
A Homozygous MAN2B1 Missense Mutation in a Doberman Pinscher Dog with Neurodegeneration, Cytoplasmic Vacuoles, Autofluorescent Storage Granules, and an α-Mannosidase Deficiency
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subject Dog
2.2. Tissue Collection, Processing, and Microscopic Analyses
2.3. Molecular Genetic Analysis
2.4. Mannosidase Enzyme Activity Assays
3. Results
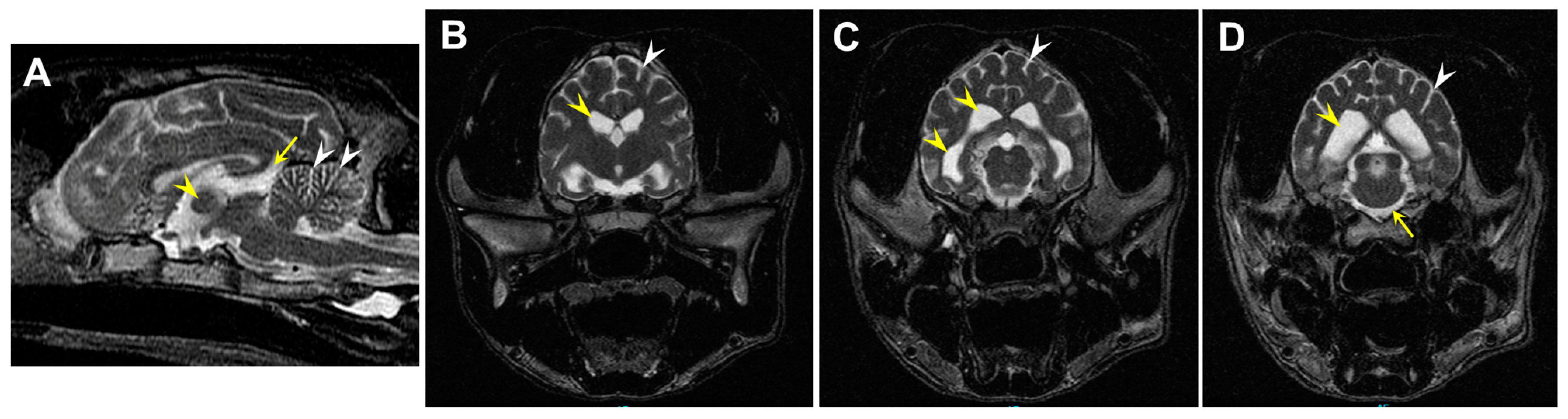
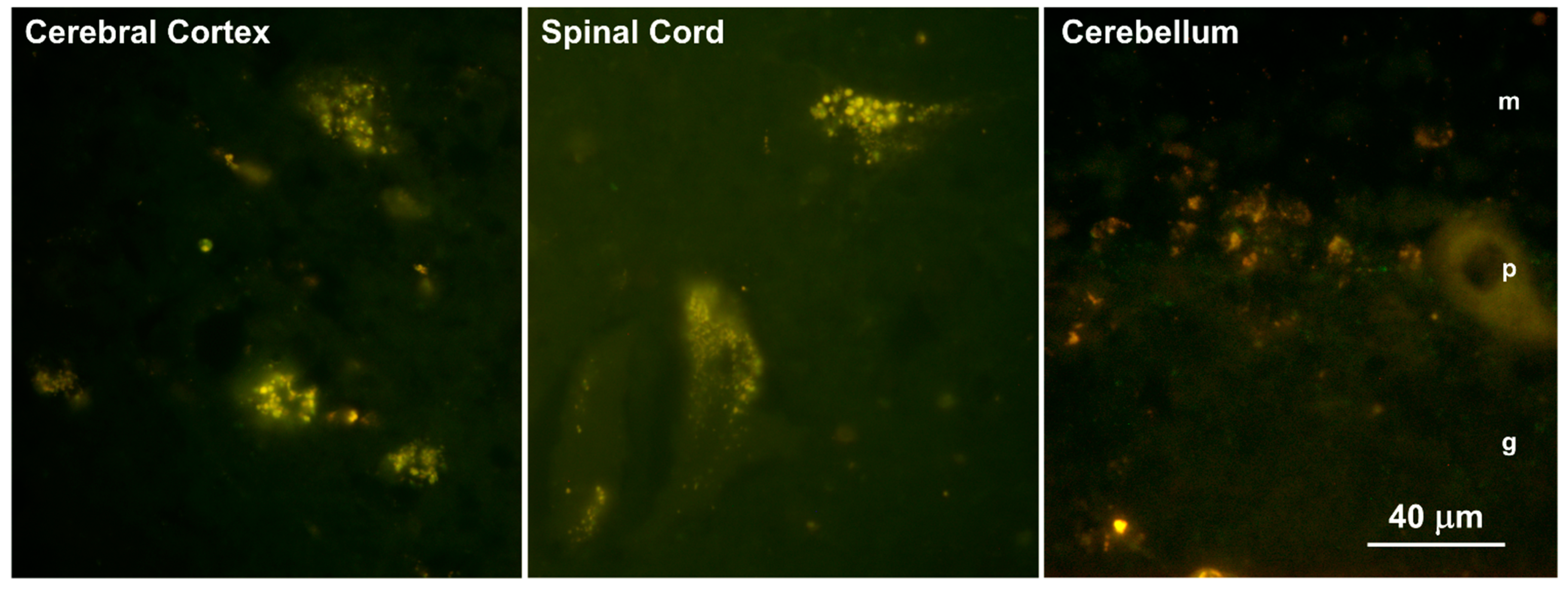
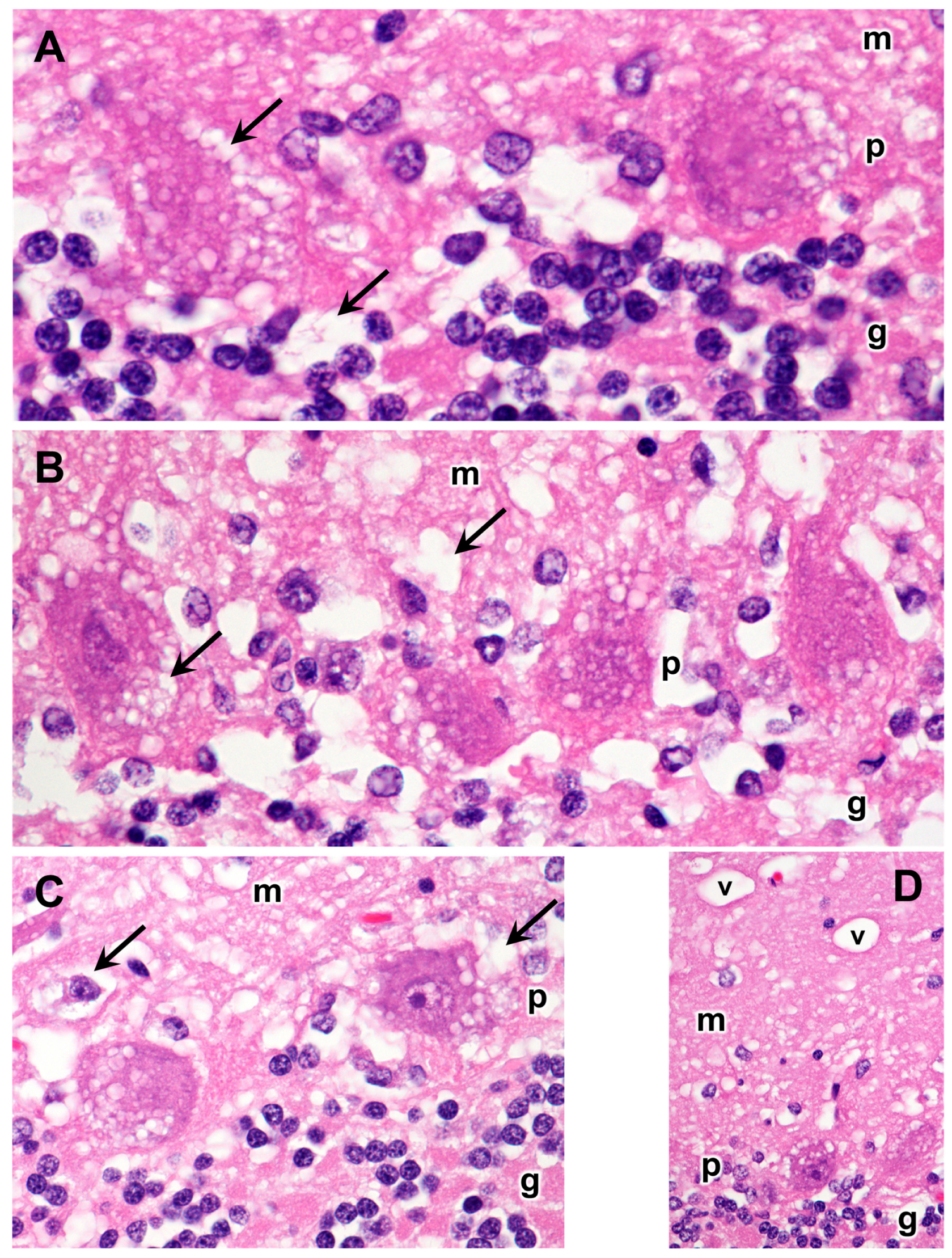
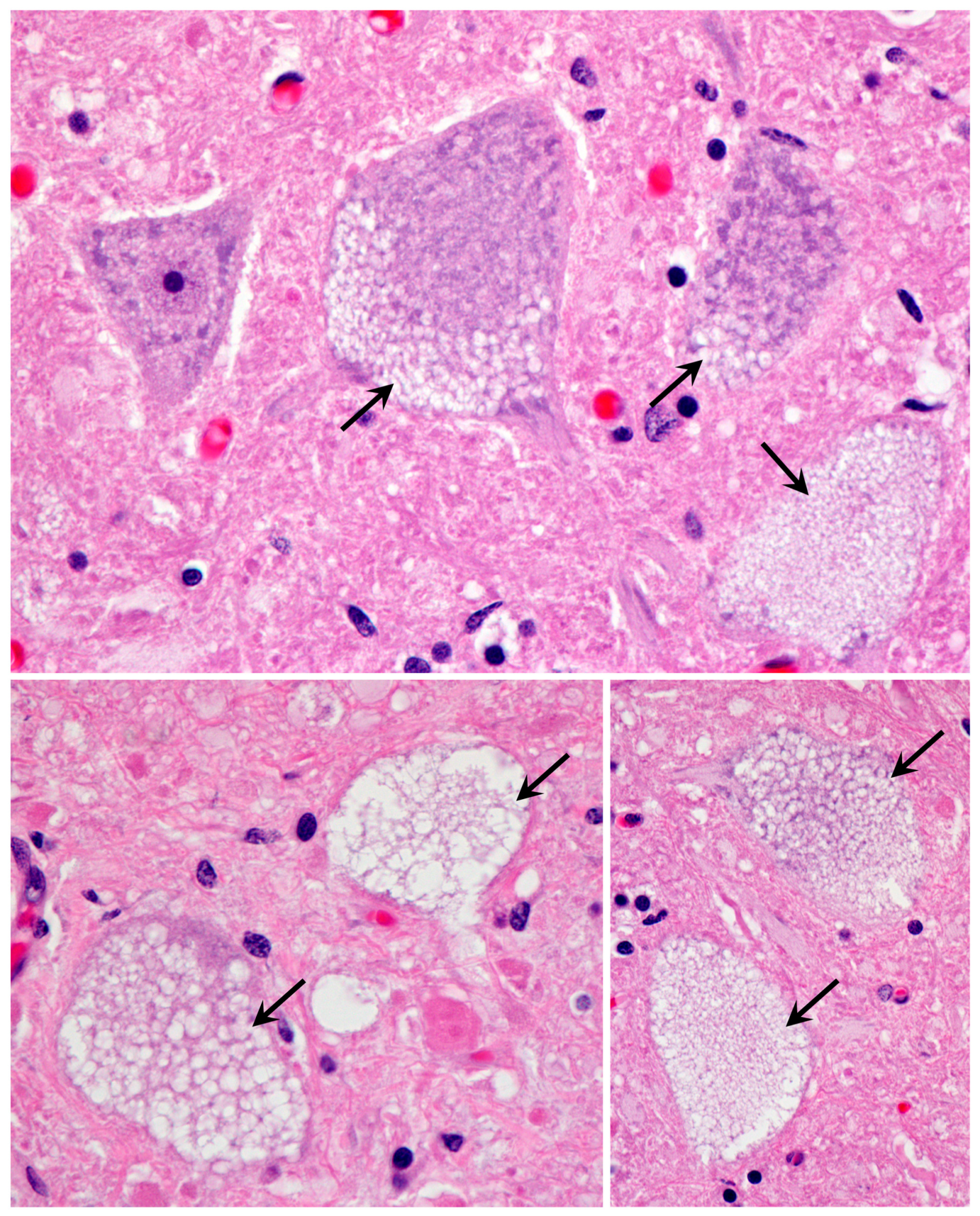
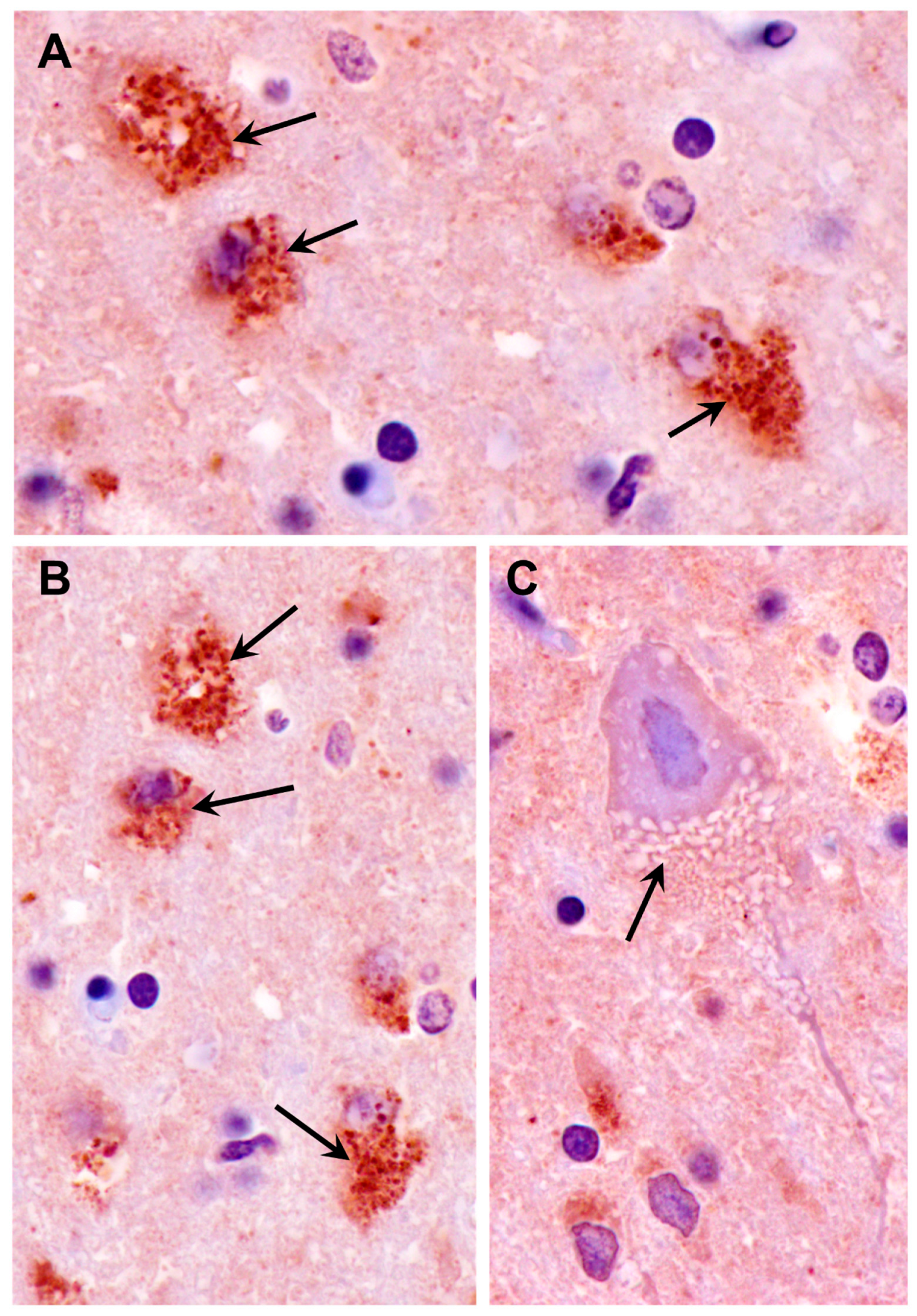
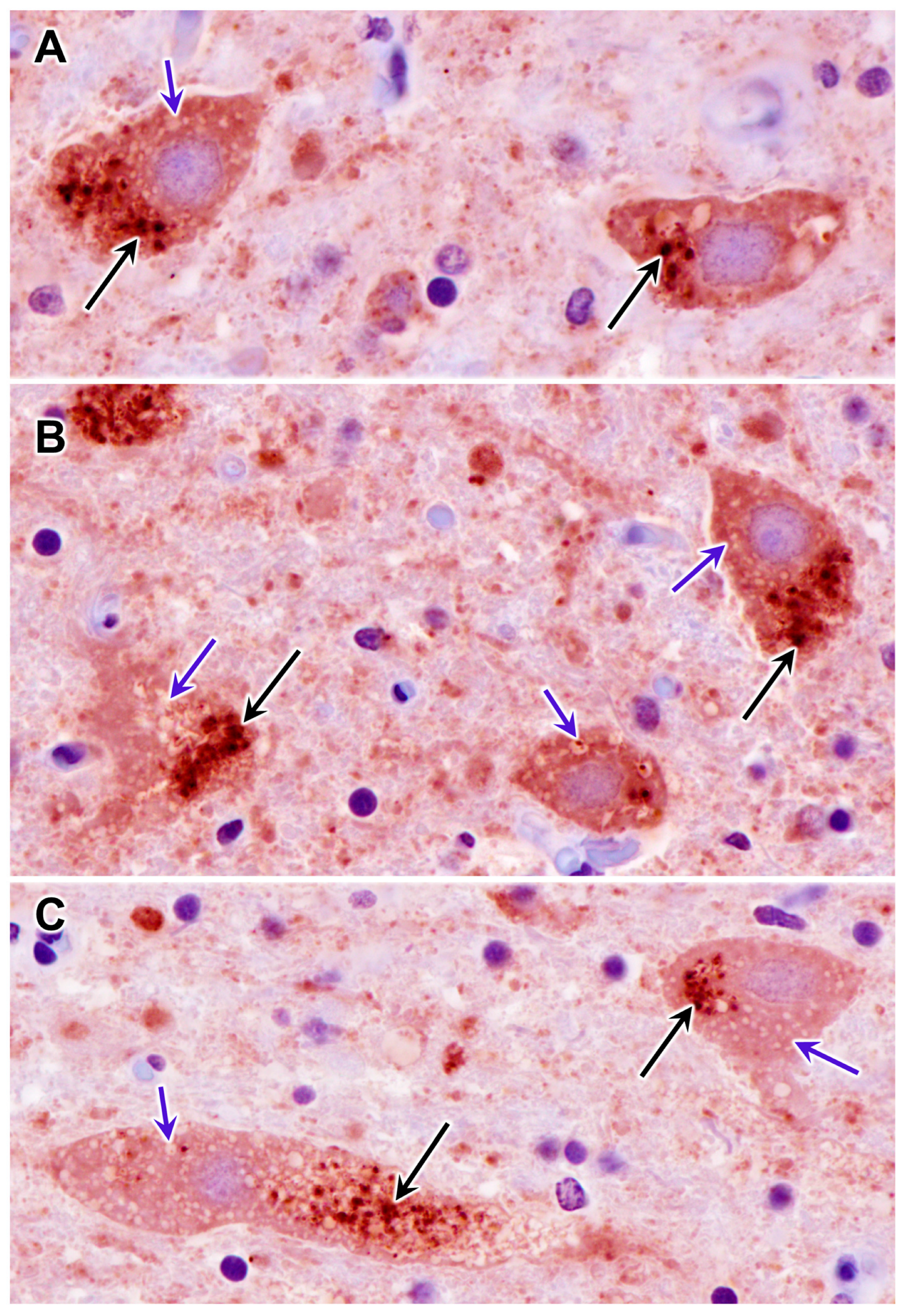
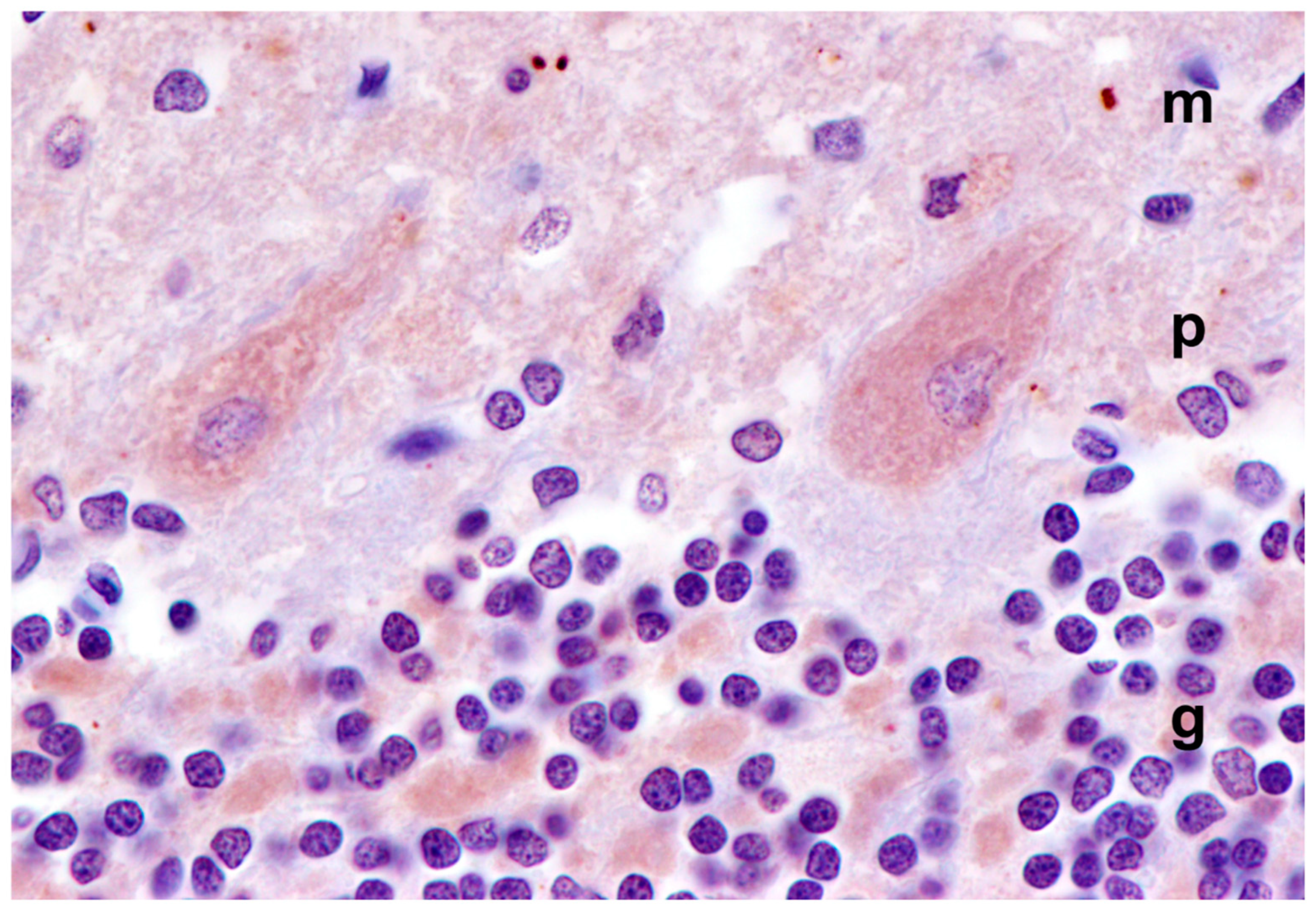
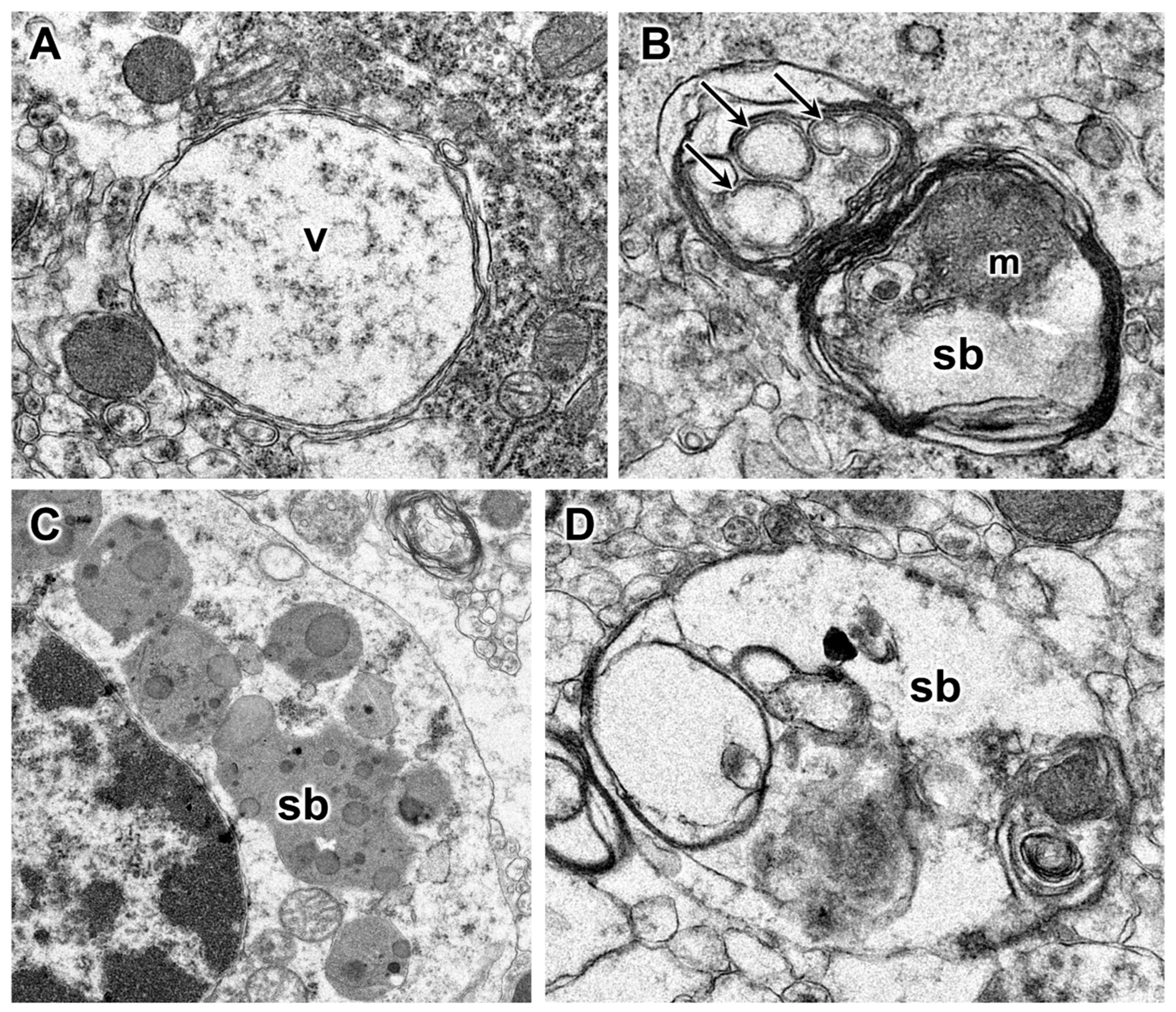
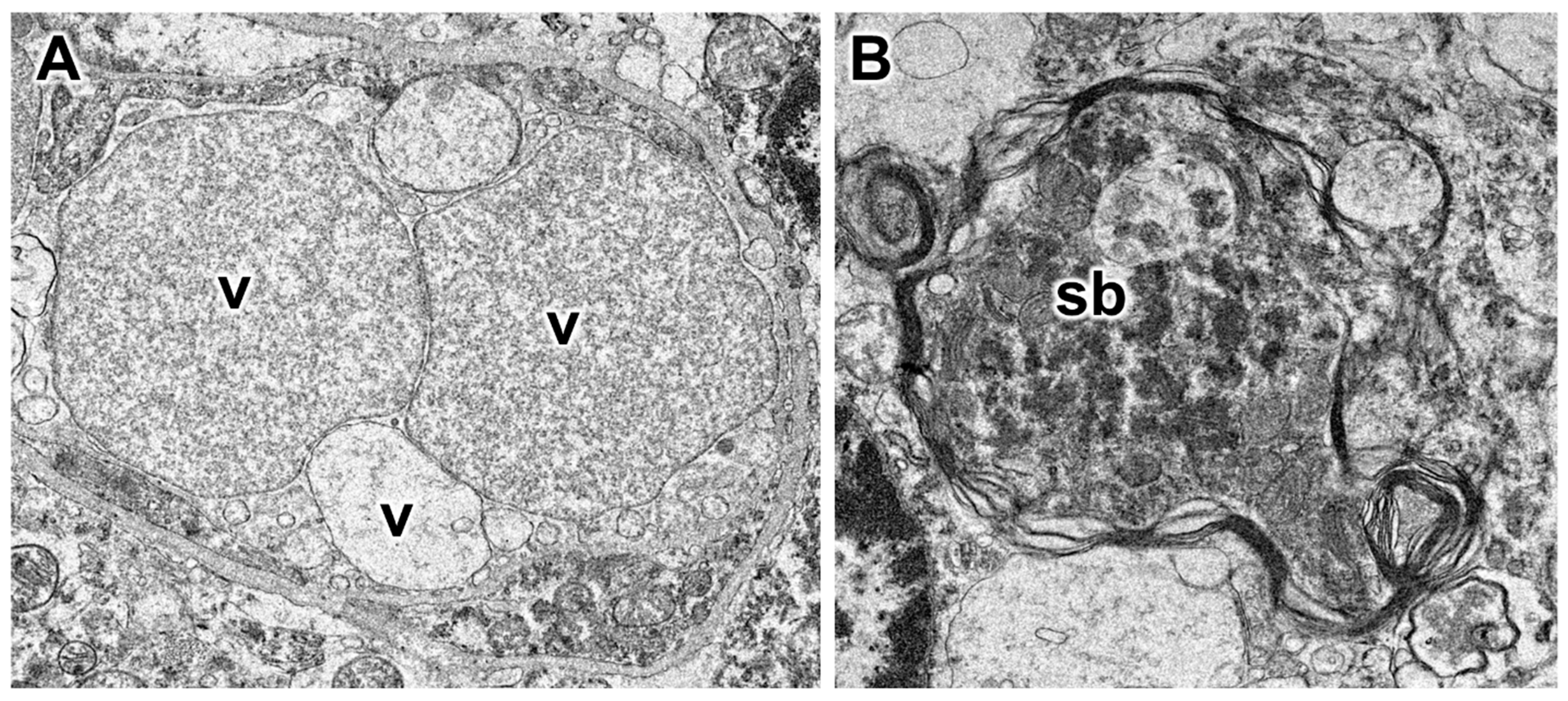
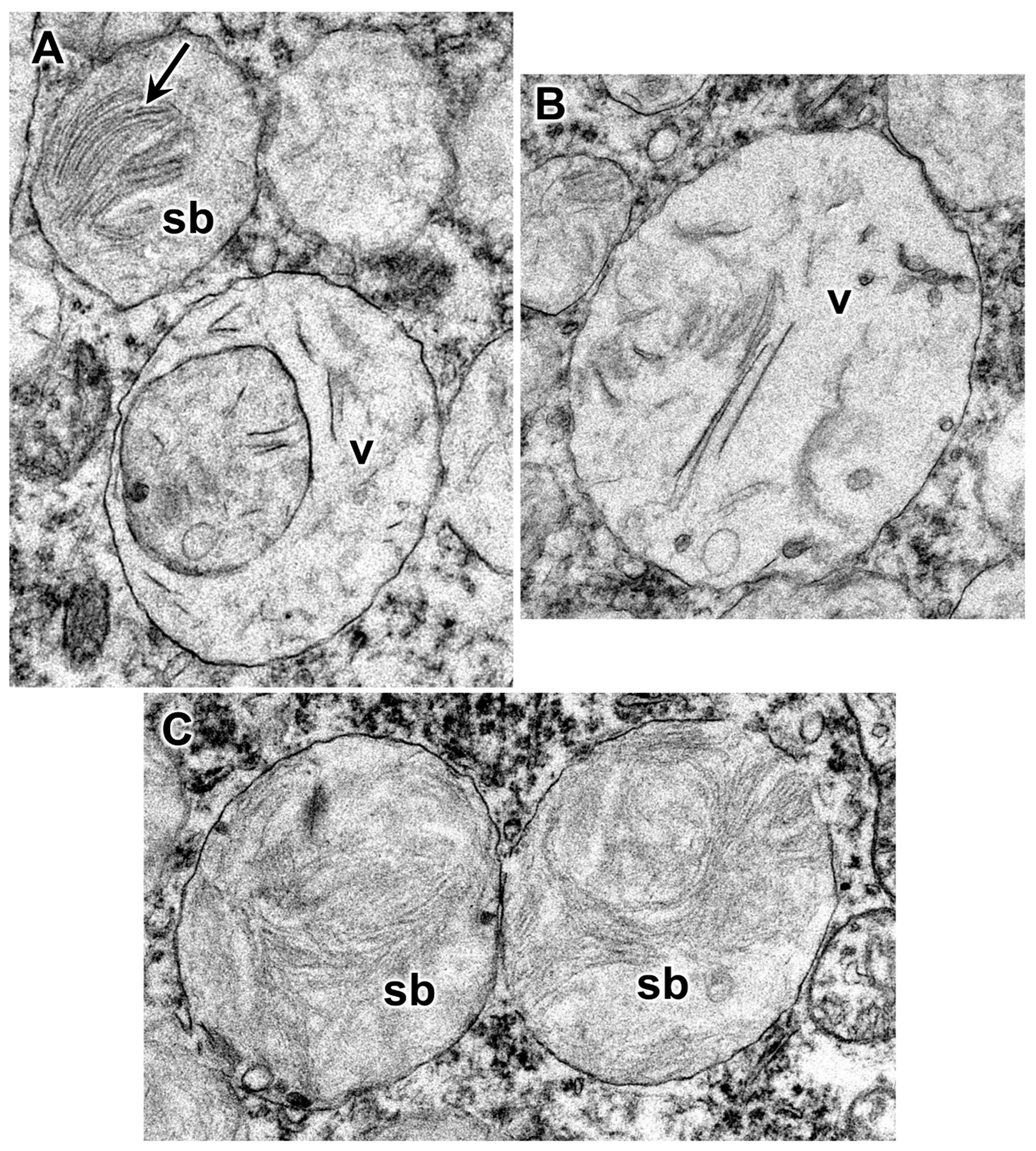
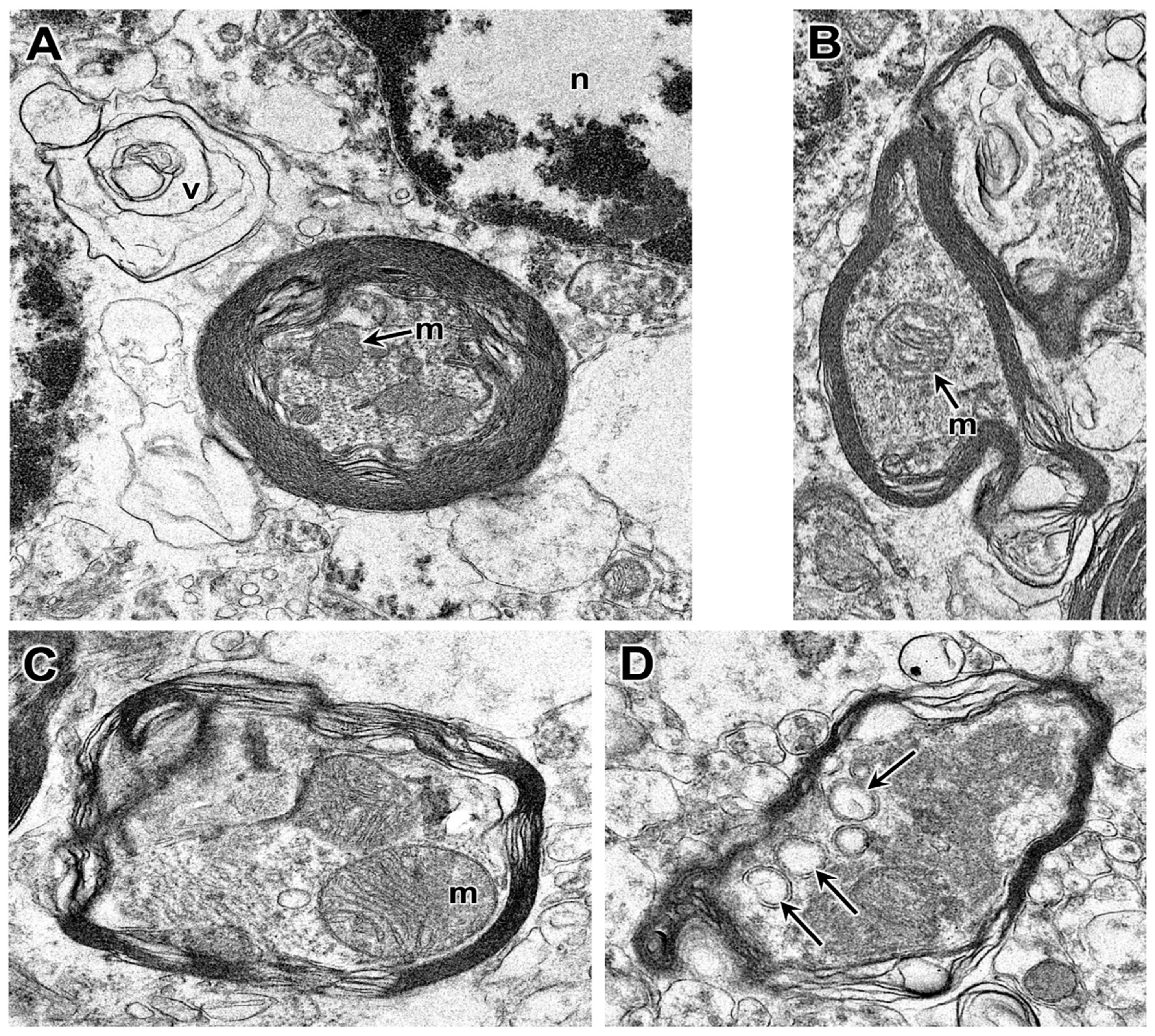
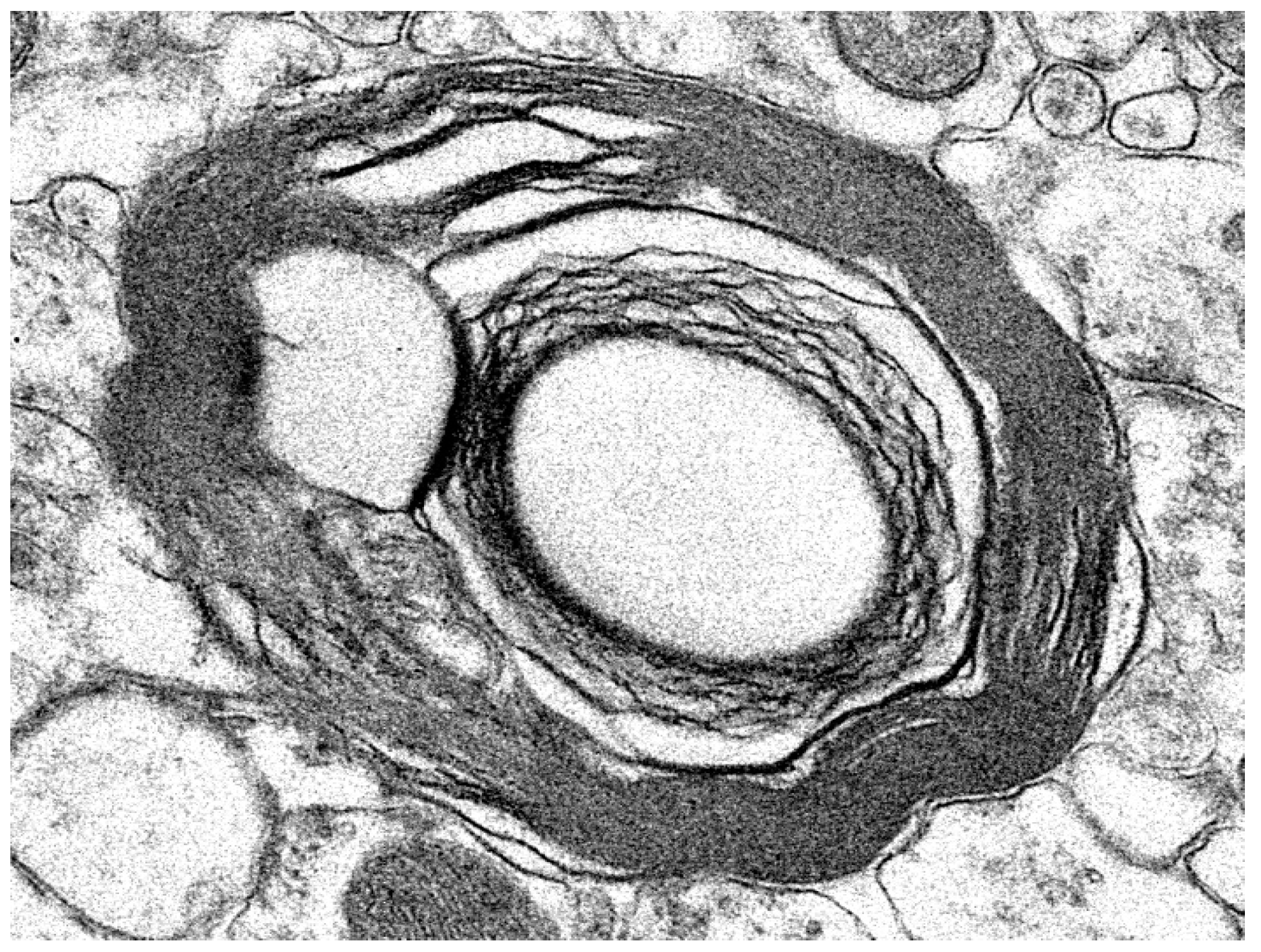
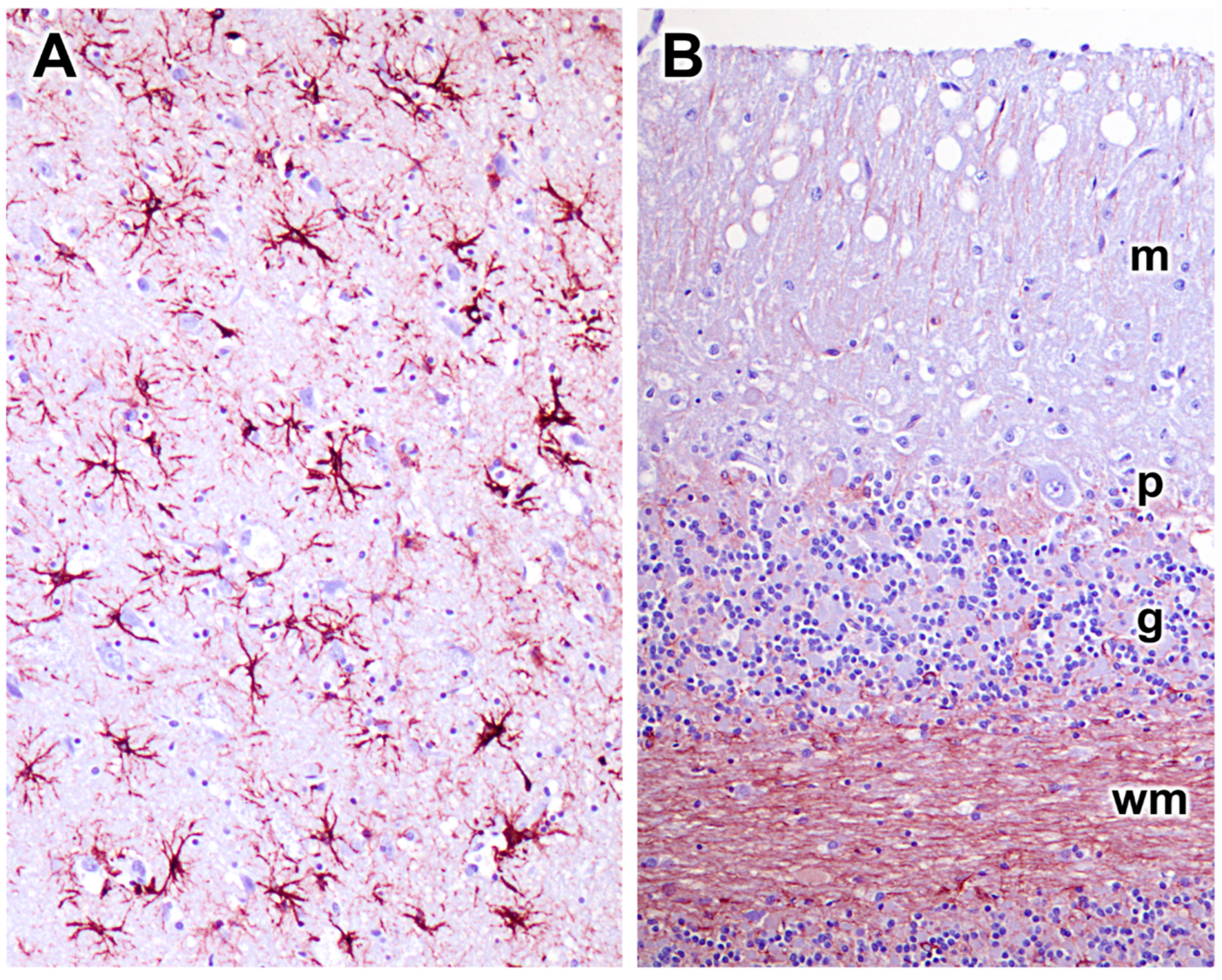
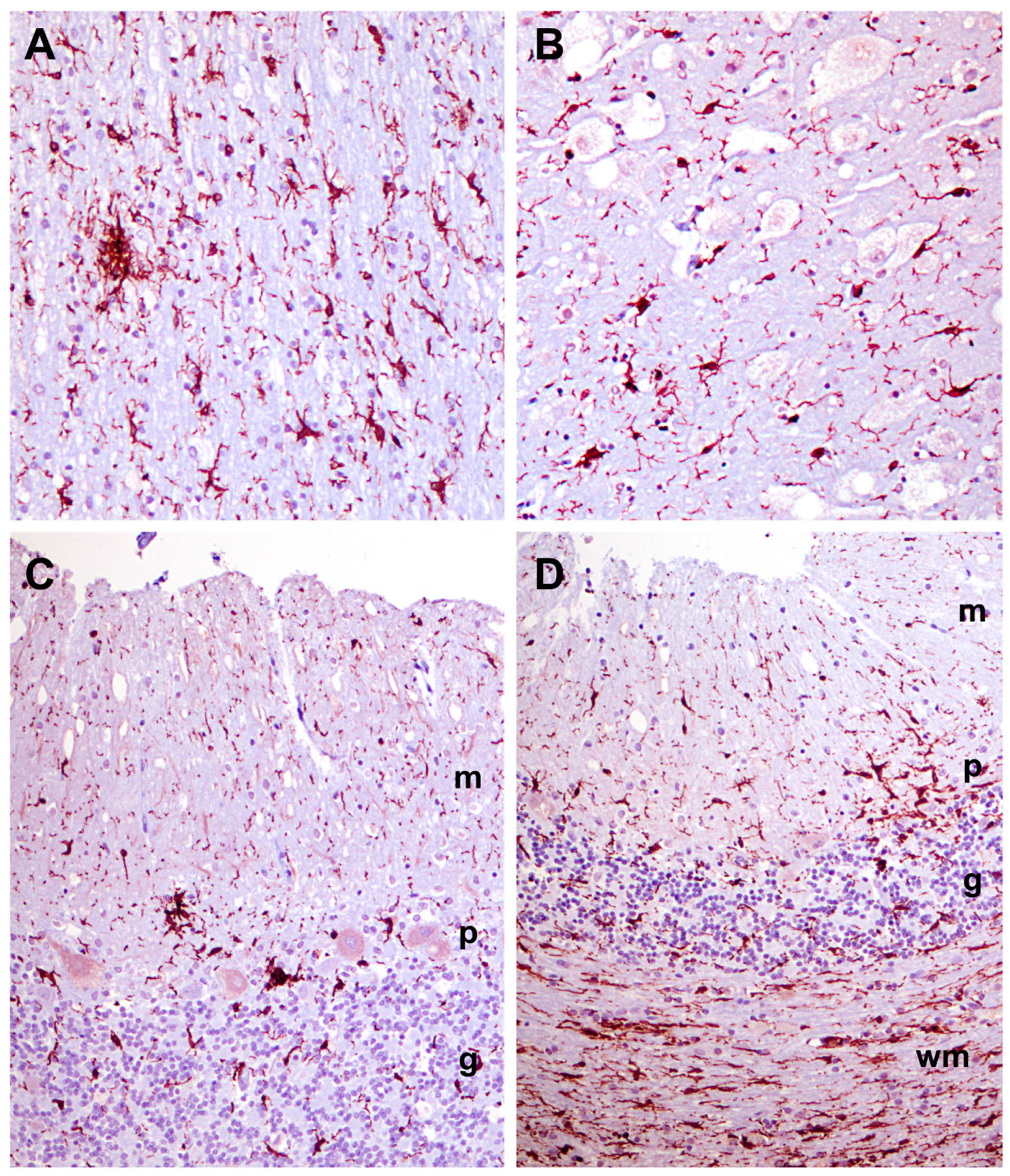
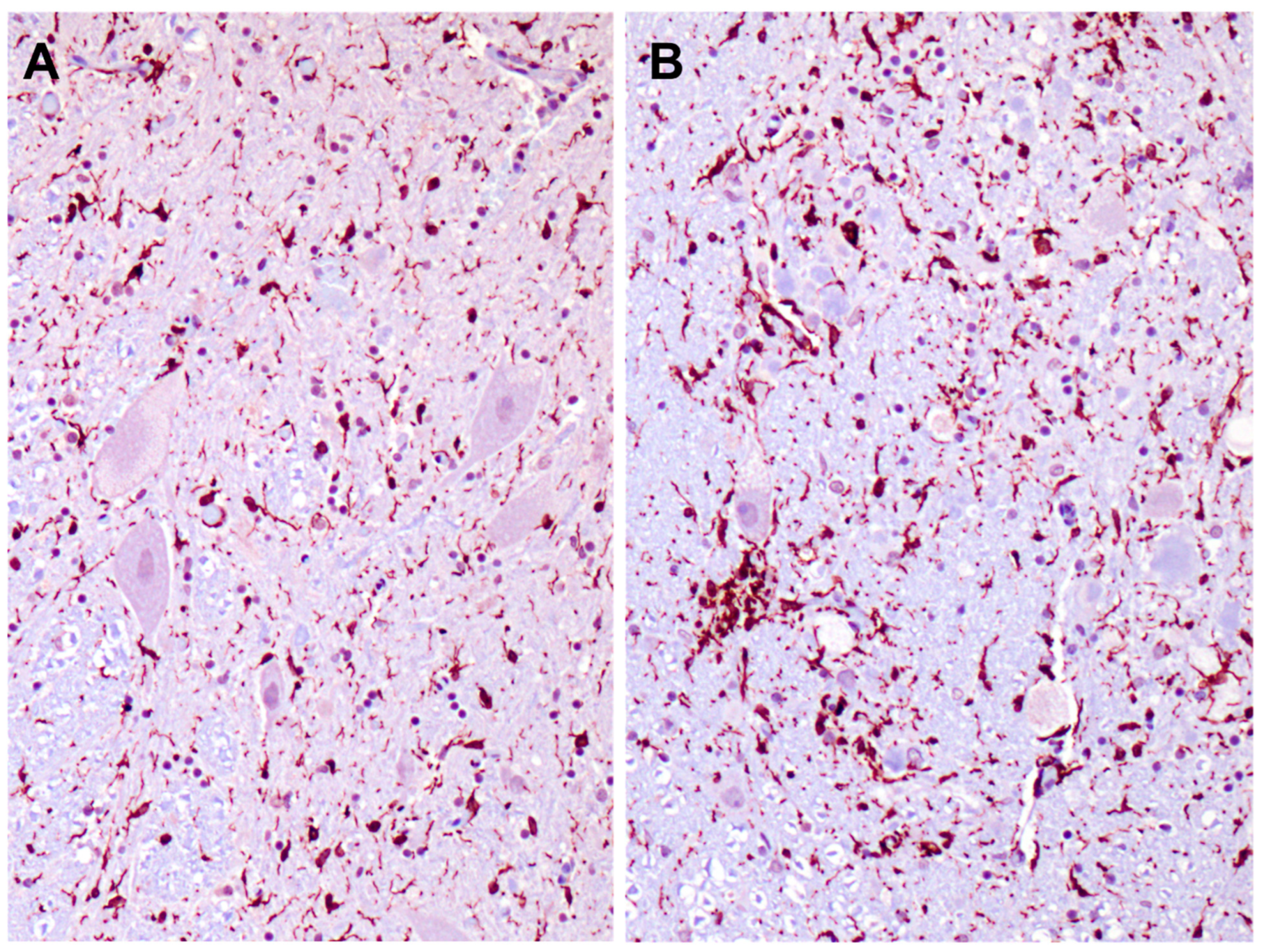
3.1. Disease Pathology
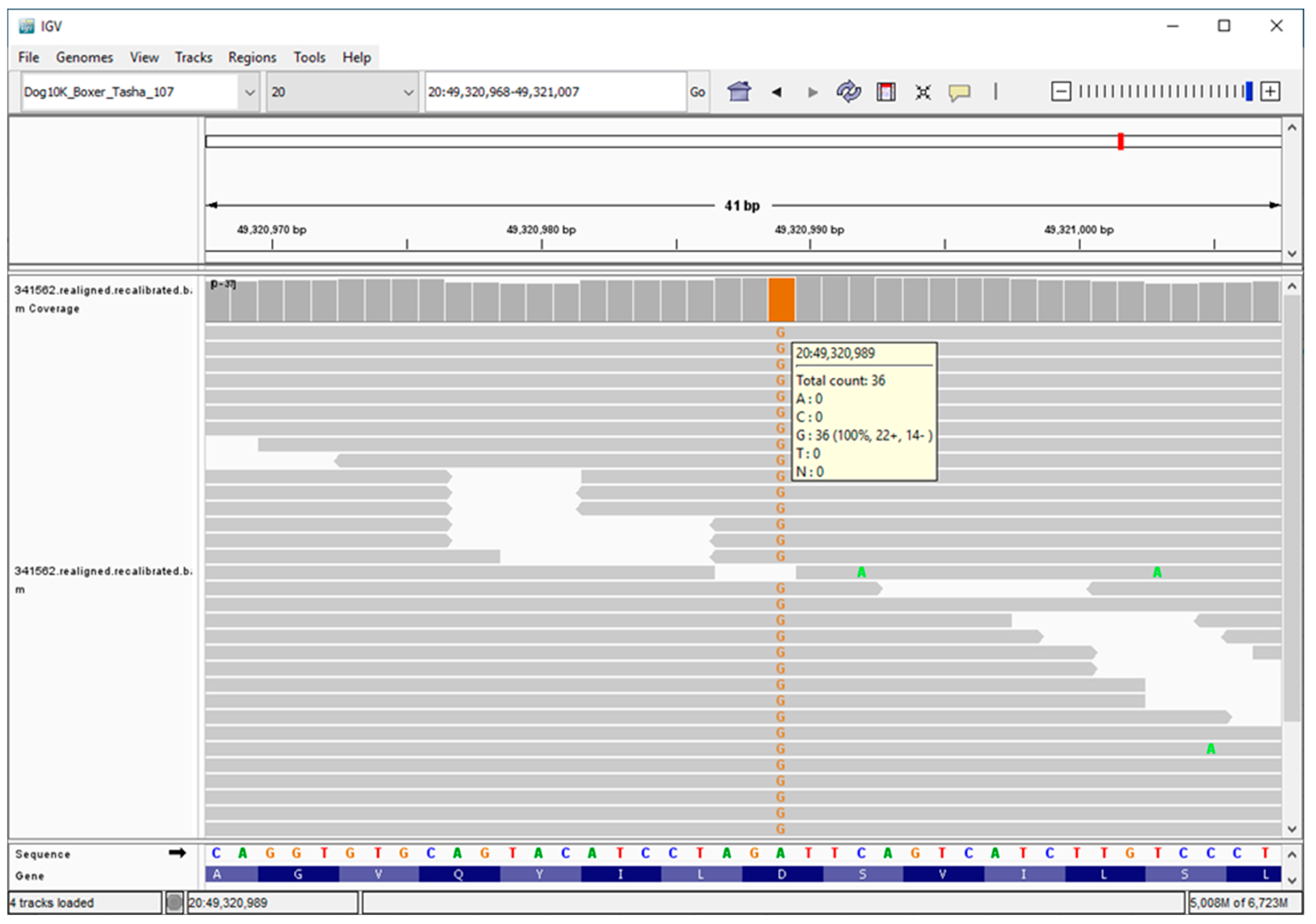
3.2. Molecular Genetics
3.3. Enzymology
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal Storage Diseases. Nat. Rev. Dis. Prim. 2018, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Sumathipala, D.; Stromme, P.; Gilissen, C.; Corominas, J.; Frengen, E.; Misceo, D. TBCK Encephaloneuropathy With Abnormal Lysosomal Storage: Use of a Structural Variant Bioinformatics Pipeline on Whole-Genome Sequencing Data Unravels a 20-Year-Old Clinical Mystery. Pediatr. Neurol. 2019, 96, 74–75. [Google Scholar] [CrossRef] [PubMed]
- Gurda, B.L.; Vite, C.H. Large Animal Models Contribute to the Development of Therapies for Central and Peripheral Nervous System Dysfunction in Patients with Lysosomal Storage Diseases. Hum. Mol. Genet. 2019, 28, R119–R131. [Google Scholar] [CrossRef]
- Nicholas, F.W. Online Mendelian Inheritance in Animals (OMIA): A Record of Advances in Animal Genet.ics, Freely Available on the Internet for 25 Years. Anim. Genet. 2021, 52, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Margolis, C.; Lin, G.; Buza, E.L.; Quick, S.; Raj, K.; Han, R.; Giger, U. Mucopolysaccharidosis Type VI in a Great Dane Caused by a Nonsense Mutation in the ARSB Gene. Vet. Pathol. 2018, 55, 286–293. [Google Scholar] [CrossRef]
- Schmutz, I.; Jagannathan, V.; Bartenschlager, F.; Stein, V.M.; Gruber, A.D.; Leeb, T.; Katz, M.L. ATP13A2 Missense Variant in Australian Cattle Dogs with Late Onset Neuronal Ceroid Lipofuscinosis. Mol. Genet. Metab. 2019, 127, 95–106. [Google Scholar] [CrossRef]
- Farias, F.H.G.; Zeng, R.; Johnson, G.S.; Wininger, F.A.; Taylor, J.F.; Schnabel, R.D.; McKay, S.D.; Sanders, D.N.; Lohi, H.; Seppälä, E.H.; et al. A Truncating Mutation in ATP13A2 Is Responsible for Adult-Onset Neuronal Ceroid Lipofuscinosis in Tibetan Terriers. Neurobiol. Dis. 2011, 42, 468–474. [Google Scholar] [CrossRef]
- Meiman, E.J.; Kick, G.R.; Jensen, C.A.; Coates, J.R.; Katz, M.L. Characterization of Neurological Disease Progression in a Canine Model of CLN5 Neuronal Ceroid Lipofuscinosis. Dev. Neurobiol. 2022, 82, 326–344. [Google Scholar] [CrossRef]
- Gilliam, D.; Kolicheski, A.; Johnson, G.S.; Mhlanga-Mutangadura, T.; Taylor, J.F.; Schnabel, R.D.; Katz, M.L. Golden Retriever Dogs with Neuronal Ceroid Lipofuscinosis Have a Two-Base-Pair Deletion and Frameshift in CLN5. Mol. Genet. Metab. 2015, 115, 101–109. [Google Scholar] [CrossRef]
- Kolicheski, A.; Johnson, G.S.; O’Brien, D.P.; Mhlanga-Mutangadura, T.; Gilliam, D.; Guo, J.; Anderson-Sieg, T.D.; Schnabel, R.D.; Taylor, J.F.; Lebowitz, A.; et al. Australian Cattle Dogs with Neuronal Ceroid Lipofuscinosis Are Homozygous for a CLN5 Nonsense Mutation Previously Identified in Border Collies. J. Vet. Intern. Med. 2016, 30, 1149–1158. [Google Scholar] [CrossRef]
- Kick, G.R.; Meiman, E.J.; Sabol, J.C.; Whiting, R.E.H.; Ota-Kuroki, J.; Castaner, L.J.; Jensen, C.A.; Katz, M.L. Visual System Pathol;ogy in a Canine Model of CLN5 Neuronal Ceroid Lipofuscinosis. Exp. Eye Res. 2021, 210, 108686. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.L.; Farias, F.H.; Sanders, D.N.; Zeng, R.; Khan, S.; Johnson, G.S.; O’Brien, D.P. A Missense Mutation in Canine CLN6 in an Australian Shepherd with Neuronal Ceroid Lipofuscinosis. J. Biomed. Biotechnol. 2011, 198042. [Google Scholar] [CrossRef]
- Katz, M.L.; Khan, S.; Awano, T.; Shahid, S.A.; Siakotos, A.N.; Johnson, G.S. A Mutation in the CLN8 Gene in English Setter Dogs with Neuronal Ceroid-Lipofuscinosis. Biochem. Biophys. Res. Commun. 2005, 327, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Johnson, G.S.; Cook, J.; Harris, O.K.; Mhlanga-Mutangadura, T.; Schnabel, R.D.; Jensen, C.A.; Katz, M.L. Neuronal Ceroid Lipofuscinosis in a German Shorthaired Pointer Associated with a Previously Reported CLN8 Nonsense Variant. Mol. Genet. Metab. Rep. 2019, 21, 100521. [Google Scholar] [CrossRef]
- Guo, J.; Johnson, G.S.; Brown, H.A.; Provencher, M.L.; da Costa, R.C.; Mhlanga-Mutangadura, T.; Taylor, J.F.; Schnabel, R.D.; O’Brien, D.P.; Katz, M.L. A CLN8 Nonsense Mutation in the Whole Genome Sequence of a Mixed Breed Dog with Neuronal Ceroid Lipofuscinosis and Australian Shepherd Ancestry. Mol. Genet. Metab. 2014, 112, 302–309. [Google Scholar] [CrossRef]
- Raj, K.; Berman-Booty, L.; Foureman, P.; Giger, U. ARSB Gene Variants Causing Mucopolysaccharidosis VI in Miniature Pinscher and Miniature Schnauzer Dogs. Anim. Genet. 2020, 51, 982–986. [Google Scholar] [CrossRef]
- Mansour, T.A.; Woolard, K.D.; Vernau, K.L.; Ancona, D.M.; Thomasy, S.M.; Sebbag, L.; Moore, B.A.; Knipe, M.F.; Seada, H.A.; Cowan, T.M.; et al. Whole Genome Sequencing for Mutation Discovery in a Single Case of Lysosomal Storage Disease (MPS Type 1) in the Dog. Sci. Rep. 2020, 10, 6558. [Google Scholar] [CrossRef]
- Faller, K.M.E.; Ridyard, A.E.; Gutierrez-Quintana, R.; Rupp, A.; Kun-Rodrigues, C.; Orme, T.; Tylee, K.L.; Church, H.J.; Guerreiro, R.; Bras, J. A Deletion of IDUA Exon 10 in a Family of Golden Retriever Dogs with an Attenuated Form of Mucopolysaccharidosis Type I. J. Vet. Intern. Med. 2020, 34, 1813–1824. [Google Scholar] [CrossRef]
- Menon, K.P.; Tieu, P.T.; Neufeld, E.F. Architecture of the Canine IDUA Gene and Mutation Underlying Canine Mucopolysaccharidosis I. Genomics 1992, 14, 763–768. [Google Scholar] [CrossRef]
- Sanders, D.N.; Zeng, R.; Wenger, D.A.; Johnson, G.S.; Johnson, G.C.; Decker, J.E.; Katz, M.L.; Platt, S.R.; O’Brien, D.P. GM2 Gangliosidosis Associated with a HEXA Missense Mutation in Japanese Chin Dogs: A Potential Model for Tay Sachs Disease. Mol. Genet. Metab. 2013, 108, 70–75. [Google Scholar] [CrossRef]
- Ashwini, A.; D’Angelo, A.; Yamato, O.; Giordano, C.; Cagnotti, G.; Harcourt-Brown, T.; Mhlanga-Mutangadura, T.; Guo, J.; Johnson, G.S.; Katz, M.L. Neuronal Ceroid Lipofuscinosis Associated with an MFSD8 Mutation in Chihuahuas. Mol. Genet. Metab. 2016, 118, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; O’Brien, D.P.; Mhlanga-Mutangadura, T.; Olby, N.J.; Taylor, J.F.; Schnabel, R.D.; Katz, M.L.; Johnson, G.S. A Rare Homozygous MFSD8 Single-Base-Pair Deletion and Frameshift in the Whole Genome Sequence of a Chinese Crested Dog with Neuronal Ceroid Lipofuscinosis. BMC Vet. Res. 2014, 10, 960. [Google Scholar] [CrossRef] [PubMed]
- Raj, K.; Ellinwood, N.M.; Giger, U. An Exonic Insertion in the NAGLU Gene Causing Mucopolysaccharidosis IIIB in Schipperke Dogs. Sci. Rep. 2020, 10, 3170. [Google Scholar] [CrossRef]
- Bolfa, P.; Wang, P.; Nair, R.; Rajeev, S.; Armien, A.G.; Henthorn, P.S.; Wood, T.; Thrall, M.A.; Giger, U. Hereditary β-Mannosidosis in a Dog: Clinicopathological and Molecular Genet.ic Characterization. Mol. Genet. Metab. 2019, 128, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Jolly, R.D.; Dittmer, K.E.; Garrick, D.J.; Chernyavtseva, A.; Hemsley, K.M.; King, B.; Fietz, M.; Shackleton, N.M.; Fairley, R.; Wylie, K. β-Mannosidosis in German Shepherd Dogs. Vet. Pathol. 2019, 56, 743–748. [Google Scholar] [CrossRef]
- Kolicheski, A.; Barnes Heller, H.L.; Arnold, S.; Schnabel, R.D.; Taylor, J.F.; Knox, C.A.; Mhlanga-Mutangadura, T.; O’Brien, D.P.; Johnson, G.S.; Dreyfus, J.; et al. Homozygous PPT1 Splice Donor Mutation in a Cane Corso Dog With Neuronal Ceroid Lipofuscinosis. J. Vet. Intern. Med. 2017, 31, 149–157. [Google Scholar] [CrossRef]
- Sanders, D.N.; Farias, F.H.; Johnson, G.S.; Chiang, V.; Cook, J.R.; O’Brien, D.P.; Hofmann, S.L.; Lu, J.-Y.; Katz, M.L. A Mutation in Canine PPT1 Causes Early Onset Neuronal Ceroid Lipofuscinosis in a Dachshund. Mol. Genet. Metab. 2010, 100, 349–356. [Google Scholar] [CrossRef]
- Awano, T.; Katz, M.L.; O’Brien, D.P.; Sohar, I.; Lobel, P.; Coates, J.R.; Khan, S.; Johnson, G.C.; Giger, U.; Johnson, G.S. A Frame Shift Mutation in Canine TPP1 (the Ortholog of Human CLN2) in a Juvenile Dachshund with Neuronal Ceroid Lipofuscinosis. Mol. Genet. Metab. 2006, 89, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Yogalingam, G.; Pollard, T.; Gliddon, B.; Jolly, R.D.; Hopwood, J.J. Identification of a Mutation Causing Mucopolysaccharidosis Type IIIA in New Zealand Huntaway Dogs. Genomics 2002, 79, 150–153. [Google Scholar] [CrossRef]
- Abitbol, M.; Thibaud, J.-L.; Olby, N.J.; Hitte, C.; Puech, J.-P.; Maurer, M.; Pilot-Storck, F.; Hedan, B.; Dreano, S.; Brahimi, S.; et al. A Canine Arylsulfatase G (ARSG) Mutation Leading to a Sulfatase Deficiency Is Associated with Neuronal Ceroid Lipofuscinosis. Proc. Natl. Acad. Sci. USA 2010, 107, 14775–14780. [Google Scholar] [CrossRef]
- Bullock, G.; Johnson, G.S.; Mhlanga-Mutangadura, T.; Petesch, S.C.; Thompson, S.; Goebbels, S.; Katz, M.L. Lysosomal Storage Disease Associated with a CNP Sequence Variant in Dalmatian Dogs. Gene 2022, 830, 146513. [Google Scholar] [CrossRef]
- Khateb, S.; Kowalewski, B.; Bedoni, N.; Damme, M.; Pollack, N.; Saada, A.; Obolensky, A.; Ben-Yosef, T.; Gross, M.; Dierks, T.; et al. A Homozygous Founder Missense Variant in Arylsulfatase G Abolishes Its Enzymatic Activity Causing Atypical Usher Syndrome in Humans. Genet. Med. 2018, 20, 1004–1012. [Google Scholar] [CrossRef]
- Peter, V.G.; Quinodoz, M.; Sadio, S.; Held, S.; Rodrigues, M.; Soares, M.; Sousa, A.B.; Santos, L.C.; Damme, M.; Rivolta, C. New Clinical and Molecular Evidence Linking Mutations in ARSG to Usher Syndrome Type IV. Hum. Mutat. 2021, 42, 261–271. [Google Scholar] [CrossRef]
- Velde, H.M.; Reurink, J.; Held, S.; Li, C.H.Z.; Yzer, S.; Oostrik, J.; Weeda, J.; Haer-Wigman, L.; Yntema, H.G.; Roosing, S.; et al. Usher Syndrome Type IV: Clinically and Molecularly Confirmed by Novel ARSG Variants. Hum. Genet. 2022, 141, 1723–1738. [Google Scholar] [CrossRef]
- Al-Abdi, L.; Al Murshedi, F.; Elmanzalawy, A.; Al Habsi, A.; Helaby, R.; Ganesh, A.; Ibrahim, N.; Patel, N.; Alkuraya, F.S. CNP Deficiency Causes Severe Hypomyelinating Leukodystrophy in Humans. Hum. Genet. 2020, 139, 615–622. [Google Scholar] [CrossRef]
- Abad-Morales, V.; Navarro, R.; Burés-Jelstrup, A.; Pomares, E. Identification of a Novel Homozygous ARSG Mutation as the Second Cause of Usher Syndrome Type 4. Am. J. Ophthalmol. Case Rep. 2020, 19, 100736. [Google Scholar] [CrossRef]
- Berg, T.; Tollersrud, O.K.; Walkley, S.U.; Siegel, D.; Nilssen, O. Purification of Feline Lysosomal α-Mannosidase, Determination of Its CDNA Sequence and Identification of a Mutation Causing α-Mannosidosis in Persian Cats. Biochem. J. 1997, 328 Pt 3, 863–870. [Google Scholar] [CrossRef]
- Tollersrud, O.K.; Berg, T.; Healy, P.; Evjen, G.; Ramachandran, U.; Nilssen, O. Purification of Bovine Lysosomal α-Mannosidase, Characterization of Its Gene and Determination of Two Mutations That Cause α-Mannosidosis. Eur. J. Biochem. 1997, 246, 410–419. [Google Scholar] [CrossRef]
- Berg, T.; Hopwood, J.J. α-Mannosidosis in the Guinea Pig: Cloning of the Lysosomal α-Mannosidase CDNA and Identification of a Missense Mutation Causing α-Mannosidosis. Biochim. Biophys. Acta 2002, 1586, 169–176. [Google Scholar] [CrossRef]
- Nilssen, O.; Berg, T.; Riise, H.M.F.; Ramachandran, U.; Evjen, G.; Hansen, G.M.; Malm, D.; Tranebjaerg, L.; Tollersrud, O.K. Mannosidosis: Functional Cloning of the Lysosomal-Mannosidase CDNA and Identification of a Mutation in Two Affected Siblings. Hum. Mol. Genet. 1997, 6, 717–726. [Google Scholar] [CrossRef]
- Morgan, B.R.; Coates, J.R.; Johnson, G.C.; Shelton, G.D.; Katz, M.L. Characterization of Thoracic Motor and Sensory Neurons and Spinal Nerve Roots in Canine Degenerative Myelopathy, a Potential Disease Model of Amyotrophic Lateral Sclerosis. J. Neurosci. Res. 2014, 92, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Villani, N.A.; Bullock, G.; Michaels, J.R.; Yamato, O.; O’Brien, D.P.; Mhlanga-Mutangadura, T.; Johnson, G.S.; Katz, M.L. A Mixed Breed Dog with Neuronal Ceroid Lipofuscinosis Is Homozygous for a CLN5 Nonsense Mutation Previously Identified in Border Collies and Australian Cattle Dogs. Mol. Genet. Metab. 2019, 127, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.L.; Parker, K.R.; Handelman, G.J.; Bramel, T.L.; Dratz, E.A. Effects of Antioxidant Nutrient Deficiency on the Retina and Retinal Pigment Epithelium of Albino Rats: A Light and Electron Microscopic Study. Exp. Eye Res. 1982, 34, 339–369. [Google Scholar] [CrossRef]
- Katz, M.L.; Eldred, G.E.; Siakotos, A.N.; Koppang, N. Characterization of Disease-Specific Brain Fluorophores in Ceroid-Lipofuscinosis. Am. J. Med. Genet. Suppl. 1988, 5, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Mole, S.E.; Williams, R.E.; Goebel, H.H. The Neuronal Ceroid Lipofuscinoses (Batten Disease), 2nd ed.; Mole, S.E., Willimas, R.E., Goebel, H.H., Eds.; Oxford University Press: Oxford, UK, 2011. [Google Scholar]
- Nanayakkara, R.; Gurung, R.; Rodgers, S.J.; Eramo, M.J.; Ramm, G.; Mitchell, C.A.; McGrath, M.J. Autophagic Lysosome Reformation in Health and Disease. Autophagy 2023, 19, 1378–1395. [Google Scholar] [CrossRef]
- Zhen, Y.; Stenmark, H. Autophagosome Biogenesis. Cells 2023, 12, 668. [Google Scholar] [CrossRef]
- Koike, M.; Nakanishi, H.; Saftig, P.; Ezaki, J.; Isahara, K.; Ohsawa, Y.; Schulz-Schaeffer, W.; Watanabe, T.; Waguri, S.; Kametaka, S.; et al. Cathepsin D Deficiency Induces Lysosomal Storage with Ceroid Lipofuscin in Mouse CNS Neurons. J. Neurosci. 2000, 20, 6898–6906. [Google Scholar] [CrossRef]
- Sarkar, C.; Sadhukhan, T.; Bagh, M.B.; Appu, A.P.; Chandra, G.; Mondal, A.; Saha, A.; Mukherjee, A.B. Cln1-Mutations Suppress Rab7-RILP Interaction and Impair Autophagy Contributing to Neuropathology in a Mouse Model of Infantile Neuronal Ceroid Lipofuscinosis. J. Inherit. Metab. Dis. 2020, 43, 1082–1101. [Google Scholar] [CrossRef]
- Cook, R.W.; Jolly, R.D.; Palmer, D.N.; Tammen, I.; Broom, M.F.; McKinnon, R. Neuronal Ceroid Lipofuscinosis in Merino Sheep. Aust. Vet. J. 2002, 80, 292–297. [Google Scholar] [CrossRef]
- Ju, W.; Wronska, A.; Moroziewicz, D.N.; Zhong, R.; Wisniewski, N.; Jurkiewicz, A.; Fiory, M.; Wisniewski, K.E.; Johnston, L.; Brown, W.T.; et al. Genotype-Phenotype Analyses of Classic Neuronal Ceroid Lipofuscinosis (NCLs): Genet.ic Predictions from Clinical and Pathological Findings. Beijing Da Xue Xue Bao. Yi Xue Ban J. Peking Univ. Health Sci. 2006, 38, 41–48. [Google Scholar]
- Kopra, O.; Vesa, J.; von Schantz, C.; Manninen, T.; Minye, H.; Fabritius, A.-L.; Rapola, J.; van Diggelen, O.P.; Saarela, J.; Jalanko, A.; et al. A Mouse Model for Finnish Variant Late Infantile Neuronal Ceroid Lipofuscinosis, CLN5, Reveals Neuropathology Associated with Early Aging. Hum. Mol. Genet. 2004, 13, 2893–2906. [Google Scholar] [CrossRef] [PubMed]
- Jadav, R.H.; Sinha, S.; Yasha, T.C.; Aravinda, H.; Gayathri, N.; Rao, S.; Bindu, P.S.; Satishchandra, P. Clinical, Electrophysiological, Imaging, and Ultrastructural Description in 68 Patients with Neuronal Ceroid Lipofuscinoses and Its Subtypes. Pediatr. Neurol. 2014, 50, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Craiu, D.; Dragostin, O.; Dica, A.; Hoffman-Zacharska, D.; Gos, M.; Bastian, A.E.; Gherghiceanu, M.; Rolfs, A.; Nahavandi, N.; Craiu, M.; et al. Rett-like Onset in Late-Infantile Neuronal Ceroid Lipofuscinosis (CLN7) Caused by Compound Heterozygous Mutation in the MFSD8 Gene and Review of the Literature Data on Clinical Onset Signs. Eur. J. Paediatr. Neurol. 2015, 19, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.A.; Georgiou, M.; Robson, A.G.; Ali, N.; Kalhoro, A.; Holthaus, S.K.; Pontikos, N.; Oluonye, N.; de Carvalho, E.R.; Neveu, M.M.; et al. Juvenile Batten Disease (CLN3): Detailed Ocular Phenotype, Novel Observations, Delayed Diagnosis, Masquerades, and Prospects for Therapy. Ophthalmol. Retin. 2020, 4, 433–445. [Google Scholar] [CrossRef]
- Pesaola, F.; Kohan, R.; Cismondi, I.A.; Guelbert, N.; Pons, P.; Oller-Ramirez, A.M.; Noher de Halac, I. Congenital CLN8 Disease of Neuronal Ceroid Lipofuscinosis: A Novel Phenotype. Rev. Neurol. 2019, 68, 155–159. [Google Scholar]
- Berkovic, S.F.; Oliver, K.L.; Canafoglia, L.; Krieger, P.; Damiano, J.A.; Hildebrand, M.S.; Morbin, M.; Vears, D.F.; Sofia, V.; Giuliano, L.; et al. Kufs Disease Due to Mutation of CLN6: Clinical, Pathological and Molecular Genetic Features. Brain 2019, 142, 59–69. [Google Scholar] [CrossRef]
- Brandenstein, L.; Schweizer, M.; Sedlacik, J.; Fiehler, J.; Storch, S. Lysosomal Dysfunction and Impaired Autophagy in a Novel Mouse Model Deficient for the Lysosomal Membrane Protein Cln7. Hum. Mol. Genet. 2016, 25, 777–791. [Google Scholar] [CrossRef]
- Mandel, H.; Cohen Katsanelson, K.; Khayat, M.; Chervinsky, I.; Vladovski, E.; Iancu, T.C.; Indelman, M.; Horovitz, Y.; Sprecher, E.; Shalev, S.A.; et al. Clinico-Pathological Manifestations of Variant Late Infantile Neuronal Ceroid Lipofuscinosis (VLINCL) Caused by a Novel Mutation in MFSD8 Gene. Eur. J. Med. Genet. 2014, 57, 607–612. [Google Scholar] [CrossRef]
- Ranta, S.; Savukoski, M.; Santavuori, P.; Haltia, M. Studies of Homogenous Populations: CLN5 and CLN8. Adv. Genet. 2001, 45, 123–140. [Google Scholar]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Bendl, J.; Stourac, J.; Salanda, O.; Pavelka, A.; Wieben, E.D.; Zendulka, J.; Brezovsky, J.; Damborsky, J. PredictSNP: Robust and Accurate Consensus Classifier for Prediction of Disease-Related Mutations. PLoS Comput. Biol. 2014, 10, e1003440. [Google Scholar] [CrossRef]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.-J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. Inferring the Molecular and Phenotypic Impact of Amino Acid Variants with MutPred2. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.L.; Rustad, E.; Robinson, G.O.; Whiting, R.E.H.; Student, J.T.; Coates, J.R.; Narfstrom, K. Canine Neuronal Ceroid Lipofuscinoses: Promising Models for Preclinical Testing of Therapeutic Interventions. Neurobiol. Dis. 2017, 108, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Kolicheski, A.; Johnson, G.S.; Villani, N.A.; O’Brien, D.P.; Mhlanga-Mutangadura, T.; Wenger, D.A.; Mikoloski, K.; Eagleson, J.S.; Taylor, J.F.; Schnabel, R.D.; et al. GM2 Gangliosidosis in Shiba Inu Dogs with an In-Frame Deletion in HEXB. J. Vet. Intern. Med. 2017, 31, 1520–1526. [Google Scholar] [CrossRef] [PubMed]
- Jolly, R.D.; Walkley, S.U. Lysosomal Storage Diseases of Animals: An Essay in Comparative Pathology. Vet. Pathol. 1997, 34, 527–548. [Google Scholar] [CrossRef] [PubMed]
- Gotoda, Y.; Wakamatsu, N.; Kawai, H.; Nishida, Y.; Matsumoto, T. Missense and Nonsense Mutations in the Lysosomal α-Mannosidase Gene (MANB) in Severe and Mild Forms of α-Mannosidosis. Am. J. Hum. Genet. 1998, 63, 1015–1024. [Google Scholar] [CrossRef]
- Sleat, D.E.; Wiseman, J.A.; Sohar, I.; El-Banna, M.; Zheng, H.; Moore, D.F.; Lobel, P. Proteomic Analysis of Mouse Models of Niemann-Pick C Disease Reveals Alterations in the Steady-State Levels of Lysosomal Proteins within the Brain. Proteomics 2012, 12, 3499–3509. [Google Scholar] [CrossRef]
- Sleat, D.E.; Wiseman, J.A.; El-Banna, M.; Zheng, H.; Zhao, C.; Soherwardy, A.; Moore, D.F.; Lobel, P. Analysis of Brain and Cerebrospinal Fluid from Mouse Models of the Three Major Forms of Neuronal Ceroid Lipofuscinosis Reveals Changes in the Lysosomal Proteome. Mol. Cell. Proteom. 2019, 18, 2244–2261. [Google Scholar] [CrossRef] [PubMed]
- Ockerman, P.A. Mannosidosis: Isolation of Oligosaccharide Storage Material from Brain. J. Pediatr. 1969, 75, 360–365. [Google Scholar] [CrossRef]
- Kjellman, B.; Gamstorp, I.; Brun, A.; Ockerman, P.A.; Palmgren, B. Mannosidosis: A Clinical and Histopathologic Study. J. Pediatr. 1969, 75, 366–373. [Google Scholar] [CrossRef]
- Hocking, J.D.; Jolly, R.D.; Batt, R.D. Deficiency of α-Mannosidase in Angus Cattle. An Inherited Lysosomal Storage Disease. Biochem. J. 1972, 128, 69–78. [Google Scholar] [CrossRef]
- Burditt, L.J.; Chotai, K.; Hirani, S.; Nugent, P.G.; Winchester, B.G.; Blakemore, W.F. Biochemical Studies on a Case of Feline Mannosidosis. Biochem. J. 1980, 189, 467–473. [Google Scholar] [CrossRef]
- Walkley, S.U.; Blakemore, W.F.; Purpura, D.P. Alterations in Neuron Morphology in Feline Mannosidosis. A Golgi Study. Acta Neuropathol. 1981, 53, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Vandevelde, M.; Fankhauser, R.; Bichsel, P.; Wiesmann, U.; Herschkowitz, N. Hereditary Neurovisceral Mannosidosis Associated with α-Mannosidase Deficiency in a Family of Persian Cats. Acta Neuropathol. 1982, 58, 64–68. [Google Scholar] [CrossRef]
- Muntz, F.H.; Bonning, L.E.; Carey, W.F. α-Mannosidosis in a Guinea Pig. Lab Anim. Sci. 1999, 49, 424–426. [Google Scholar] [PubMed]
- Borgwardt, L.; Lund, A.M.; Dali, C.I. α-Mannosidosis-a Review of Genet.ic, Clinical Findings and Options of Treatment. Pediatr. Endocrinol. Rev. 2014, 12 (Suppl. S1), 185–191. [Google Scholar] [PubMed]
- Govender, R.; Mubaiwa, L. α-Mannosidosis: A Report of 2 Siblings and Review of the Literature. J. Child Neurol. 2014, 29, 131–134. [Google Scholar] [CrossRef]
- Berg, T.; Riise, H.M.; Hansen, G.M.; Malm, D.; Tranebjaerg, L.; Tollersrud, O.K.; Nilssen, O. Spectrum of Mutations in α-Mannosidosis. Am. J. Hum. Genet. 1999, 64, 77–88. [Google Scholar] [CrossRef]
- Borgwardt, L.; Stensland, H.; Olsen, K.; Wibrand, F.; Klenow, H.; Beck, M.; Amraoui, Y.; Arash, L.; Fogh, J.; Nilssen, O.; et al. α-Mannosidosis: Correlation between Phenotype, Genotype and Mutant MAN2B1 Subcellular Localisation. Orphanet. J. Rare Dis. 2015, 10, 70. [Google Scholar] [CrossRef]
- Healy, P.J.; Harper, P.A.; Dennis, J.A. Phenotypic Variation in Bovine α-Mannosidosis. Res. Vet. Sci. 1990, 49, 82–84. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Chapman, M.; Evans, K.; Azevedo, L.; Hayden, M.; Heywood, S.; Millar, D.S.; Phillips, A.D.; et al. The Human Gene Mutation Database (HGMD R): Optimizing Its Use in a Clinical Diagnostic or Research Setting. Hum. Genet. 2020, 139, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- Riise Stensland, H.M.F.; Frantzen, G.; Kuokkanen, E.; Buvang, E.K.; Klenow, H.B.; Heikinheimo, P.; Malm, D.; Nilssen, O. Amamutdb.No: A Relational Database for MAN2B1 Allelic Variants That Compiles Genotypes, Clinical Phenotypes, and Biochemical and Structural Data of Mutant MAN2B1 in α-Mannosidosis. Hum. Mutat. 2015, 36, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Riise Stensland, H.M.F.; Klenow, H.B.; Van Nguyen, L.; Hansen, G.M.; Malm, D.; Nilssen, O. Identification of 83 Novel α-Mannosidosis-Associated Sequence Variants: Functional Analysis of MAN2B1 Missense Mutations. Hum. Mutat. 2012, 33, 511–520. [Google Scholar] [CrossRef]
- Kuokkanen, E.; Riise Stensland, H.M.F.; Smith, W.; Kjeldsen Buvang, E.; Van Nguyen, L.; Nilssen, O.; Heikinheimo, P. Molecular and Cellular Characterization of Novel {α}-Mannosidosis Mutations. Hum. Mol. Genet. 2011, 20, 2651–2661. [Google Scholar] [CrossRef] [PubMed]
- Khan, J.M.; Ranganathan, S. A Multi-Species Comparative Structural Bioinformatics Analysis of Inherited Mutations in α-D-Mannosidase Reveals Strong Genotype-Phenotype Correlation. BMC Genom. 2009, 10 (Suppl. S3), S33. [Google Scholar] [CrossRef] [PubMed]
- Pittis, M.G.; Montalvo, A.L.E.; Heikinheimo, P.; Sbaragli, M.; Balducci, C.; Persichetti, E.; Van Maldergem, L.; Filocamo, M.; Bembi, B.; Beccari, T. Funtional Characterization of Four Novel MAN2B1 Mutations Causing Juvenile Onset α-Mannosidosis. Clin. Chim. Acta 2007, 375, 136–139. [Google Scholar] [CrossRef]
- Leipold, H.W.; Smith, J.E.; Jolly, R.D.; Eldridge, F.E. Mannosidosis of Angus Calves. J. Am. Vet. Med. Assoc. 1979, 175, 457–459. [Google Scholar]
- Crawley, A.C.; Walkley, S.U. Developmental Analysis of CNS Pathology in the Lysosomal Storage Disease α-Mannosidosis. J. Neuropathol. Exp. Neurol. 2007, 66, 687–697. [Google Scholar] [CrossRef]
- Cummings, J.F.; Wood, P.A.; de Lahunta, A.; Walkley, S.U.; Le Boeuf, L. The Clinical and Pathologic Heterogeneity of Feline α-Mannosidosis. J. Vet. Intern. Med. 1988, 2, 163–170. [Google Scholar] [CrossRef]
- Jolly, R.D.; Thompson, K.G. The Pathology of Bovine Mannosidosis. Vet. Pathol 1978, 15, 141–152. [Google Scholar] [CrossRef]
- Sung, J.H.; Hayano, M.; Desnick, R.J. Mannosidosis: Pathology of the Nervous System. J. Neuropathol. Exp. Neurol. 1977, 36, 807–820. [Google Scholar] [CrossRef] [PubMed]
- DeGasperi, R.; al Daher, S.; Daniel, P.F.; Winchester, B.G.; Jeanloz, R.W.; Warren, C.D. The Substrate Specificity of Bovine and Feline Lysosomal α-D-Mannosidases in Relation to α-Mannosidosis. J. Biol. Chem. 1991, 266, 16556–16563. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.N. The Relevance of the Storage of Subunit c of ATP Synthase in Different Forms and Models of Batten Disease (NCLs). Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 2287–2291. [Google Scholar] [CrossRef]
- Katz, M.L.; Coates, J.R.; Cooper, J.J.; O’Brien, D.P.; Jeong, M.; Narfström, K. Retinal Pathology in a Canine Model of Late Infantile Neuronal Ceroid Lipofuscinosis. Invest. Ophthalmol. Vis. Sci. 2008, 49, 2686–2695. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.; Katz, M.L.; Levesque, D.; Shelton, G.D.; De Lahunta, A.; O’Brien, D. A Variant Form of Neuronal Ceroid Lipofuscinosis in American Bulldogs. J. Vet. Intern. Med. 2005, 19, 44–51. [Google Scholar] [CrossRef]
- Katz, M.L.; Johnson, G.C.; Leach, S.B.; Williamson, B.G.; Coates, J.R.; Whiting, R.E.H.; Vansteenkiste, D.P.; Whitney, M.S. Extraneuronal Pathology in a Canine Model of CLN2 Neuronal Ceroid Lipofuscinosis after Intracerebroventricular Gene Therapy That Delays Neurological Disease Progression. Gene Ther. 2017, 24, 215–223. [Google Scholar] [CrossRef]
- Awano, T.; Katz, M.L.; O’Brien, D.P.; Taylor, J.F.; Evans, J.; Khan, S.; Sohar, I.; Lobel, P.; Johnson, G.S. A Mutation in the Cathepsin D Gene (CTSD) in American Bulldogs with Neuronal Ceroid Lipofuscinosis. Mol. Genet. Metab. 2006, 87, 341–348. [Google Scholar] [CrossRef]
- Katz, M.L.; Buckley, R.M.; Biegen, V.; O’Brien, D.P.; Johnson, G.C.; Warren, W.C.; Lyons, L.A. Neuronal Ceroid Lipofuscinosis in a Domestic Cat Associated with a DNA Sequence Variant That Creates a Premature Stop Codon in CLN6. G3 (Bethesda) 2020, 10, 2741–2751. [Google Scholar] [CrossRef]
- Goodman, L.A.; Livingston, P.O.; Walkley, S.U. Ectopic Dendrites Occur Only on Cortical Pyramidal Cells Containing Elevated GM2 Ganglioside in α-Mannosidosis. Proc. Natl. Acad. Sci. USA 1991, 88, 11330–11334. [Google Scholar] [CrossRef]
- Breiden, B.; Sandhoff, K. Mechanism of Secondary Ganglioside and Lipid Accumulation in Lysosomal Disease. Int. J. Mol. Sci. 2020, 21, 2566. [Google Scholar] [CrossRef]
- Walkley, S.U.; Vanier, M.T. Secondary Lipid Accumulation in Lysosomal Disease. Biochim. Biophys. Acta 2009, 1793, 726–736. [Google Scholar] [CrossRef]
- Scerra, G.; De Pasquale, V.; Scarcella, M.; Caporaso, M.G.; Pavone, L.M.; D’Agostino, M. Lysosomal Positioning Diseases: Beyond Substrate Storage. Open Biol. 2022, 12, 220155. [Google Scholar] [CrossRef]
- Myerowitz, R.; Puertollano, R.; Raben, N. Impaired Autophagy: The Collateral Damage of Lysosomal Storage Disorders. EBioMedicine 2021, 63, 103166. [Google Scholar] [CrossRef] [PubMed]
- Hagopian, K. Preparative Electrophoretic Method for the Purification of a Hydrophobic Membrane Protein: Subunit c of the Mitochondrial ATP Synthase from Rat Liver. Anal. Biochem. 1999, 273, 240–251. [Google Scholar] [CrossRef]
- Lieberman, A.P.; Puertollano, R.; Raben, N.; Slaugenhaupt, S.; Walkley, S.U.; Ballabio, A. Autophagy in Lysosomal Storage Disorders. Autophagy 2012, 8, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Seranova, E.; Connolly, K.J.; Zatyka, M.; Rosenstock, T.R.; Barrett, T.; Tuxworth, R.I.; Sarkar, S. Dysregulation of Autophagy as a Common Mechanism in Lysosomal Storage Diseases. Essays Biochem. 2017, 61, 733–749. [Google Scholar] [CrossRef] [PubMed]
- Koike, M.; Shibata, M.; Waguri, S.; Yoshimura, K.; Tanida, I.; Kominami, E.; Gotow, T.; Peters, C.; von Figura, K.; Mizushima, N.; et al. Participation of Autophagy in Storage of Lysosomes in Neurons from Mouse Models of Neuronal Ceroid-Lipofuscinoses (Batten Disease). Am. J. Pathol. 2005, 167, 1713–1728. [Google Scholar] [CrossRef]
- Liao, G.; Yao, Y.; Liu, J.; Yu, Z.; Cheung, S.; Xie, A.; Liang, X.; Bi, X. Cholesterol Accumulation Is Associated with Lysosomal Dysfunction and Autophagic Stress in Npc1-/- Mouse Brain. Am. J. Pathol. 2007, 171, 962–975. [Google Scholar] [CrossRef]
- Thelen, M.; Damme, M.; Schweizer, M.; Hagel, C.; Wong, A.M.S.; Cooper, J.D.; Braulke, T.; Galliciotti, G. Disruption of the Autophagy-Lysosome Pathway Is Involved in Neuropathology of the Nclf Mouse Model of Neuronal Ceroid Lipofuscinosis. PLoS ONE 2012, 7, e35493. [Google Scholar] [CrossRef]
- Baudot, A.D.; Wang, V.M.-Y.; Leach, J.D.; O’Prey, J.; Long, J.S.; Paulus-Hock, V.; Lilla, S.; Thomson, D.M.; Greenhorn, J.; Ghaffar, F.; et al. Glycan Degradation Promotes Macroautophagy. Proc. Natl. Acad. Sci. USA 2022, 119, e2111506119. [Google Scholar] [CrossRef]
- Michalski, J.C.; Klein, A. Glycoprotein Lysosomal Storage Disorders: α- and β-Mannosidosis, Fucosidosis and α-N-Acetylgalactosaminidase Deficiency. Biochim. Biophys. Acta 1999, 1455, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Levin, S.W.; Baker, E.H.; Zein, W.M.; Zhang, Z.; Quezado, Z.M.N.; Miao, N.; Gropman, A.; Griffin, K.J.; Bianconi, S.; Chandra, G.; et al. Oral Cysteamine Bitartrate and N-Acetylcysteine for Patients with Infantile Neuronal Ceroid Lipofuscinosis: A Pilot Study. Lancet Neurol. 2014, 13, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Wang, F.; Lotfi, P.; Sardiello, M.; Segatori, L. 2-Hydroxypropyl-β-Cyclodextrin Promotes Transcription Factor EB-Mediated Activation of Autophagy: Implications for Therapy. J. Biol. Chem. 2014, 289, 10211–10222. [Google Scholar] [CrossRef]
- Scotto Rosato, A.; Krogsaeter, E.K.; Jaslan, D.; Abrahamian, C.; Montefusco, S.; Soldati, C.; Spix, B.; Pizzo, M.T.; Grieco, G.; Bock, J.; et al. TPC2 Rescues Lysosomal Storage in Mucolipidosis Type IV, Niemann-Pick Type C1, and Batten Disease. EMBO Mol. Med. 2022, 14, e15377. [Google Scholar] [CrossRef]
- Jana, M.; Dutta, D.; Poddar, J.; Pahan, K. Activation of PPAR alpha Exhibits Therapeutic Efficacy in a Mouse Model of Juvenile Neuronal Ceroid Lipofuscinosis. J. Neurosci. 2023, 43, 1814–1829. [Google Scholar] [CrossRef]
- Klein, M.; Kaleem, A.; Oetjen, S.; Wunkhaus, D.; Binkle, L.; Schilling, S.; Gjorgjieva, M.; Scholz, R.; Gruber-Schoffnegger, D.; Storch, S.; et al. Converging Roles of PSENEN/PEN2 and CLN3 in the Autophagy-Lysosome System. Autophagy 2022, 18, 2068–2085. [Google Scholar] [CrossRef] [PubMed]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Prediction Tool | Variant Score/Score for Deleterious Prediction | Prediction of Pathogenicity |
|---|---|---|
| PredictSNP * | 87% | Deleterious * |
| MutPred2 | 0.717 | Likely pathogenic |
| Enzyme | Dog | Tissue | Activity * |
|---|---|---|---|
| α-mannosidase | Proband | Cerebral Cortex | 0 |
| α-mannosidase | CLN2 Dachshund (n = 3) | Cerebral Cortex | 137 † |
| α-mannosidase | Normal Control | Cerebral Cortex | 89 |
| α-mannosidase | Proband | Cerebellum | 0 |
| α-mannosidase | CLN2 Dachshund (n = 3) | Cerebellum | 141 † |
| α-mannosidase | Normal Control | Cerebellum | 59 |
| β-mannosidase | Proband | Cerebral Cortex | 147 |
| β-mannosidase | CLN2 Dachshund (n = 3) | Cerebral Cortex | 71 † |
| β-mannosidase | Normal Control | Cerebral Cortex | 90 |
| β-mannosidase | Proband | Cerebellum | 152 |
| β-mannosidase | CLN2 Dachshund (n = 3) | Cerebellum | 111 † |
| β-mannosidase | Normal Control | Cerebellum | 65 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bullock, G.; Johnson, G.S.; Pattridge, S.G.; Mhlanga-Mutangadura, T.; Guo, J.; Cook, J.; Campbell, R.S.; Vite, C.H.; Katz, M.L. A Homozygous MAN2B1 Missense Mutation in a Doberman Pinscher Dog with Neurodegeneration, Cytoplasmic Vacuoles, Autofluorescent Storage Granules, and an α-Mannosidase Deficiency. Genes 2023, 14, 1746. https://doi.org/10.3390/genes14091746
Bullock G, Johnson GS, Pattridge SG, Mhlanga-Mutangadura T, Guo J, Cook J, Campbell RS, Vite CH, Katz ML. A Homozygous MAN2B1 Missense Mutation in a Doberman Pinscher Dog with Neurodegeneration, Cytoplasmic Vacuoles, Autofluorescent Storage Granules, and an α-Mannosidase Deficiency. Genes. 2023; 14(9):1746. https://doi.org/10.3390/genes14091746
Chicago/Turabian StyleBullock, Garrett, Gary S. Johnson, Savannah G. Pattridge, Tendai Mhlanga-Mutangadura, Juyuan Guo, James Cook, Rebecca S. Campbell, Charles H. Vite, and Martin L. Katz. 2023. "A Homozygous MAN2B1 Missense Mutation in a Doberman Pinscher Dog with Neurodegeneration, Cytoplasmic Vacuoles, Autofluorescent Storage Granules, and an α-Mannosidase Deficiency" Genes 14, no. 9: 1746. https://doi.org/10.3390/genes14091746