
Transcriptomic Profiling of Shoot Apical Meristem Aberrations in the Multi-Main-Stem Mutant (ms) of Brassica napus L.
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods and Materials
2.1. Plant Materials and Growth Conditions
2.2. Tissue Section
2.2.1. Paraffin Section Preparation
2.2.2. Paraffin Section Safranin O-Fast Green Staining
2.3. Scanning Electron Microscopy
2.4. Quantification of Cytokinins
2.4.1. Sample Preparation
2.4.2. HPLC-MS/MS Analysis
2.5. RNA Extraction, Sequencing, and Analysis
2.6. Real-Time Quantitative PCR (RT-qPCR)
2.7. Statistical Analysis
3. Results
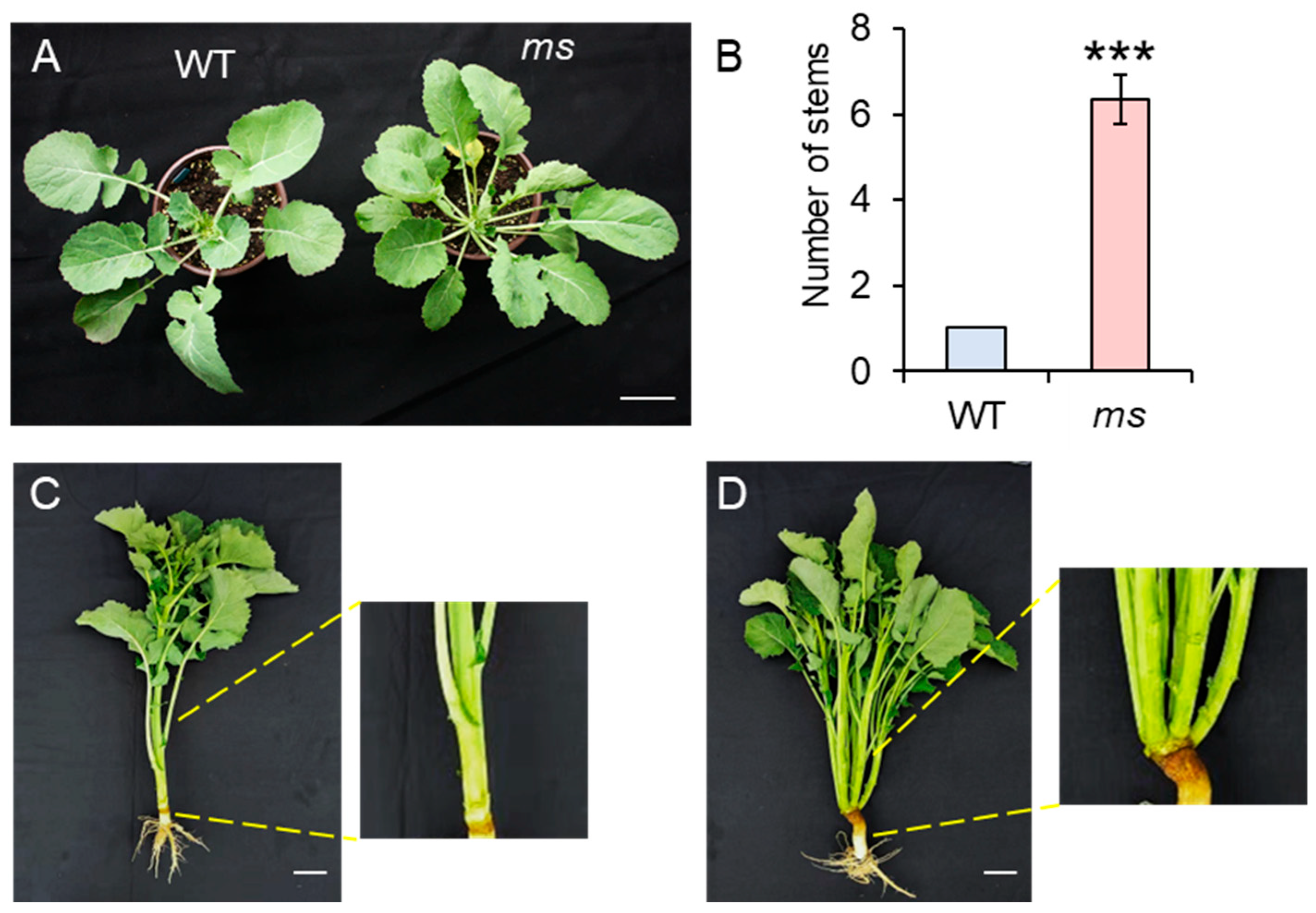
3.1. Phenotype Investigation of MS Mutant
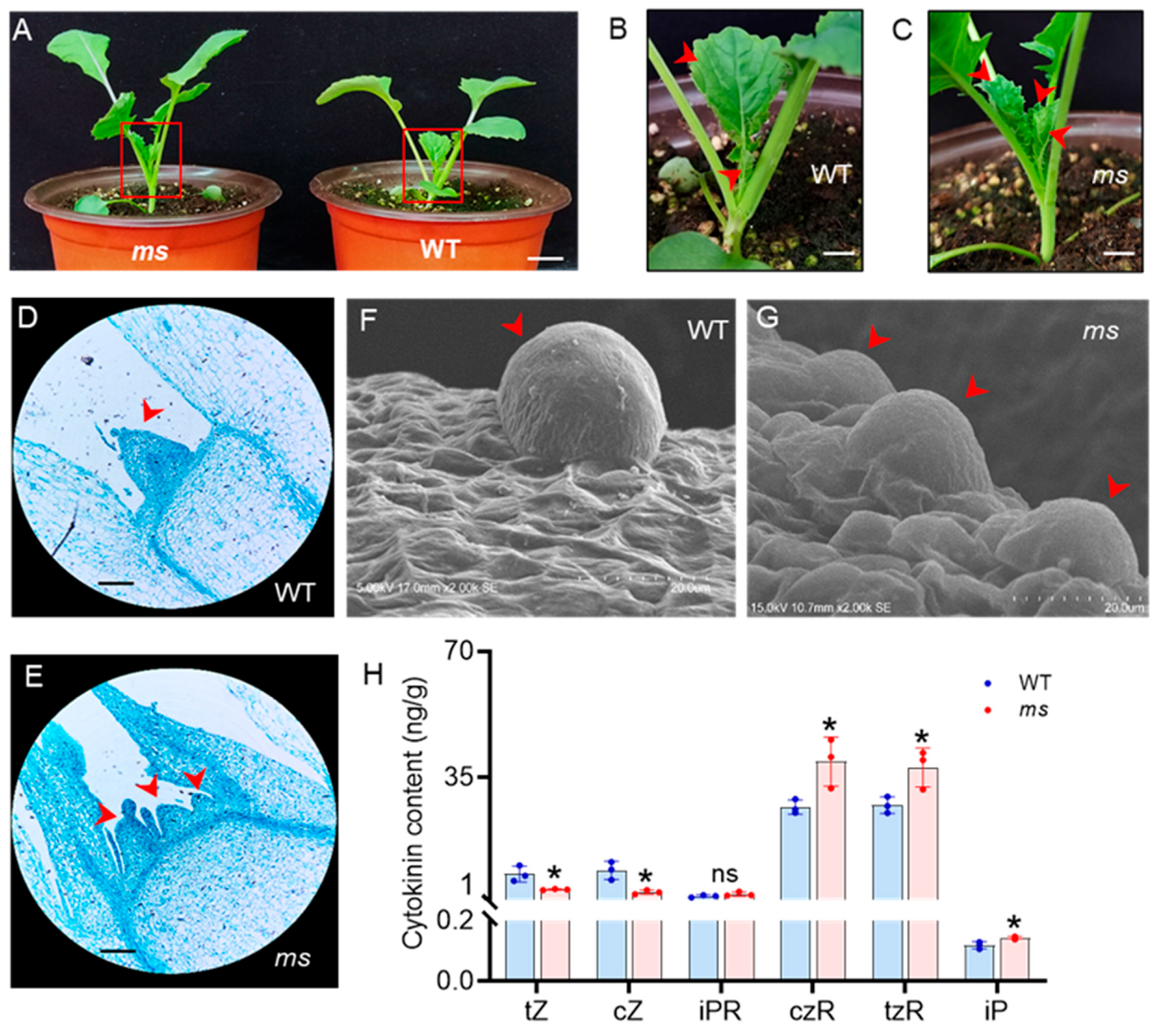
3.2. Abnormal Development of the SAM in MS Mutant
3.3. Changes in Content of CKs in the SAM of the ms Mutant
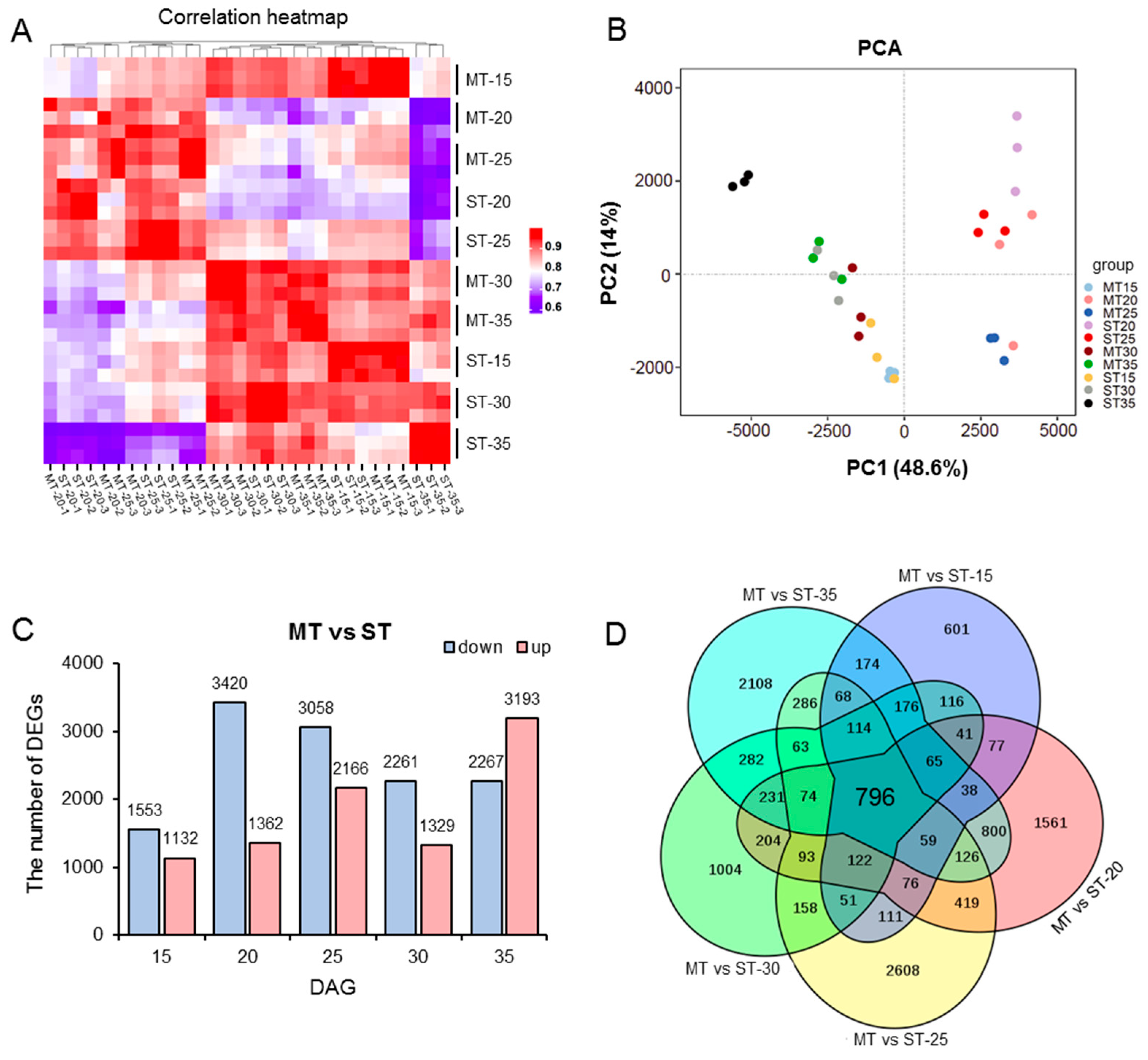
3.4. RNA-seq Analysis of the SAM in MS Mutant
3.5. Differential Transcriptional in Different Developmental Stages in the SAM of MS Mutant
3.6. GO and KEGG Analysis of All DEGs
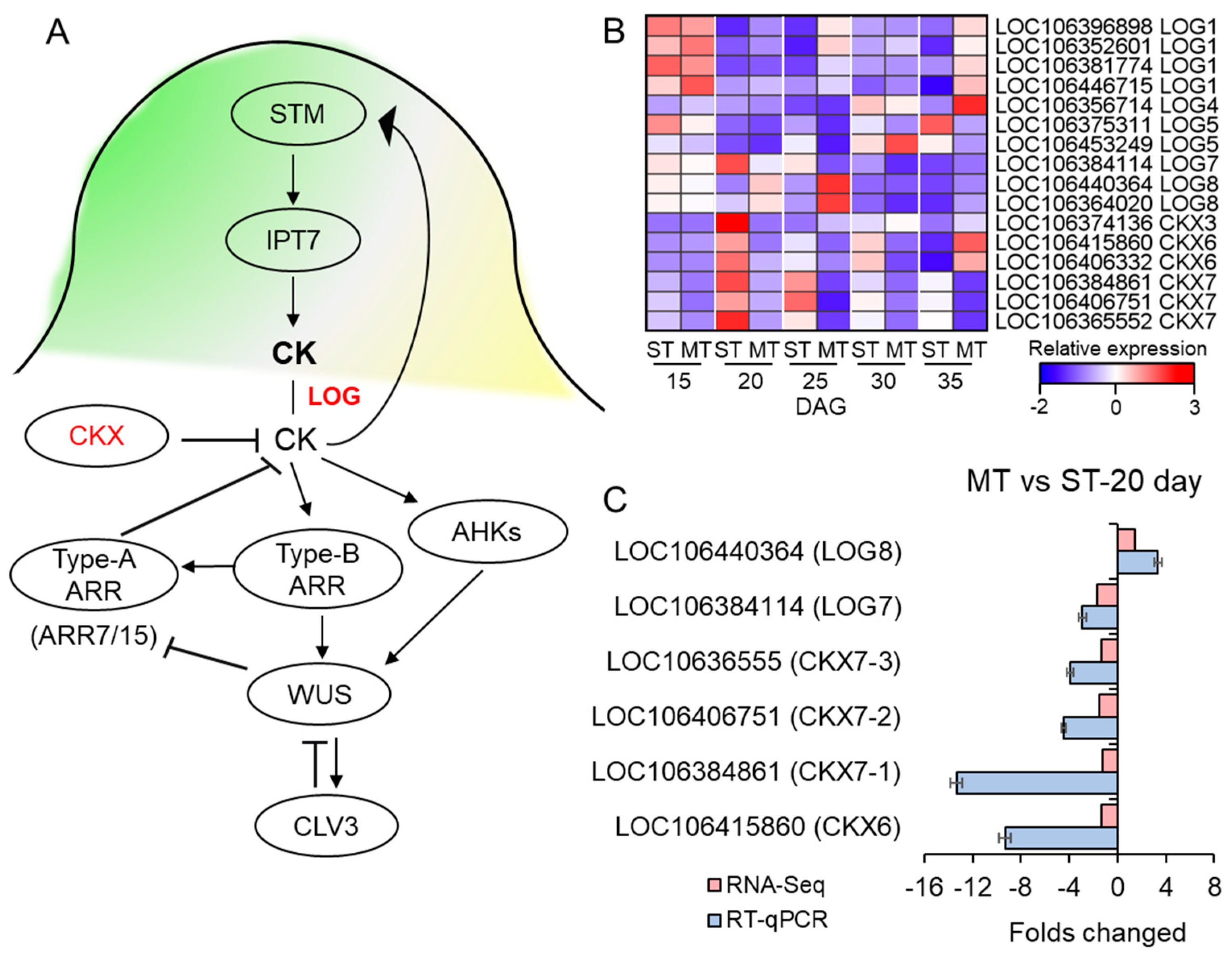
3.7. The Expression of CK-Related Genes Was Affected in the SAM of MS Mutant
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mänd, M.; Williams, I.H.; Viik, E.; Karise, R. Oilseed Rape, Bees and Integrated Pest Management. In Biocontrol-Based Integrated Management of Oilseed Rape Pests; Williams, I.H., Ed.; Springer: Dordrecht, The Netherlands, 2010; pp. 357–379. [Google Scholar] [CrossRef]
- Raboanatahiry, N.; Li, H.; Yu, L.; Li, M. Rapeseed (Brassica napus): Processing, Utilization, and Genetic Improvement. Agronomy 2021, 11, 1776. [Google Scholar] [CrossRef]
- Shimizu-Sato, S.; Mori, H. Control of outgrowth and dormancy in axillary buds. Plant Physiol. 2001, 127, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- McSteen, P.; Leyser, O. Shoot branching. Annu. Rev. Plant Biol. 2005, 56, 353–374. [Google Scholar] [CrossRef] [PubMed]
- Aichinger, E.; Kornet, N.; Friedrich, T.; Laux, T. Plant stem cell niches. Annu. Rev. Plant Biol. 2012, 63, 615–636. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.K. Twenty years on: The inner workings of the shoot apical meristem, a developmental dynamo. Dev. Biol. 2010, 341, 95–113. [Google Scholar] [CrossRef] [Green Version]
- Barbier, F.F.; Dun, E.A.; Kerr, S.C.; Chabikwa, T.G.; Beveridge, C.A. An Update on the Signals Controlling Shoot Branching. Trends Plant Sci. 2019, 24, 220–236. [Google Scholar] [CrossRef]
- Wang, B.; Smith, S.M.; Li, J. Genetic Regulation of Shoot Architecture. Annu. Rev. Plant Biol. 2018, 69, 437–468. [Google Scholar] [CrossRef]
- Shani, E.; Yanai, O.; Ori, N. The role of hormones in shoot apical meristem function. Curr. Opin. Plant Biol. 2006, 9, 484–489. [Google Scholar] [CrossRef]
- Sakakibara, H. Cytokinins: Activity, biosynthesis, and translocation. Annu. Rev. Plant Biol. 2006, 57, 431–449. [Google Scholar] [CrossRef] [Green Version]
- Mok, D.W.; Mok, M.C. Cytokinin Metabolism and Action. Annu. Rev. Plant Physiol. Plant Mol. Biol. 2001, 52, 89–118. [Google Scholar] [CrossRef]
- Sp Chal, L.X. Cytokinins—Recent news and views of evolutionally old molecules. Funct. Plant Biol. 2012, 39, 267–284. [Google Scholar] [CrossRef] [Green Version]
- Lomin, S.N.; Krivosheev, D.M.; Steklov, M.Y.; Arkhipov, D.V.; Osolodkin, D.I.; Schmulling, T.; Romanov, G.A. Plant membrane assays with cytokinin receptors underpin the unique role of free cytokinin bases as biologically active ligands. J. Exp. Bot. 2015, 66, 1851–1863. [Google Scholar] [CrossRef] [Green Version]
- Bajguz, A.; Piotrowska, A. Conjugates of auxin and cytokinin. Phytochemistry 2009, 70, 957–969. [Google Scholar] [CrossRef]
- Raspor, M.; Motyka, V.; Ninkovic, S.; Dobrev, P.I.; Malbeck, J.; Cosic, T.; Cingel, A.; Savic, J.; Tadic, V.; Dragicevic, I.C. Endogenous levels of cytokinins, indole-3-acetic acid and abscisic acid in in vitro grown potato: A contribution to potato hormonomics. Sci. Rep. 2020, 10, 3437. [Google Scholar] [CrossRef] [Green Version]
- Del Rosario Cardenas-Aquino, M.; Sarria-Guzman, Y.; Martinez-Antonio, A. Review: Isoprenoid and aromatic cytokinins in shoot branching. Plant Sci. 2022, 319, 111240. [Google Scholar] [CrossRef]
- Tarkowska, D.; Dolezal, K.; Tarkowski, P.; Astot, C.; Holub, J.; Fuksova, K.; Schmulling, T.; Sandberg, G.; Strnad, M. Identification of new aromatic cytokinins in Arabidopsis thaliana and Populus x canadensis leaves by LC-(+)ESI-MS and capillary liquid chromatography/frit-fast atom bombardment mass spectrometry. Physiol. Plant 2003, 117, 579–590. [Google Scholar] [CrossRef]
- Nishimura, C.; Ohashi, Y.; Sato, S.; Kato, T.; Tabata, S.; Ueguchi, C. Histidine kinase homologs that act as cytokinin receptors possess overlapping functions in the regulation of shoot and root growth in Arabidopsis. Plant Cell 2004, 16, 1365–1377. [Google Scholar] [CrossRef] [Green Version]
- Tantikanjana, T.; Yong, J.W.; Letham, D.S.; Griffith, M.; Hussain, M.; Ljung, K.; Sandberg, G.; Sundaresan, V. Control of axillary bud initiation and shoot architecture in Arabidopsis through the SUPERSHOOT gene. Genes. Dev. 2001, 15, 1577–1588. [Google Scholar] [CrossRef] [Green Version]
- Giulini, A.; Wang, J.; Jackson, D. Control of phyllotaxy by the cytokinin-inducible response regulator homologue ABPHYL1. Nature 2004, 430, 1031–1034. [Google Scholar] [CrossRef]
- Kieber, J.J.; Schaller, G.E. Cytokinin signaling in plant development. Development 2018, 145, dev149344. [Google Scholar] [CrossRef] [Green Version]
- Snipes, S.A.; Rodriguez, K.; DeVries, A.E.; Miyawaki, K.N.; Perales, M.; Xie, M.; Reddy, G.V. Cytokinin stabilizes WUSCHEL by acting on the protein domains required for nuclear enrichment and transcription. PLoS Genet. 2018, 14, e1007351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, W.J.; Cheng, Z.J.; Sang, Y.L.; Zhang, M.M.; Rong, X.F.; Wang, Z.W.; Tang, Y.Y.; Zhang, X.S. Type-B ARABIDOPSIS RESPONSE REGULATORs Specify the Shoot Stem Cell Niche by Dual Regulation of WUSCHEL. Plant Cell 2017, 29, 1357–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, M.; Chen, H.; Huang, L.; O’Neil, R.C.; Shokhirev, M.N.; Ecker, J.R. A B-ARR-mediated cytokinin transcriptional network directs hormone cross-regulation and shoot development. Nat. Commun. 2018, 9, 1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Andersen, S.U.; Ljung, K.; Dolezal, K.; Miotk, A.; Schultheiss, S.J.; Lohmann, J.U. Hormonal control of the shoot stem-cell niche. Nature 2010, 465, 1089–1092. [Google Scholar] [CrossRef] [PubMed]
- Leibfried, A.; To, J.P.; Busch, W.; Stehling, S.; Kehle, A.; Demar, M.; Kieber, J.J.; Lohmann, J.U. WUSCHEL controls meristem function by direct regulation of cytokinin-inducible response regulators. Nature 2005, 438, 1172–1175. [Google Scholar] [CrossRef] [PubMed]
- Werner, T.; Köllmer, I.; Bartrina, I.; Holst, K.; Schmülling, T. New insights into the biology of cytokinin degradation. Plant Biol. 2006, 8, 371–381. [Google Scholar] [CrossRef]
- Werner, T.; Motyka, V.; Laucou, V.; Smets, R.; Van Onckelen, H.; Schmülling, T. Cytokinin-deficient transgenic Arabidopsis plants show multiple developmental alterations indicating opposite functions of cytokinins in the regulation of shoot and root meristem activity. Plant Cell 2003, 15, 2532–2550. [Google Scholar] [CrossRef] [Green Version]
- Motyka, V.; Faiss, M.; Strand, M.; Kaminek, M.; Schmulling, T. Changes in Cytokinin Content and Cytokinin Oxidase Activity in Response to Derepression of ipt Gene Transcription in Transgenic Tobacco Calli and Plants. Plant Physiol. 1996, 112, 1035–1043. [Google Scholar] [CrossRef] [Green Version]
- Morris, R.O.; Bilyeu, K.D.; Laskey, J.G.; Cheikh, N.N. Isolation of a gene encoding a glycosylated cytokinin oxidase from maize. Biochem. Biophys. Res. Commun. 1999, 255, 328–333. [Google Scholar] [CrossRef]
- Ashikari, M.; Sakakibara, H.; Lin, S.; Yamamoto, T.; Takashi, T.; Nishimura, A.; Angeles, E.R.; Qian, Q.; Kitano, H.; Matsuoka, M. Cytokinin oxidase regulates rice grain production. Science 2005, 309, 741–745. [Google Scholar] [CrossRef]
- Mameaux, S.; Cockram, J.; Thiel, T.; Steuernagel, B.; Stein, N.; Taudien, S.; Jack, P.; Werner, P.; Gray, J.C.; Greenland, A.J.; et al. Molecular, phylogenetic and comparative genomic analysis of the cytokinin oxidase/dehydrogenase gene family in the Poaceae. Plant Biotechnol. J. 2012, 10, 67–82. [Google Scholar] [CrossRef]
- Le, D.T.; Nishiyama, R.; Watanabe, Y.; Vankova, R.; Tanaka, M.; Seki, M.; Ham Le, H.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Tran, L.S. Identification and expression analysis of cytokinin metabolic genes in soybean under normal and drought conditions in relation to cytokinin levels. PLoS ONE 2012, 7, e42411. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Zhang, C.; Ma, J.Q.; Zhang, L.Y.; Yang, B.; Tang, X.Y.; Huang, L.; Zhou, X.T.; Lu, K.; Li, J.N. Genome-Wide Identification and Expression Profiling of Cytokinin Oxidase/Dehydrogenase (CKX) Genes Reveal Likely Roles in Pod Development and Stress Responses in Oilseed Rape (Brassica napus L.). Genes 2018, 9, 168. [Google Scholar] [CrossRef] [Green Version]
- Kurakawa, T.; Ueda, N.; Maekawa, M.; Kobayashi, K.; Kojima, M.; Nagato, Y.; Sakakibara, H.; Kyozuka, J. Direct control of shoot meristem activity by a cytokinin-activating enzyme. Nature 2007, 445, 652–655. [Google Scholar] [CrossRef]
- Kuroha, T.; Tokunaga, H.; Kojima, M.; Ueda, N.; Ishida, T.; Nagawa, S.; Fukuda, H.; Sugimoto, K.; Sakakibara, H. Functional analyses of LONELY GUY cytokinin-activating enzymes reveal the importance of the direct activation pathway in Arabidopsis. Plant Cell 2009, 21, 3152–3169. [Google Scholar] [CrossRef] [Green Version]
- Tokunaga, H.; Kojima, M.; Kuroha, T.; Ishida, T.; Sugimoto, K.; Kiba, T.; Sakakibara, H. Arabidopsis lonely guy (LOG) multiple mutants reveal a central role of the LOG-dependent pathway in cytokinin activation. Plant J. Cell Mol. Biol. 2012, 69, 355–365. [Google Scholar] [CrossRef]
- Lu, Z.; Shao, G.; Xiong, J.; Jiao, Y.; Wang, J.; Liu, G.; Meng, X.; Liang, Y.; Xiong, G.; Wang, Y.; et al. MONOCULM 3, an ortholog of WUSCHEL in rice, is required for tiller bud formation. J. Genet. Genom. 2015, 42, 71–78. [Google Scholar] [CrossRef]
- Hu, Y.S.; Ren, T.H.; Li, Z.; Tang, Y.Z.; Ren, Z.L.; Yan, B.J. Molecular mapping and genetic analysis of a QTL controlling spike formation rate and tiller number in wheat. Gene 2017, 634, 15–21. [Google Scholar] [CrossRef]
- Zhao, W.; Chao, H.; Zhang, L.; Ta, N.; Zhao, Y.; Li, B.; Zhang, K.; Guan, Z.; Hou, D.; Chen, K.; et al. Integration of QTL Mapping and Gene Fishing Techniques to Dissect the Multi-Main Stem Trait in Rapeseed (Brassica napus L.). Front. Plant Sci. 2019, 10, 1152. [Google Scholar] [CrossRef] [Green Version]
- Xu, A.; Wei, C. Comprehensive comparison and applications of different sections in investigating the microstructure and histochemistry of cereal kernels. Plant Methods 2020, 16, 8. [Google Scholar] [CrossRef]
- Kong, Y.; Ebrahimpour, P.; Liu, Y.; Yang, C.; Alonso, L.C. Pancreatic Islet Embedding for Paraffin Sections. J. Vis. Exp. 2018, 136, 57931. [Google Scholar] [CrossRef]
- Tarkowski, P.; Ge, L.; Yong, J.W.H.; Tan, S.N. Analytical methods for cytokinins. TrAC Trends Anal. Chem. 2009, 28, 323–335. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Yang, H.; Liu, J.; Huang, S.; Guo, T.; Deng, L.; Hua, W. Selection and evaluation of novel reference genes for quantitative reverse transcription PCR (qRT-PCR) based on genome and transcriptome data in Brassica napus L. Gene 2014, 538, 113–122. [Google Scholar] [CrossRef]
- Shi, B.; Vernoux, T. Hormonal control of cell identity and growth in the shoot apical meristem. Curr. Opin. Plant Biol. 2022, 65, 102111. [Google Scholar] [CrossRef]
- Takei, K.; Sakakibara, H.; Sugiyama, T. Identification of genes encoding adenylate isopentenyltransferase, a cytokinin biosynthesis enzyme, in Arabidopsis thaliana. J. Biol. Chem. 2001, 276, 26405–26410. [Google Scholar] [CrossRef] [Green Version]
- Erb, M.; Meldau, S.; Howe, G.A. Role of phytohormones in insect-specific plant reactions. Trends Plant Sci. 2012, 17, 250–259. [Google Scholar] [CrossRef] [Green Version]
- Koo, A.J.; Howe, G.A. The wound hormone jasmonate. Phytochemistry 2009, 70, 1571–1580. [Google Scholar] [CrossRef] [Green Version]
- Zhu, K.-M.; Xu, S.; Li, K.-X.; Chen, S.; Zafar, S.; Cao, W.; Wang, Z.; Ding, L.-N.; Yang, Y.-H.; Li, Y.-M.; et al. Transcriptome analysis of the irregular shape of shoot apical meristem in dt (dou tou) mutant of Brassica napus L. Mol. Breed. 2019, 39, 39. [Google Scholar] [CrossRef] [Green Version]
- Kuderova, A.; Gallova, L.; Kuricova, K.; Nejedla, E.; Curdova, A.; Micenkova, L.; Plihal, O.; Smajs, D.; Spichal, L.; Hejatko, J. Identification of AHK2- and AHK3-like cytokinin receptors in Brassica napus reveals two subfamilies of AHK2 orthologues. J. Exp. Bot. 2015, 66, 339–353. [Google Scholar] [CrossRef] [Green Version]
- Hutchison, C.E.; Li, J.; Argueso, C.; Gonzalez, M.; Lee, E.; Lewis, M.W.; Maxwell, B.B.; Perdue, T.D.; Schaller, G.E.; Alonso, J.M.; et al. The Arabidopsis histidine phosphotransfer proteins are redundant positive regulators of cytokinin signaling. Plant Cell 2006, 18, 3073–3087. [Google Scholar] [CrossRef] [Green Version]
- Mason, M.G.; Mathews, D.E.; Argyros, D.A.; Maxwell, B.B.; Kieber, J.J.; Alonso, J.M.; Ecker, J.R.; Schaller, G.E. Multiple type-B response regulators mediate cytokinin signal transduction in Arabidopsis. Plant Cell 2005, 17, 3007–3018. [Google Scholar] [CrossRef] [Green Version]
- Müller, D.; Waldie, T.; Miyawaki, K.; To, J.P.C.; Melnyk, C.W.; Kieber, J.J.; Kakimoto, T.; Leyser, O. Cytokinin is required for escape but not release from auxin mediated apical dominance. Plant J. Cell Mol. Biol. 2015, 82, 874–886. [Google Scholar] [CrossRef] [Green Version]
- Skoog, F.; Thimann, K.V. Further Experiments on the Inhibition of the Development of Lateral Buds by Growth Hormone. Proc. Natl. Acad. Sci. USA 1934, 20, 480–485. [Google Scholar] [CrossRef]
- Stirnberg, P.; van De Sande, K.; Leyser, H.M. MAX1 and MAX2 control shoot lateral branching in Arabidopsis. Development 2002, 129, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Chen, Z.; Zhang, S.; Zhang, W.; Jiang, G.; Zhao, X.; Zhai, W.; Pan, X.; Zhu, L. Characterizations and fine mapping of a mutant gene for high tillering and dwarf in rice (Oryza sativa L.). Planta 2005, 222, 604–612. [Google Scholar] [CrossRef]
- Beveridge, C.A. Long-distance signalling and a mutational analysis of branching in pea. Plant Growth Regul. 2000, 32, 193–203. [Google Scholar] [CrossRef]
- Gomez-Roldan, V.; Fermas, S.; Brewer, P.B.; Puech-Pages, V.; Dun, E.A.; Pillot, J.P.; Letisse, F.; Matusova, R.; Danoun, S.; Portais, J.C.; et al. Strigolactone inhibition of shoot branching. Nature 2008, 455, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Hayward, A.; Stirnberg, P.; Beveridge, C.; Leyser, O. Interactions between auxin and strigolactone in shoot branching control. Plant Physiol. 2009, 151, 400–412. [Google Scholar] [CrossRef]
- Domagalska, M.A.; Leyser, O. Signal integration in the control of shoot branching. Nat. Rev. Mol. Cell Biol. 2011, 12, 211–221. [Google Scholar] [CrossRef]
- Tang, M.; Tong, C.; Liang, L.; Du, C.; Zhao, J.; Xiao, L.; Tong, J.; Dai, X.; Helal, M.; Dai, W.; et al. A recessive high-density pod mutant resource of Brassica napus. Plant Sci. 2020, 293, 110411. [Google Scholar] [CrossRef]
- Salam, B.B.; Malka, S.K.; Zhu, X.; Gong, H.; Ziv, C.; Teper-Bamnolker, P.; Ori, N.; Jiang, J.; Eshel, D. Etiolated Stem Branching Is a Result of Systemic Signaling Associated with Sucrose Level. Plant Physiol. 2017, 175, 734–745. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Q.; Bai, X.; Zhang, C.; He, Y. Advanced high-throughput plant phenotyping techniques for genome-wide association studies: A review. J. Adv. Res. 2022, 35, 215–230. [Google Scholar] [CrossRef]
- Brachi, B.; Morris, G.P.; Borevitz, J.O. Genome-wide association studies in plants: The missing heritability is in the field. Genome Biol. 2011, 12, 232. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.U.; Saeed, S.; Khan, M.H.U.; Fan, C.; Ahmar, S.; Arriagada, O.; Shahzad, R.; Branca, F.; Mora-Poblete, F. Advances and Challenges for QTL Analysis and GWAS in the Plant-Breeding of High-Yielding: A Focus on Rapeseed. Biomolecules 2021, 11, 1516. [Google Scholar] [CrossRef]
- Sun, F.; Liu, J.; Hua, W.; Sun, X.; Wang, X.; Wang, H. Identification of stable QTLs for seed oil content by combined linkage and association mapping in Brassica napus. Plant Sci. 2016, 252, 388–399. [Google Scholar] [CrossRef]
- Wang, T.; Wei, L.; Wang, J.; Xie, L.; Li, Y.Y.; Ran, S.; Ren, L.; Lu, K.; Li, J.; Timko, M.P.; et al. Integrating GWAS, linkage mapping and gene expression analyses reveals the genetic control of growth period traits in rapeseed (Brassica napus L.). Biotechnol. Biofuels 2020, 13, 134. [Google Scholar] [CrossRef]
- Zhao, C.; Xie, M.; Liang, L.; Yang, L.; Han, H.; Qin, X.; Zhao, J.; Hou, Y.; Dai, W.; Du, C.; et al. Genome-Wide Association Analysis Combined With Quantitative Trait Loci Mapping and Dynamic Transcriptome Unveil the Genetic Control of Seed Oil Content in Brassica napus L. Front. Plant Sci. 2022, 13, 929197. [Google Scholar] [CrossRef]
- Li, Y.; Shen, J.; Wang, T.; Chen, Q.; Zhang, X.; Fu, T.; Meng, J.; Tu, J.; Ma, C.J.C. QTL analysis of yield-related traits and their association with functional markers in Brassica napus L. Aust. J. Agric. Res. 2007, 58, 759–766. [Google Scholar] [CrossRef]
- Shi, J.; Li, R.; Qiu, D.; Jiang, C.; Long, Y.; Morgan, C.; Bancroft, I.; Zhao, J.; Meng, J. Unraveling the complex trait of crop yield with quantitative trait loci mapping in Brassica napus. Genetics 2009, 182, 851–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, G.; Zhao, Z.; Liao, Y.; Hu, Y.; Shi, L.; Long, Y.; Xu, F. Quantitative trait loci for seed yield and yield-related traits, and their responses to reduced phosphorus supply in Brassica napus. Ann. Bot. 2012, 109, 747–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Zhang, Y.; Liu, X.; Chen, B.; Tu, J.; Tingdong, F. Detection of QTL for six yield-related traits in oilseed rape (Brassica napus) using DH and immortalized F2 populations. Theor. Appl. Genet. 2007, 115, 849–858. [Google Scholar] [CrossRef]
- Luo, X.; Ma, C.; Yue, Y.; Hu, K.; Li, Y.; Duan, Z.; Wu, M.; Tu, J.; Shen, J.; Yi, B.; et al. Unravelling the complex trait of harvest index in rapeseed (Brassica napus L.) with association mapping. BMC Genom. 2015, 16, 379. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Chen, B.; Xu, K.; Gao, G.; Yan, G.; Qiao, J.; Li, J.; Li, H.; Li, L.; Xiao, X.; et al. A genome-wide association study of plant height and primary branch number in rapeseed (Brassica napus). Plant Sci. 2016, 242, 169–177. [Google Scholar] [CrossRef]
- He, Y.; Wu, D.; Wei, D.; Fu, Y.; Cui, Y.; Dong, H.; Tan, C.; Qian, W. GWAS, QTL mapping and gene expression analyses in Brassica napus reveal genetic control of branching morphogenesis. Sci. Rep. 2017, 7, 15971. [Google Scholar] [CrossRef] [Green Version]
- Zheng, M.; Peng, C.; Liu, H.; Tang, M.; Yang, H.; Li, X.; Liu, J.; Sun, X.; Wang, X.; Xu, J.; et al. Genome-Wide Association Study Reveals Candidate Genes for Control of Plant Height, Branch Initiation Height and Branch Number in Rapeseed (Brassica napus L.). Front. Plant Sci. 2017, 8, 1246. [Google Scholar] [CrossRef]
- Li, B.; Gao, J.; Chen, J.; Wang, Z.; Shen, W.; Yi, B.; Wen, J.; Ma, C.; Shen, J.; Fu, T.; et al. Identification and fine mapping of a major locus controlling branching in Brassica napus. Theor. Appl. Genet. 2020, 133, 771–783. [Google Scholar] [CrossRef]
- Li, P.; Su, T.; Zhang, B.; Li, P.; Xin, X.; Yue, X.; Cao, Y.; Wang, W.; Zhao, X.; Yu, Y.; et al. Identification and fine mapping of qSB.A09, a major QTL that controls shoot branching in Brassica rapa ssp. chinensis Makino. Theor. Appl. Genet. 2020, 133, 1055–1068. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Xue, N.; Sun, C.; Tao, J.; Mi, C.; Yuan, Y.; Pan, X.; Gui, M.; Long, R.; Ding, R.; et al. Transcriptomic Profiling of Shoot Apical Meristem Aberrations in the Multi-Main-Stem Mutant (ms) of Brassica napus L. Genes 2023, 14, 1396. https://doi.org/10.3390/genes14071396
Wang Q, Xue N, Sun C, Tao J, Mi C, Yuan Y, Pan X, Gui M, Long R, Ding R, et al. Transcriptomic Profiling of Shoot Apical Meristem Aberrations in the Multi-Main-Stem Mutant (ms) of Brassica napus L. Genes. 2023; 14(7):1396. https://doi.org/10.3390/genes14071396
Chicago/Turabian StyleWang, Qian, Na Xue, Chao Sun, Jing Tao, Chao Mi, Yi Yuan, Xiangwei Pan, Min Gui, Ronghua Long, Renzhan Ding, and et al. 2023. "Transcriptomic Profiling of Shoot Apical Meristem Aberrations in the Multi-Main-Stem Mutant (ms) of Brassica napus L." Genes 14, no. 7: 1396. https://doi.org/10.3390/genes14071396