390/genes15020254
Zhang G, Gao M, Chen Y, Wang Y, Gan T, Zhu F, Liu H. The First Complete Mitochondrial Genome of the Genus Litostrophus: Insights into the Rearrangement and Evolution of Mitochondrial Genomes in Diplopoda. Genes. 2024; 15(2):254. https://doi.org/10.3390/genes15020254
Chicago/Turabian StyleZhang, Gaoji, Ming Gao, Yukun Chen, Yinuo Wang, Tianyi Gan, Fuyuan Zhu, and Hongyi Liu. 2024. "The First Complete Mitochondrial Genome of the Genus Litostrophus: Insights into the Rearrangement and Evolution of Mitochondrial Genomes in Diplopoda" Genes 15, no. 2: 254. https://doi.org/10.3390/genes15020254