
Phenotypes and Genotypes in Patients with SMC1A-Related Developmental and Epileptic Encephalopathy
by
 , ,
, ,
Xiuhua L. Bozarth
1,* ,
,
Jonathan Lopez
1,
He Fang
2,
Jacqueline Lee-Eng
1,
Zhijun Duan
3,4 and
Xinxian Deng
2,* 1
Division of Neurology, Seattle Children’s Hospital, University of Washington, Seattle, WA 98105, USA
2
Department of Laboratory Medicine and Pathology, University of Washington, Seattle, WA 98195, USA
3
Division of Hematology, University of Washington, Seattle, WA 98195, USA
4
Institute for Stem Cell and Regenerative Medicine, University of Washington, Seattle, WA 98195, USA
*
Authors to whom correspondence should be addressed.
Genes 2023, 14(4), 852; https://doi.org/10.3390/genes14040852
Submission received: 17 February 2023
/
Revised: 27 March 2023
/
Accepted: 29 March 2023
/
Published: 31 March 2023
(This article belongs to the Section Human Genomics and Genetic Diseases)
Abstract
:The X-linked SMC1A gene encodes a core subunit of the cohesin complex that plays a pivotal role in genome organization and gene regulation. Pathogenic variants in SMC1A are often dominant-negative and cause Cornelia de Lange syndrome (CdLS) with growth retardation and typical facial features; however, rare SMC1A variants cause a developmental and epileptic encephalopathy (DEE) with intractable early-onset epilepsy that is absent in CdLS. Unlike the male-to-female ratio of 1:2 in those with CdLS associated with dominant-negative SMC1A variants, SMC1A-DEE loss-of-function (LOF) variants are found exclusively in females due to presumed lethality in males. It is unclear how different SMC1A variants cause CdLS or DEE. Here, we report on phenotypes and genotypes of three females with DEE and de novo SMC1A variants, including a novel splice-site variant. We also summarize 41 known SMC1A-DEE variants to characterize common and patient-specific features. Interestingly, compared to 33 LOFs detected throughout the gene, 7/8 non-LOFs are specifically located in the N/C-terminal ATPase head or the central hinge domain, both of which are predicted to affect cohesin assembly, thus mimicking LOFs. Along with the characterization of X-chromosome inactivation (XCI) and SMC1A transcription, these variants strongly suggest that a differential SMC1A dosage effect of SMC1A-DEE variants is closely associated with the manifestation of DEE phenotypes.
1. Introduction
Next-generation sequencing has accelerated the discovery of important genes enriched in pathogenic de novo variants in severe, often previously undiagnosed, developmental disorders [1]. One such gene is the X-linked SMC1A (MIM: 300040). SMC1A is an X-linked gene that escapes X-chromosome inactivation (XCI) in females with an estimate of ~30% expression from the allele on the inactive X chromosome (Xi) [2,3,4,5]. SMC1A encodes a protein dimerizing with SMC3 to form the core of cohesin, a multisubunit protein complex involved in chromosome segregation, DNA repair, and regulation of chromosomal architecture [6,7]. Pathogenic variants in SMC1A as well as other cohesin subunit-encoding and cohesin regulatory protein-encoding genes, can often cause Cornelia de Lange syndrome (CdLS), a developmental condition involving multiple systems. The common features in CdLS include intellectual disability (ID), distinctive facial features with synophrys, short nose, concave nasal ridge, ruptured nasal tip, long and/or smooth philtrum prenatal and postnatal growth retardation, and congenital upper limb anomalies ranging from small hands to severe reduction anomalies of the forearms. Some patients have heart malformations, cleft palate, and nasolacrimal duct obstruction [8]. Multiple genes are associated with CdLS, including heterozygous pathogenic variants in NIPBL, RAD21, SMC3, and BRD4, or hemizygous pathogenic variants in HDAC8 and SMC1A. NIPBL variants are the most common and associated with 60 to 70% of CdLS patients. SMC1A pathogenic variants, mostly missense and small in-frame mutations, were found in about 5% of CdLS patients with a male-to-female ratio of 1:2, often associated with mild CdLS phenotypes [9,10]. The SMC1A variants in these patients do not affect their overall expression and are protein-sparing, suggesting a dominant negative effect of these mutant SMC1A proteins that could form partially functional cohesin and result in mild CdLS [5,11].
Intriguingly, cases have been increasingly reported of females with heterozygous de novo SMC1A pathogenic variants, mostly loss-of-function mutations (LOFs). These variants are associated with a more severe clinical phenotype of developmental and epileptic encephalopathy (DEE), a devastating form of early onset intractable epilepsy beginning in infancy and associated with global developmental delay, cognitive dysfunction, and ongoing epileptiform activity [9,12,13,14,15,16,17,18,19,20,21,22,23,24]. Most reported cases did not show significant brain MRI abnormalities, but cases with holoprosencephaly have been reported [15,23]. Other findings in brain MRI include thin corpus callosum, enlarged extra-axial space, cerebral volume loss, cerebellar atrophy, small cavum septum, and mild periventricular white matter abnormalities [12,13,15]. SMC1A-DEE is clearly distinct from SMC1A-related CdLS but resembles the clinical phenotype of Rett syndrome (RTT), a severe DEE mostly caused by heterozygous mutations of the X-linked MECP2 gene, which encodes a chromatin regulator to mediate DNA methylation and gene repression in the brain [9,25]. This suggests a novel link between SMC1A and brain development in females.
It is unclear how these SMC1A LOF variants cause severe DEE instead of CdLS. Here, we report on the characterization of phenotypes and genotypes of three female patients with de novo pathogenic SMC1A-DEE variants, including a previously unreported variant affecting a transcriptional splicing site as well as the XCI patterns and SMC1A transcription. We further summarize 41 SMC1A-DEE variants reported (including the cases presented here) to characterize common and patient-specific phenotypic features and associated variants for insights related to the etiology of SMC1A-DEE.
2. Materials and Methods
Patients: Under one approved IRB (IRB #: STUDY00001445), we recruited three family trios with heterozygous SMC1A-DEE variants at Seattle Children’s Hospital (SCH, Seattle, WA, USA). Data on clinical phenotypes were collected by chart reviews and evaluations by their neurologists. Whole exome sequencing (WES) was performed for blood samples of all three patient family trios. The most representative transcript, Genbank accession NM_006306, is used when examining the location of these variants on SMC1A mRNA. Informed consent was obtained from all affected individuals or their guardians per the specifications of Seattle Children’s Hospital and the University of Washington (Seattle, WA, USA), including consent to publish photographs.
Genomic DNA isolation and confirmation of SMC1A variants: Genomic DNA (gDNA) was extracted from whole blood from patient families using the Qiagen DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA, USA). gDNA was subject to PCR with primer pairs flanking the SMC1A variants, followed by Sanger sequencing for confirmation. The primer information is in Table S1. Note that the blood sample of the father of patient 3 (P3) was not available.
RNA isolation and expression analysis: Total RNA was extracted from whole blood using the QIAamp RNA Blood Mini Kit (Qiagen, Valencia, CA, USA) with on-column DNase I digestion. cDNA was produced from reverse transcription (RT) of total RNA and used for PCR with specific primers flanking the variants to detect SMC1A transcripts. Sanger sequencing was then performed to confirm alternative transcripts in patient 1 (P1) carrying a splice-site variant or the missense transcript in patient 3. Note that to confirm the transcript with exons 4–5 skipping in P1, pre-amplified PCR product from cDNA with primers flanking exons 2–6 was subjected to HindIII digestion that only cut the wild-type transcript carrying a HindIII site in exon 5. The digested product was diluted and used for another round of PCR to only amplify the transcript with exons 4–5 skipping followed by Sanger sequencing. The primer information is in Table S1.
XCI evaluation: Methyl-sensitive PCR-based assay for the AR (androgen receptor) locus (i.e., HUMARA assay) containing the polymorphic trinucleotide repeats was performed for gDNA from blood as previously described with minor modifications [26]. In brief, DNA samples were PCR amplified for the CAG repeat region (CTG from the minus strand) in the AR exon 1. In parallel, DNA samples were digested with the methyl-sensitive restriction enzyme HpaII, followed by PCR. Sanger sequencing and comparison of the amplified product from DNA with and without digestion were used to determine the presence of polymorphic repeat in test samples and the extent of XCI skewing. For patient 2 (P2), showing no polymorphic CAG repeats of AR (i.e., both paternal and maternal alleles have the same number of repeats), a similar methyl-sensitive PCR-based assay was performed for the RP2 (retinitis pigmentosa 2) locus containing the polymorphic CAAA repeats [27]. The primer information is in Table S1.
3. Results
Clinical phenotypes (Table 1 and Table 2): P1 is currently an 11-years old Caucasian girl. She was born at full term. At birth, she weighed 2.35 kg (1.7th centile), her length was 45.7 cm (3rd centile), and her head circumference (OFC) was 32.5 cm (12th centile). She had mild neonatal jaundice that did not require treatment. She does not have facial CdLS features. She has very mild 2–3 syndactyly on both feet, severe pes planovalgus deformity, the myopia of both eyes with astigmatism, diffuse hypotonia, and mild ptosis bilaterally. She had G-tube placement at the age of 7 years. She had a history of normal development until approximately 4 months of age when she had her first seizure. At the age of 11 years, she has a profound global developmental delay. She remains nonverbal. She is able to grab objects with both hands and crawl but cannot stand or walk independently. Her weight is at the 10th centile, her height is at the 15th centile, and her OFC is at the 1.7th centile. Her seizures started at the age of 4 months. She has multiple seizure types, and seizures are refractory to treatment. Her seizure types include generalized tonic–clonic, myoclonic, atonic, atypical absence seizures, and focal onset motor and nonmotor seizures. She has frequent clusters of seizures and status epilepticus, requiring hospitalization. She also has vagal nerve stimulation placement and a trial of modified Atkin’s diet that did not show clear efficacy. She continues to have frequent seizures and is on four AEDs, including topiramate, perampanel, epidiolex, and lacosamide.
P2 is currently an 8-years old Hispanic girl. She was born full term. At birth, she weighed 3.26 kg (55th centile), her length was 48.6 cm (23rd centile), and her OFC was 33.1 cm (14th centile). She was found to have severe axial and appendicular hypotonia, neonatal hyperbilirubinemia, and feeding difficulties. She has a wide cleft palate, microform cleft lip, synophrys of her eyebrows, tapered fingers, and hirsutism. She had G-tube placement at 9 months of age. She had a heart murmur, and her echocardiogram revealed left ventricular hypertrophy at the age of 9 months. Her kidney ultrasound revealed right hydronephrosis and small bilateral cysts. She also has hyperopia and astigmatism at the age of 15 months on ophthalmological examination. She has mild cortical visual impairment on visual evoked potential test at the age of 20 months. She was found to have sleep apnea. She lacked progression of developmental milestones. At the age of 8 years, she has a profound global developmental delay. She remains nonverbal and does not have good head control. There is a paucity of spontaneous movements. Her current weight is at the 43rd centile, height is at the 0.2th centile, and OFC is <1st centile. Her seizures started at the age of 2 months, and developmental infantile spasms at the age of 9 months. Her seizure types include tonic/atonic, tonic–clonic, focal motor, and focal nonmotor seizures. She is still on two AEDs, including clobazam and levetiracetam. A modified Atkin’s diet was partially effective.
P3 is currently a 9-years old Asian descendant girl. She was born at 36 + 6/7 weeks of gestational age. At birth, she weighed 2.2 kg (<1st centile), her length was 44.5 cm (<1st centile), and her OFC was 30.5 cm (<1st centile). She has mild synophrys, a mid-low anterior hairline, a mild small jaw, small hands and feet, shortened fifth digits, and mild proximal placement of her thumbs. She has myopia, astigmatism, and mild bilateral ptosis. She had a history of gastroesophageal reflux disease as an infant. Her audiologic testing showed a mild conductive hearing loss, and BAER revealed a mild conductive high-frequency hearing loss at the age of 1 year. Her renal ultrasound study at the age of 1 year revealed bilateral minimal central pelviectasis. At the age of 9 years, she has a severe global developmental delay. She is a minimally verbal communicator who uses vocalizations, occasional word approximations, signs, gestures, and picture symbols to communicate. She is able to understand picture symbols representing common actions, objects, and people, as well as some descriptive words. She uses a combination of methods, including vocalizations, word approximations, sign language, and picture symbols, on a speech device to make a variety of requests and communicate her basic needs. She is able to walk and run independently with a mild wide-based gait. Her current weight is at the 83rd centile, height at the 71st centile, and OFC at the 10th centile. She had a history of a cluster of febrile seizures at the age of 2 years and 11 months. Her unprovoked seizures started at 3.5 years old. Her seizure types include tonic–clonic, tonic/atonic, and myoclonic. Her seizures cluster and occur from less than a month up to seven seizures per week. She did not respond to Atkin’s diet. She currently is on two AEDs, including clobazam and brivaracetam. Brivaracetam significantly reduced her seizure burden.
Diagnostic workup results: SMC1A mutations in all three patients were originally identified by WES as the only pathogenic variants linked to DEE (Table 3). WES for P1 showed heterozygous de novo likely pathogenic novel variant in the X-linked SMC1A gene at c.615+5G>A in intron 4. P1 underwent multiple genetic tests without detected abnormalities, including oligonucleotide array, single gene sequencing for SCN1A, CDKL5, SLC2A1, sequencing, deletion, and duplication testing for MECP2, and the epiSEEK panel (Courtagen, Woburn, MA, USA) that included 71 known epilepsy genes. She has had normal metabolic screenings for plasma amino acids, urine organic acids, acylcarnitine profile, transferrin isoelectric focusing to screen for congenital disorders of glycosylation, AASA, and P6C levels to screen for pyridoxine-dependent epilepsy.
WES for P2 showed a heterozygous de novo pathogenic indel variant in SMC1A at c.1636_1638del ATT. P2 had normal chromosomal microarray analysis prior to WES. She has had normal metabolic screenings for plasma amino acids, urine organic acids, acylcarnitine profile, transferrin isoelectric focusing to screen for congenital disorders of glycosylation, and urine quantitative mucopolysaccharides.
WES for P3 showed a heterozygous de novo pathogenic missense variant in SMC1A at c.1487G>A (p.R496H). P3 had normal genetic testing prior to WES, including prenatal karyotype and chromosome microarray analysis on chorionic villi sampling and postnatal NIPBL sequencing. She has had normal metabolic screenings for ammonia, lactate, pyruvate, amino acids, acylcarnitine profile, urine oligosaccharides, and urine organic acids.
Brain MRI in P1 showed a small cyst in the left anterior temporal region at the age of 7 months and at the age of 9 years with no change. Brain MRI in P2 at the age of 2 years was normal. P3 did not have a brain MRI.
EEG showed diffuse background slowing in all three patients (Table 2). P2 had a history of hypsarrhythmia on her EEG. Her EEG did not have a posterior dominant rhythm and did not have normal sleep architecture at the age of 3 years.
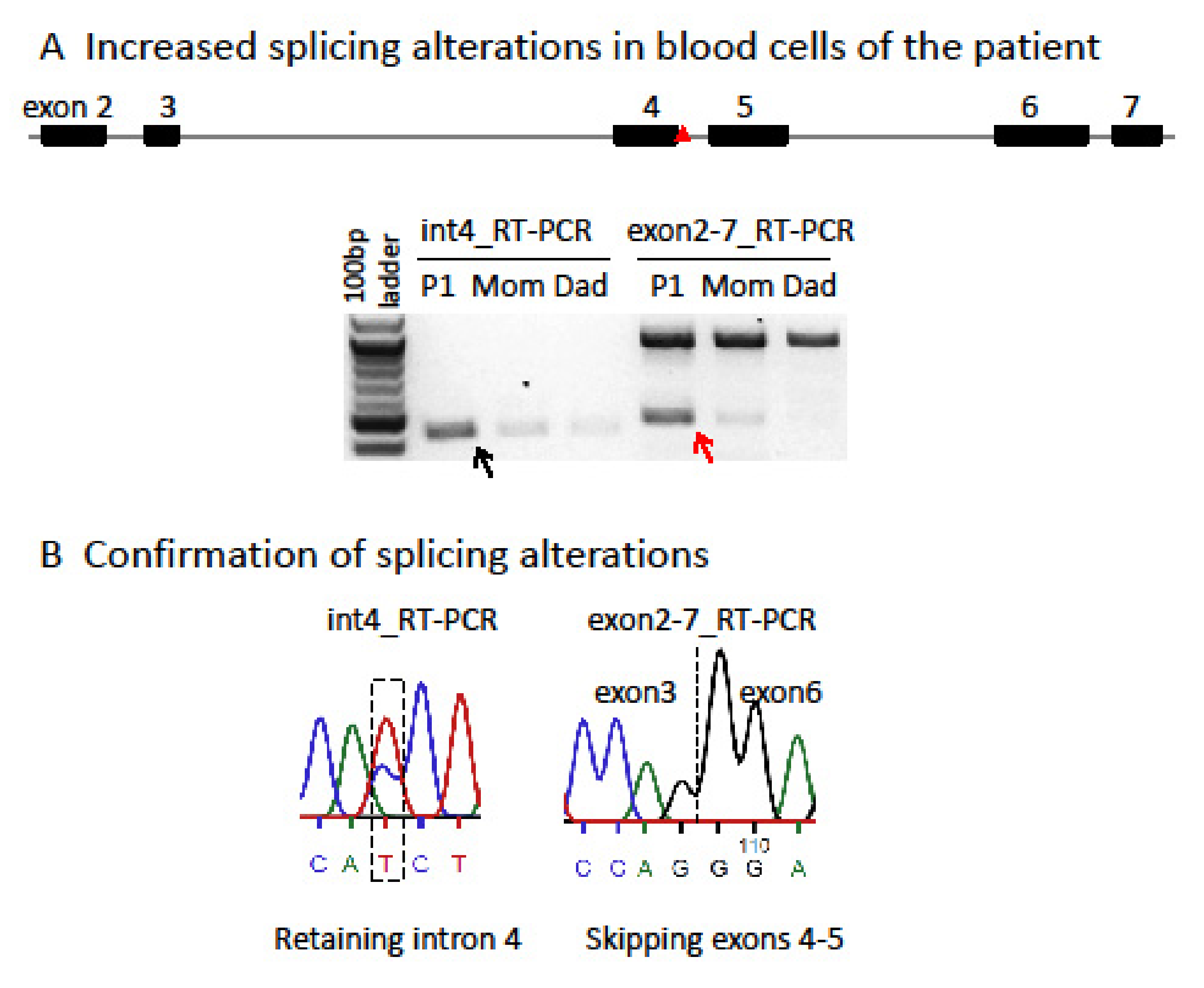
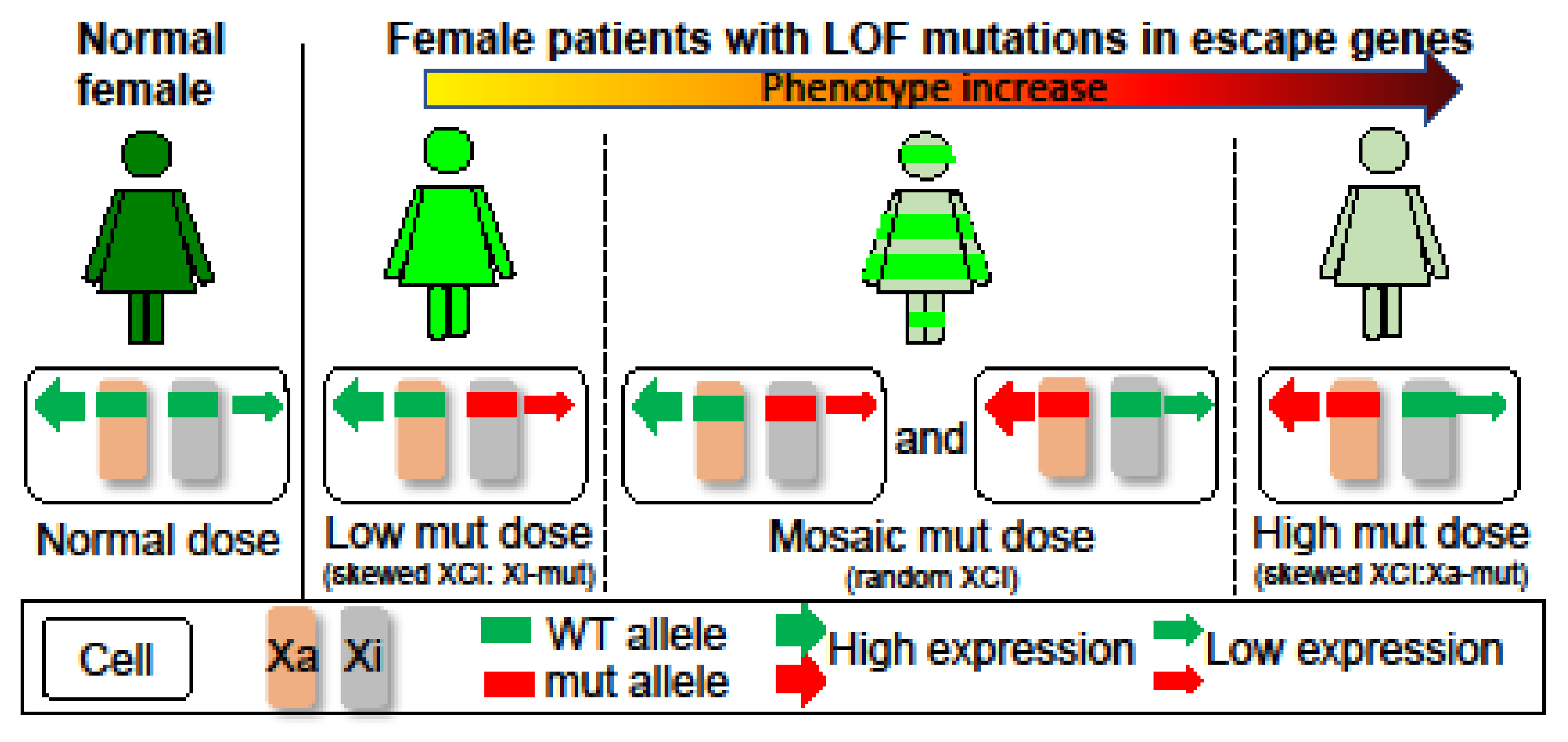
Characterization and comparison of de novo SMC1A-DEE variants in our study and literature: Sanger sequencing for PCR products flanking the variant confirmed that P1 carries an unreported de novo heterozygous splice-site mutation (c.615+5G>A) in intron 4 of SMC1A (Figure S1). Interestingly, two variants (c.615G>A and c.615+1G>C) were recently reported to affect the same splice site (Table 3) [15,19]. Note that in the report by Symonds et al. [15], case 3 is identified as a splice-site variant at c.549G>A, which is at the exon junction of the SMC1A transcript NM:001281463 but not NM_006306. Thus, it corresponds to c.615G>A in NM_006306. This variant has also been reported in a new patient (case 12) [19]. In addition, two other variants (c.616-2A>G, VCV000159962.5; c.615+2T>C, VCV000804008.1) out of a total of seven pathogenic splicing variants deposited in ClinVar also affect this splice-site but has not been reported in any literature, strongly indicating a pathogenic splicing hotspot. RT-PCR for RNA from blood using primers specific for intron 4 or spanning exons 2–7 showed two alternative transcripts in P1, which were not or only barely detected in the mother or father (Figure 1). Using a different pair of primers spanning exons 2–6 showed a similar result. Sanger sequencing confirmed these alternative transcripts. This is consistent with the prediction of splice-site interference from this variant. Both abnormally spliced transcripts cause frameshifts and premature translational stop codons and thus cannot produce functional proteins consistent with LOF (Table S2 and Figure 2). We performed a methyl-sensitive PCR-based assay for XCI evaluation and found that P1 has a random XCI pattern in blood, i.e., an equal chance of the maternal or paternal X being the Xi (Figure S2). Given that SMC1A escapes XCI with ~30% of expression levels from the allele on the Xi [5], random XCI in P1 would lead to a mosaic dosage effect at the tissue level (Figure 3), i.e., expression levels of this variant are ~two-fold lower in cells when it is located on the Xi versus in cells when it is located on the active X chromosome (Xa).
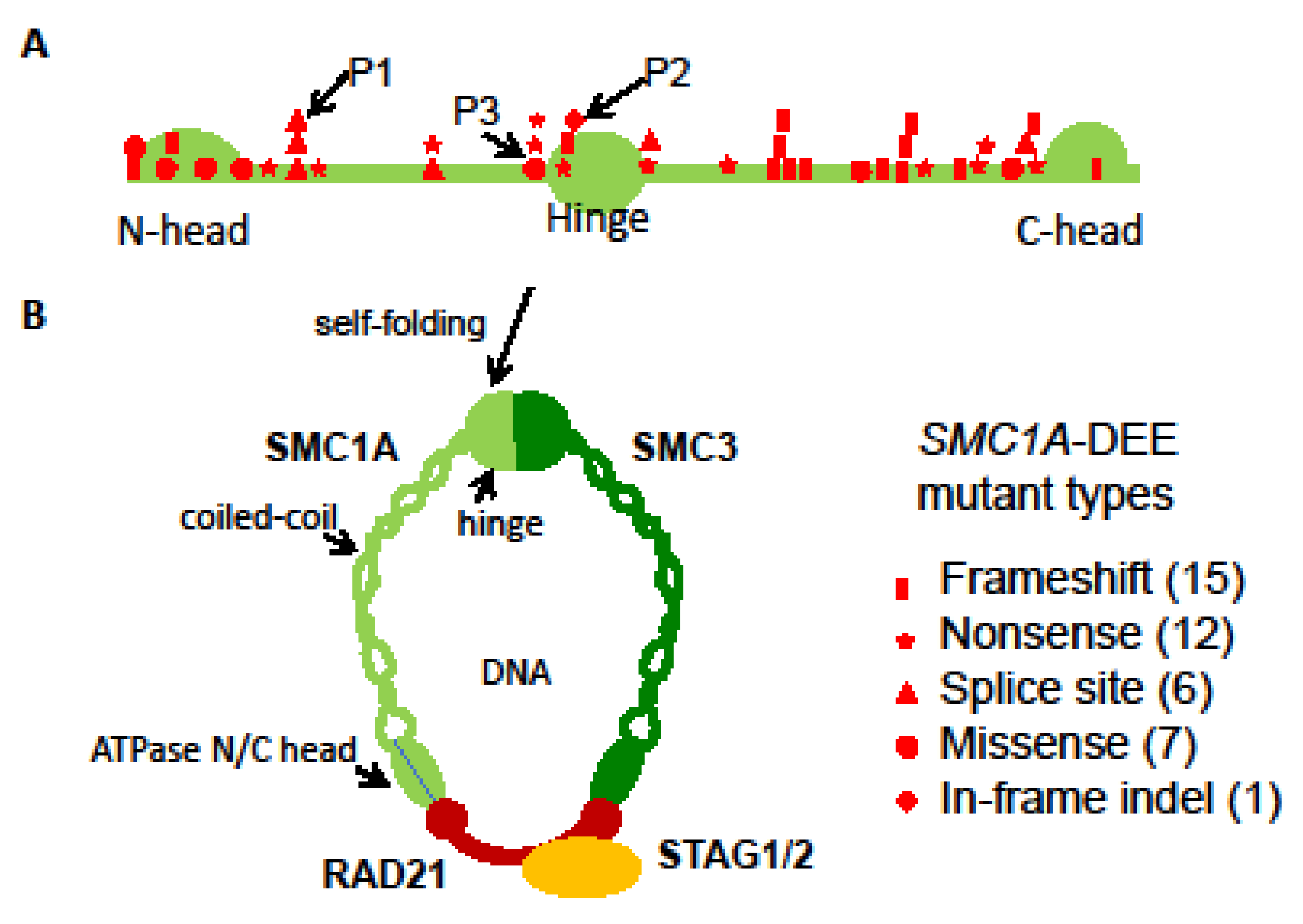
A de novo heterozygous in-frame indel (c.1636-1638delATT) was previously reported in P2 by Seattle Children’s Hospital in 2016 (Table 3) [21] and confirmed by Sanger sequencing (Figure S1). This variant results in missing an isoleucine residue at position 546 (p.Ile546del) in the hinge. This residue is highly conserved across species and is important for hinge domain dimerization with SMC3 (Figure 2) [9,28,29]. We hypothesize that this variant affects the assembly of the cohesin complex and thus has a similar dosage effect as those LOF ones (Figure 2). P2 also has a random XCI pattern in blood, again suggesting a mosaic dosage effect (Figure 3 and Figure S3).
P3 is a previously unreported patient carrying a de novo missense variant (c.1487G>A; Table 3), which has been reported in three other unrelated female probands previously classified as mild CdLS cases with considerable growth and development variability [28]. Sanger sequencing confirmed this variant in P3 but not in the mom. This mutation leads to an arginine to histidine substitution (p. R496H) at the junction between the coiled coil and the hinge (Figure 2). This region is also highly conserved and predicted to be vital for hinge domain dimerization [28]. Thus, similar to the variant in P2, this missense variant could have a dosage effect as those LOFs. Interestingly, seizures have also been noticed in these patients, including our P3. P3 had an increased frequency of seizures starting at the age of 3 years but not as severe as in P1 and P2 (Table 1). XCI testing showed a completely skewed inactivation of the paternal X in P3 blood (Figure S4), allowing examination of the SMC1A escape status. RT-PCR using cDNA-specific primers flanking this variant followed by Sanger sequencing reveals expression of both wild-type (WT) and variant alleles in blood cells with skewed XCI, consistent with the escape of SMC1A from XCI (Figure S5). In addition, the peak ratio between the WT allele (i.e., G) and the variant (A) is increased by ~4-fold from the cDNA-derived PCR product compared to that from the gDNA-derived PCR product, suggesting most of SMC1A expression is from the WT allele. Given that the allele on the Xa is expressed at a higher level than that on the Xi, we can infer that this variant is on the paternal Xi, and the expression level of the variant (i.e., the escape level) is about 25% of that of the WT allele on the maternal Xa. This suggests a mild dosage effect and is consistent with an alleviated DEE phenotype in P3 (Figure 3).
Our characterization of these three heterozygous SMC1A-DEE variants and their XCI patterns suggests a possible link between SMC1A dosage reduction to the severity of phenotype (Figure 3). To further test this, we surveyed 41 known SMC1A-DEE variants, including ours and one unreported, and characterized common and patient-specific features (Table 3) [9,12,13,14,15,16,17,18,19,20,21,22,23,24]. Clearly, SMC1A-DEE variants are mostly LOF caused by nonsense, frameshift, or splice-site interference (Figure 2). Seven of the eight non-LOF variants, including two from our cohort (variants in P2 and P3) and one unreported (c.3241A>T) for which the patient will be included in our cohort in the future, are located in the N/C-terminal ATPase head or in the central hinge domain. These regions are predicted to disrupt interactions with SMC3 or RAD21, respectively, and to prevent cohesin assembly, thus probably mimicking LOF (Figure 2) [9,28,29]. All 41 variants occurred in heterozygous girls, suggesting lethality in males. These variants result in intractable early-onset epilepsy (with some features similar to PCDH19 clustering epilepsy), progressive neurodevelopmental impairment, and (in some) regression. Six of these SMC1A LOFs have even been linked to the most severe midline brain defect, holoprosencephaly (HPE) [23]. Interestingly, two of the HPE variants were also reported in unrelated patients with a minor phenotype, including late-onset DEE and no midline brain defects [17,24]. Overall, ten variants are associated with patients, including our P3, who show a lesser phenotype: seizure onset between birth to 15 months with variable response to antiseizure medication and less severe GDD with independent walking and limited expressive speech. This is consistent with the study by Chinen et al. [24], indicating that the age of seizure onset at 15 months or later in six cases is associated with better motor development. When the XCI status is available, these patients with less severe DEE are more often with skewed XCI. Overall, these suggest that different XCI status probably contributes to a patient-specific manifestation of the disease as proposed by our dosage model (Figure 3).
4. Discussion
The phenotype of SMC1A-DEE is clearly distinct from CdLS but resembles MECP2-linked RTT: early onset intractable epilepsy and severe or profound intellectual disability. In addition, heterozygous variants of other chromatin regulators (e.g., HDAC8, SATB2, and HNRNPU) also often cause RTT-like phenotypes [25]. These clinical and genetic observations strongly suggest a dosage-sensitive role of chromatin components in early brain development. Considering that RTT-like phenotypes occur in the early postnatal period, mutations of these master chromatin regulators could disrupt multiple pathways critical for the early development of complicated neuronal networks, such as synapse formation that peaks sometime in the first year of life and is the basis of much of the learning [30]. Indeed, it has been reported that in mice heterozygous for a knockout of SMC3, which encodes the SMC1A partner and the core subunit of cohesin, altered neural gene expression during cortical synapse formation leads to defective synapse development and anxiety-related behavior [31].
The etiology of SMC1A-DEE has yet to be studied due to limited patient numbers and the inaccessibility of brain tissue. A phenotype and genotype comparison of SMC1A-DEE patients in our study suggests an X-specific differential dosage effect that is influenced by the XCI status (Figure 3) [32]. Differential SMC1A insufficiency in SMC1A-DEE individuals with random XCI could further lead to cellular interference (i.e., interference between cells with differential levels of SMC1A) at tissue levels, similar to the proposed for the etiology of PCDH19 clustering epilepsy [33]. Interestingly, SMC1A-DEE and PCDH19 clustering epilepsy share some epilepsy-related features, e.g., drug resistance and seizure clustering [15]. In contrast, SMC1A-DEE individuals with skewed XCI toward the LOF variant, such as our P3, would have a minor deficiency compared to that in normal females with expression from both alleles and thus show a less severe phenotype (Figure 3). Additional products from an expression of the allele of XCI-escape genes on the Xi have been proposed to underlie sex differences between males and females, including brain development, and contribute to phenotypes associated with X aneuploidy [32,34]. Interestingly, the ortholog of SMC1A in mice does not escape XCI in the brain [35], and thus, studies of SMC1A-DEE in mouse animal models could fail to recapitulate the differential dosage effect of SMC1A in humans. In vitro neural models using patient-derived induced pluripotent stem cells carrying the SMC1A-DEE variant on the Xi or Xa could be more applicable in functional studies of SMC1A-DEE. Our collections of blood cells from rare SMC1A-DEE patients will be valuable for such studies.
5. Conclusions
Characterization of SMC1A-DEE variants as LOFs suggests a dosage-sensitive role of SMC1A in early brain function and development. The XCI status in female SMC1A-DEE patients plays an important role in the manifestation of DEE phenotypes.
6. Future Directions
There is no effective treatment for this patient population. Understanding the disease mechanism has the potential to provide new therapeutic targets. In vitro neural models using patient-derived induced pluripotent cells could be an important tool to understand the pathology, while animal models will provide further in vivo insights for this condition. Both disease models could provide new revenues for personalized therapeutic treatments. Additionally, understanding the natural history of SMC1A-DEE will lay the important groundwork for clinical trial readiness by defining disease severity and change in the symptoms over time. It is especially crucial for disease-modifying treatment, such as gene therapy for increasing SMC1A expression. Thus, multicenter studies and international collaboration are important steps toward understanding this rare genetic disorder and uncovering new therapies.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/genes14040852/s1. Figure S1, Sanger sequencing confirms de novo SMC1A-DEE variants in three heterozygous female patients; Figure S2, random XCI patterns in the blood of the heterozygous SMC1A-DEE patient 1; Figure S3, random XCI patterns in the blood of the heterozygous SMC1A-DEE patient 2; Figure S4, skewed XCI patterns in the blood of the heterozygous SMC1A-DEE patient 3; Figure S5, the SMC1A-DEE variant escapes from skewed XCI in the blood of the heterozygous patient 3; Table S1, primers used in this study; Table S2, truncated protein sequence predicted from alternative transcripts of SMC1A in patient 1.
Author Contributions
Conceptualization, X.L.B. and X.D.; Methodology, X.L.B., J.L., H.F. and X.D.; Validation and Data Analysis, H.F. and X.D.; Writing—Review and Editing, X.L.B., J.L., Z.D. and X.D.; Project Administration, J.L.-E.; Funding Acquisition, X.L.B. and X.D. All authors have read and agreed to the published version of the manuscript.
Funding
The research reported in this publication was supported in part by the American Epilepsy Society Seed Grant (419380040101). This work is also supported in part by grants from the National Institutes of Health (GM127327 to X.D. and GM131745 to C.M.D).
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Institutional Review Board of Seattle Children’s Hospital (IRB #: STUDY00001445 and approval date: 11 July 2018).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
All data supporting the conclusions of this manuscript are provided in the text and figures. Web Resources—The URLs for data presented include: OMIM: http://omim.org/ (accessed on 1 January 2023); ExAC: http://exac.broadinstitute.org/ (accessed on 5 December 2022); gnomAD: http://gnomad.broadinstitute.org (accessed on 5 December 2022); ClinVar: https://www.ncbi.nlm.nih.gov/clinvar/ (accessed on 5 December 2022).
Acknowledgments
We thank the patients’ families for participating and sharing their stories.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017, 542, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Miller, A.P.; Carrel, L.; Rupert, J.L.; Davies, K.E.; Willard, H.F. The DXS423E gene in Xp11.21 escapes X chromosome inactivation. Hum. Mol. Genet. 1995, 4, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Carrel, L.; Willard, H.F. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 2005, 434, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Balaton, B.P.; Cotton, A.M.; Brown, C.J. Derivation of consensus inactivation status for X-linked genes from genome-wide studies. Biol. Sex Differ. 2015, 6, 35. [Google Scholar] [CrossRef] [Green Version]
- Parenti, I.; Rovina, D.; Masciadri, M.; Cereda, A.; Azzollini, J.; Picinelli, C.; Limongelli, G.; Finelli, P.; Selicorni, A.; Russo, S.; et al. Overall and allele-specific expression of the SMC1A gene in female Cornelia de Lange syndrome patients and healthy controls. Epigenetics 2014, 9, 973–979. [Google Scholar] [CrossRef] [Green Version]
- Davidson, I.F.; Peters, J.-M. Genome folding through loop extrusion by SMC complexes. Nat. Rev. Mol. Cell Biol. 2021, 22, 445–464. [Google Scholar] [CrossRef]
- Oldenkamp, R.; Rowland, B.D. A walk through the SMC cycle: From catching DNAs to shaping the genome. Mol. Cell 2022, 82, 1616–1630. [Google Scholar] [CrossRef] [PubMed]
- Selicorni, A.; Mariani, M.; Lettieri, A.; Massa, V. Cornelia de Lange Syndrome: From a Disease to a Broader Spectrum. Genes 2021, 12, 1075. [Google Scholar] [CrossRef] [PubMed]
- Huisman, S.; Mulder, P.A.; Redeker, E.; Bader, I.; Bisgaard, A.-M.; Brooks, A.; Cereda, A.; Cinca, C.; Clark, D.; Cormier-Daire, V.; et al. Phenotypes genotypes in individuals with SMC1A variants. Am. J. Med. Genet. A 2017, 173, 2108–2125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyle, M.I.; Jespersgaard, C.; Brøndum-Nielsen, K.; Bisgaard, A.-M.; Tümer, Z. Cornelia de Lange syndrome. Clin. Genet. 2015, 88, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Feldman, R.; Zhang, Z.; Deardorff, M.A.; Haverfield, E.V.; Kaur, M.; Li, J.R.; Clark, D.; Kline, A.D.; Waggoner, D.J.; et al. SMC1A expression and mechanism of pathogenicity in probands with X-Linked Cornelia de Lange syndrome. Hum. Mutat. 2009, 30, 1535–1542. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, J.H.R.; Tim-Aroon, T.; Shieh, J.; Merrill, M.; Deeb, K.K.; Zhang, S.; Bass, N.E.; Bedoyan, J.K. Novel SMC1A frameshift mutations in children with developmental delay and epilepsy. Eur. J. Med. Genet. 2015, 58, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Jansen, S.; Kleefstra, T.; Willemsen, M.H.; de Vries, P.; Pfundt, R.; Hehir-Kwa, J.Y.; Gilissen, C.; Veltman, J.A.; de Vries, B.B.A.; Vissers, L.E.L.M. De novo loss-of-function mutations in X-linked SMC1A cause severe ID and therapy-resistant epilepsy in females: Expanding the phenotypic spectrum. Clin. Genet. 2016, 90, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Lebrun, N.; Lebon, S.; Jeannet, P.-Y.; Jacquemont, S.; Billuart, P.; Bienvenu, T. Early-onset encephalopathy with epilepsy associated with a novel splice site mutation in SMC1A. Am. J. Med. Genet. A 2015, 167A, 3076–3081. [Google Scholar] [CrossRef] [PubMed]
- Symonds, J.D.; Joss, S.; Metcalfe, K.A.; Somarathi, S.; Cruden, J.; Devlin, A.M.; Donaldson, A.; DiDonato, N.; Fitzpatrick, D.; Kaiser, F.J.; et al. Heterozygous truncation mutations of the SMC1A gene cause a severe early onset epilepsy with cluster seizures in females: Detailed phenotyping of 10 new cases. Epilepsia 2017, 58, 565–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorman, K.M.; Forman, E.; Conroy, J.; Allen, N.M.; Shahwan, A.; Lynch, S.A.; Ennis, S.; King, M.D. Novel SMC1A variant and epilepsy of infancy with migrating focal seizures: Expansion of the phenotype. Epilepsia 2017, 58, 1301–1302. [Google Scholar] [CrossRef] [Green Version]
- Oguni, H.; Nishikawa, A.; Sato, Y.; Otani, Y.; Ito, S.; Nagata, S.; Kato, M.; Hamanaka, K.; Miyatake, S.; Matsumoto, N. A missense variant of SMC1A causes periodic pharmaco-resistant cluster seizures similar to PCDH19-related epilepsy. Epilepsy Res. 2019, 155, 106149. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Zhang, X.; Xiao, B.; Zhang, L.; Long, H. A de novo mutation in SMC1A gene identified in a Chinese infant with nonclassical Cornelia de Lange syndrome and drug-resistant epilepsy. Neurol. Sci. 2021, 42, 329–331. [Google Scholar] [CrossRef]
- Barañano, K.W.; Kimball, A.; Fong, S.L.; Egense, A.S.; Hudon, C.; Kline, A.D. Further Characterization of SMC1A Loss of Function Epilepsy Distinct From Cornelia de Lange Syndrome. J. Child. Neurol. 2022, 37, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Elwan, M.; Fowkes, R.; Lewis-Smith, D.; Winder, A.; Baker, M.R.; Thomas, R.H. Late-onset cluster seizures and intellectual disability associated with a novel truncation variant in SMC1A. Epilepsy Behav. Rep. 2022, 19, 100556. [Google Scholar] [CrossRef]
- Wenger, T.L.; Chow, P.; Randle, S.C.; Rosen, A.; Birgfeld, C.; Wrede, J.; Javid, P.; King, D.; Manh, V.; Hing, A.V.; et al. Novel findings of left ventricular non-compaction cardiomyopathy, microform cleft lip and poor vision in patient with SMC1A-associated Cornelia de Lange syndrome. Am. J. Med. Genet. A 2017, 173, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Mulder, P.A.; Huisman, S.; Landlust, A.M.; Moss, J.; SMC1A Consortium; Piening, S.; Hennekam, R.C.; van Balkom, I.D.C. Development, behaviour and autism in individuals with SMC1A variants. J. Child. Psychol. Psychiatry 2019, 60, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Kruszka, P.; Berger, S.I.; Casa, V.; Dekker, M.R.; Gaesser, J.; Weiss, K.; Martinez, A.F.; Murdock, D.R.; Louie, R.J.; Prijoles, E.J.; et al. Cohesin complex-associated holoprosencephaly. Brain. J. Neurol. 2019, 142, 2631–2643. [Google Scholar] [CrossRef] [Green Version]
- Chinen, Y.; Nakamura, S.; Kaneshi, T.; Nakayashiro, M.; Yanagi, K.; Kaname, T.; Naritomi, K.; Nakanishi, K. A novel nonsense SMC1A mutation in a patient with intractable epilepsy and cardiac malformation. Hum. Genome Var. 2019, 6, 23. [Google Scholar] [CrossRef] [Green Version]
- Spagnoli, C.; Fusco, C.; Pisani, F. Rett Syndrome Spectrum in Monogenic Developmental-Epileptic Encephalopathies and Epilepsies: A Review. Genes 2021, 12, 1157. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.C.; Zoghbi, H.Y.; Moseley, A.B.; Rosenblatt, H.M.; Belmont, J.W. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am. J. Hum. Genet. 1992, 51, 1229–1239. [Google Scholar]
- Machado, F.B.; Machado, F.B.; Faria, M.A.; Lovatel, V.L.; Alves da Silva, A.F.; Radic, C.P.; De Brasi, C.D.; Rios, Á.F.L.; de Sousa Lopes, S.M.C.; da Silveira, L.S.; et al. 5meCpG epigenetic marks neighboring a primate-conserved core promoter short tandem repeat indicate X-chromosome inactivation. PLoS ONE 2014, 9, e103714. [Google Scholar] [CrossRef] [PubMed]
- Deardorff, M.A.; Kaur, M.; Yaeger, D.; Rampuria, A.; Korolev, S.; Pie, J.; Gil-Rodríguez, C.; Arnedo, M.; Loeys, B.; Kline, A.D.; et al. Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of cornelia de Lange syndrome with predominant mental retardation. Am. J. Hum. Genet. 2007, 80, 485–494. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, M.; Scheinost, J.C.; Petela, N.J.; Gligoris, T.G.; Wissler, M.; Ogushi, S.; Collier, J.E.; Voulgaris, M.; Kurze, A.; Chan, K.-L.; et al. The Cohesin Ring Uses Its Hinge to Organize DNA Using Non-topological as well as Topological Mechanisms. Cell 2018, 173, 1508–1519.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tierney, A.L.; Nelson, C.A. Brain Development and the Role of Experience in the Early Years. Zero Three 2009, 30, 9–13. [Google Scholar] [PubMed]
- Fujita, Y.; Masuda, K.; Bando, M.; Nakato, R.; Katou, Y.; Tanaka, T.; Nakayama, M.; Takao, K.; Miyakawa, T.; Tanaka, T.; et al. Decreased cohesin in the brain leads to defective synapse development and anxiety-related behavior. J. Exp. Med. 2017, 214, 1431–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, H.; Deng, X.; Disteche, C.M. X-factors in human disease: Impact of gene content and dosage regulation. Hum. Mol. Genet. 2021, 30, R285–R295. [Google Scholar] [CrossRef] [PubMed]
- Moncayo, J.A.; Ayala, I.N.; Argudo, J.M.; Aguirre, A.S.; Parwani, J.; Pachano, A.; Ojeda, D.; Cordova, S.; Mora, M.G.; Tapia, C.M.; et al. Understanding Protein Protocadherin-19 (PCDH19) Syndrome: A Literature Review of the Pathophysiology. Cureus 2022, 14, e25808. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Cobos, M.J.; Balaton, B.P.; Brown, C.J. Genes that escape from X-chromosome inactivation: Potential contributors to Klinefelter syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Berletch, J.B.; Ma, W.; Yang, F.; Shendure, J.; Noble, W.S.; Disteche, C.M.; Deng, X. Escape from X inactivation varies in mouse tissues. PLoS Genet. 2015, 11, e1005079. [Google Scholar] [CrossRef]
Figure 1.
The SMC1A splice-site variant in P1 impacts transcriptional splicing. (A) c.615+5G>A (red triangle) in intron 4 strongly increases two alternative splicing events in P1, retaining intron 4 (black arrow; 438 bp) and skipping exons 4–5 (red arrow; 479 bp), as shown by RT-PCR using primer pairs specific for intron 4 transcript or spanning exons 2–7 and RNA from whole blood of the P1 family trio. The large band for the wild-type transcript containing exons 4–5 detected by primer pairs spanning exons 2–7 is 922 bp. (B) Sanger sequencing of RT-PCR products confirms these two alternative splicing events. Both abnormally spliced transcripts cause frameshifts and premature translational stop codons and thus cannot produce functional proteins, consistent with loss of function (see Table S2).
Figure 1.
The SMC1A splice-site variant in P1 impacts transcriptional splicing. (A) c.615+5G>A (red triangle) in intron 4 strongly increases two alternative splicing events in P1, retaining intron 4 (black arrow; 438 bp) and skipping exons 4–5 (red arrow; 479 bp), as shown by RT-PCR using primer pairs specific for intron 4 transcript or spanning exons 2–7 and RNA from whole blood of the P1 family trio. The large band for the wild-type transcript containing exons 4–5 detected by primer pairs spanning exons 2–7 is 922 bp. (B) Sanger sequencing of RT-PCR products confirms these two alternative splicing events. Both abnormally spliced transcripts cause frameshifts and premature translational stop codons and thus cannot produce functional proteins, consistent with loss of function (see Table S2).

Figure 2.
SMC1A-DEE variants. (A) Location and types of 41 SMC1A-DEE variants, including 39 reported and 2 new ones shown on the SMC1A protomer (see Table 1). Variants of our patients 1–3 are marked. (B) After self-folding of the coiled-coil, SMC1A and SMC3 form a hinge domain at one end and bind to RAD21 at the other end (N/C head). Finally, RAD21 interacts with STAG1/2 and forms a ring-shaped cohesin complex to fold chromatin. The number of assembled cohesin would be reduced by these LOFs.
Figure 2.
SMC1A-DEE variants. (A) Location and types of 41 SMC1A-DEE variants, including 39 reported and 2 new ones shown on the SMC1A protomer (see Table 1). Variants of our patients 1–3 are marked. (B) After self-folding of the coiled-coil, SMC1A and SMC3 form a hinge domain at one end and bind to RAD21 at the other end (N/C head). Finally, RAD21 interacts with STAG1/2 and forms a ring-shaped cohesin complex to fold chromatin. The number of assembled cohesin would be reduced by these LOFs.

Figure 3.
Impact of XCI on X-linked disorders due to heterozygous LOF mutations in escape genes. Normal females have a dose of products from wild-type (WT) alleles on the Xa and Xi. Patients with skewed XCI have a differential dose reduction depending on which X carries the mutation (mut). This would lead to a mild phenotype in patients with Xi-mut, whereas a most severe phenotype in patients with Xa-mut. Thus, skewed XCI towards the mutant allele could alleviate the phenotype. In contrast, patients with random XCI have a mosaic reduction due to mixtures of cells expressing Xa- or Xi-mut, leading to a phenotype between patients with Xi-mut or Xa-mut. In addition, XCI mosaicism could cause cellular interference at tissue levels and lead to a different phenotype, as is the case for PCDH19 clustering epilepsy.
Figure 3.
Impact of XCI on X-linked disorders due to heterozygous LOF mutations in escape genes. Normal females have a dose of products from wild-type (WT) alleles on the Xa and Xi. Patients with skewed XCI have a differential dose reduction depending on which X carries the mutation (mut). This would lead to a mild phenotype in patients with Xi-mut, whereas a most severe phenotype in patients with Xa-mut. Thus, skewed XCI towards the mutant allele could alleviate the phenotype. In contrast, patients with random XCI have a mosaic reduction due to mixtures of cells expressing Xa- or Xi-mut, leading to a phenotype between patients with Xi-mut or Xa-mut. In addition, XCI mosaicism could cause cellular interference at tissue levels and lead to a different phenotype, as is the case for PCDH19 clustering epilepsy.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Patients’ general characteristics.
| Patient 1 | Patient 2 | Patient 3 | |
|---|---|---|---|
| Full term | Yes | Yes | No, 36 + 6/7 wk |
| Congenital microcephaly | No | No | Yes |
| Postnatal microcephaly | Yes | Yes | Yes initially |
| Hypotonia | Yes | Yes | Yes |
| Expressive language | None | None | A few words |
| Encephalopathy | Profound | Profound | Severe |
| G-tube | Yes (4 yr) | Yes (9 M) | No |
| CDLS features | No | Minor | Minor |
| Independent walking | No | No | Yes |
| Precocious puberty | Yes | No | No |
| Cardiac abnormalities | No | Left ventricular cardiomyopathy | No |
| MRI: | A small cyst in left anterior temporal region (age 7 M and 9 yr) | Normal (2 yr) | No |
M = month; yr = years; wk = weeks.
Table 2.
Patients’ characteristics in epilepsy.
| Patient 1 | Patient 2 | Patient 3 | |
|---|---|---|---|
| Age of seizure onset (M) | 4 | 2 | 35 |
| Intractable epilepsy | Yes | Yes | Yes |
| Epilepsy syndrome | |||
| LGS | Yes | No | Yes |
| Epileptic spasms | No | Yes (9 M) | No |
| History of status epilepticus | Yes | Yes | No |
| Seizure burden | Weekly and up to daily | Weekly and up to daily | Less than monthly |
| Seizure Types | |||
| Tonic/atonic | Yes | Yes | Yes |
| Tonic clonic | Yes | Yes | Yes |
| Myoclonic | Yes | No | Yes |
| Focal onset, motor | Yes | Yes | No |
| Focal onset, nonmotor | Yes | Yes | No |
| Atypcial absence | Yes | No | No |
| Cluster seizures | Yes | Yes | Yes |
| Age starting AED | 4 M | 2M | 47M |
| Current AED: | Topiramate, Perampanel, Epidiolex, Lacosamide | Clobazam Levetiracetam | Brivaracetam Rufinamide Clobazam |
| Past AED | Zonisamide, Valproic acid, Clobazam Oxcarbazepine, Levetiracetam, Rufinamide, Lamotrigene, Brivaracetam | Phenobarbital, Oxcarbazepine, ACTH, Viagabatrin, Diazepam, Zonisamide | Valproci acid, Levetiracetam |
| AEDs: severe adverse effects | No | No | No |
| Ketogenic/modified Atkin’s diet | Yes, no efficacy | Yes, partial efficacy | Yes, no efficacy |
| Vagal nerve stimulator | Yes, unclear efficacy | No | No |
| Epilepsy resection surgery | No | No | No |
| EEG | |||
| Normal Yes/No | No | No | No |
| Posterior dominant rhythm | Yes, slow for age | No | Yes, slow for age |
| EEG background | Diffuse slow | Diffuse slow, history of hypsarrhythmia | Diffuse slow |
| Sleep architecture | No | No | Yes |
| Interictal pattern | Generalized; multifocal, more posteriorly | Multifocal | Generalized spike wave |
| GPFA | Yes | No | No |
| Slow Spike wave | No | No | Yes |
AED = antiseizure medication; PDR = posterior dominant rhythm; Ruf = rufinamide; GPFA = Generalized paroxysmal fast activity; M = months.
Table 3.
Information of 41 SMC1A-DEE variants.
| Variant # | Nucleotide Changes | Amino Acid Changes | Domain | Type | XCI | Ref. | Age of Sz Onset | Speech | ID | Walking | MRI |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | C.20_23del | Ile7Argfs*42 | P1 head | Frameshift | Moderately skewed 81:19 | Barañano, 2022 [19] (P6) | 15 M | none | yes | No | n/a |
| 2 | c.31A>T | Asn11Tyr | P1 head | Missense | n/a | Huisman, 2017 [9] (P1) | 2.5 M | n/a | yes | normal | |
| 3 | c.140T>G | Phe47Cys | P1 head | Missense | Random 74:26 | Barañano, 2022 [19] (P3) | 3 M | none | yes | No | n/a |
| 4 | c.157dup | Thr53AsnfsX34 | P1 head | Frameshift | n/a | Huisman, 2017 [9] (P2) | 5 M | n/a | yes | n/a | cerebral volume loss |
| 5 | c.287G>C | Arg96Pro | P1 head | Missense | Random | Barañano, 2022 [19] (P11) | 18 M | n/a | yes | n/a | n/a |
| 6 | c.421G>A | Glu141Lys | P1 head | Missense | highly Skewed 100:0 | Barañano, 2022 [19] (P1) | 2.5 M | none | yes | n/a | n/a |
| 7 | c.511C>T | Arg171Ter | Coil | Nonsense | Random | Symonds, 2017 [15] (P7) | 4 Weeks | none | yes | No | normal |
| 8 | c.615G>A | Glu205fs* | Coil | Splice-site | N/A | (NM:00128146:c.549G>A) in Symonds, 2017 [15] (P3); P12 in Barañano, 2022 [19] | P3 Symonds-4 M; P12 Barañano-13 M | none | yes | No | P3 Symonds-small hemorrhage along posterior falx and tentorium; P12-Barañano-n/a |
| 9 | c.615G+1G>C | Glu205fs* | Coil | Splice-site | Moderately skewed 83:17 | Barañano, 2022 [19] (P9) | 1 M | n/a | yes | No | n/a |
| 10 (P1 in our cohort) | c.615+5G>A | Glu205fs* | Coil | Splice-site | Random | Our cohort P1 | 4 M | none | yes | no | normal |
| 11 | c.694G>T | Glu232Ter | Coil | Nonsense | n/a | Huisman, 2017 [9] (P7) | 4 M | n/a | yes | n/a | n/a |
| 12 | c.1113+1G>A | Gln371fs* | Coil | Splice-site | n/a | Gorman 2017 [16] (P2) | 7 Weeks | none | yes | n/a | n/a |
| 13 | c.1114delG | Val372Ter | Coil | Nonsense | n/a | Gorman 2017 [16] (P1) | 7 Weeks | none | yes | no | n/a |
| 14 (P3 in our cohot) | c.1487G>A | Arg496His | Close to hinge | Missense | Skewed in our patient P3; N/A in 5 patients from Deardorff | Our cohort P3; Deardorff, 2007 [28] (P3) | 35 M | limited | yes | Yes | n/a |
| 15 | c.1489C>T | Arg497Ter | Close to hinge | Nonsense | n/a | Fang, 2021 [18] | 4 M | none | yes | No | normal |
| 16 | c.1495C>T | Arg499Ter | Close to hinge | Nonsense | n/a | Chinen, 2019 [24]; Kruszka, 2019 [23] (P8) | 40 M-Chinen, 2019; N/A- | none | yes | Chinen-Yes | ChinenP8- microform of HPE, Kruszka P8-Triventricular ectasia |
| 17 | c.1591C>T | Gln531Ter | Hinge | Nonsense | Moderately skewed | Symonds, 2017 [15] (P1) | 15 M | none | yes | Yes | normal |
| 18 | c.1609delG | Val537Phefs*42 | Hinge | Frameshift | highly skewed | Barañano, 2022 [19] (P8) | 5 M | none | yes | No | n/a |
| 19 (P2 in our cohort) | c.1636_1638delATT | Ilu546del | Hinge | In-frame | Random | reported in Wenger, 2016 [21] | 2 M | none | yes | No | normal |
| 20 | c.1900C>T | Gln634Ter | Close to hinge | Nonsense | Moderately skewed 86:14, Mosaic | Barañano, 2022 [19] (P2) | 3 M | n/a | yes | n/a | n/a |
| 21 | c.1911+1G>T | Thr638Valfs*48 | Close to hinge | Splice-site | n/a | Lebrun, 2015 [14] | Neonate | none | yes | no | small frontal lobe, thin CC |
| 22 | c.2197G>T | Glu733Ter | Coil | Nonsense | Random | Symonds, 2017 [15] (P4) | 5 M | none | yes | No | volume loss |
| 23 | c.2364del | Asn788Lysfs*10 | Coil | Frameshift | Random | Jansen, 2016 [16] (P1) | 9 M | none | yes | No | slightly enlarged ventricles, hypotrophy cerebellar vermis |
| 24 | c.2394delA | Lys798Asnfs*31 | Coil | Frameshift | n/a | Kruszka, 2019 [23] (P9) | n/a | n/a | yes | semi-lobar HPE | |
| 25 | c.2394dupA | Arg799fs | Coil | Frameshift | n/a | Barañano, 2022 [19] (P5) | 4 M | none | yes | No | n/a |
| 26 | c.2421_2562del | Leu808Arg-fs*6 | Coil | Frameshift | Moderately skewed 85:15 | Jansen, 2016 [16] (P2) | 2 M | none | yes | None | mild periventricular white matter abnormalities |
| 27 | c.2477delA | p825fs | Coil | Frameshift | n/a | Symonds, 2017 [15] (P8, P9) | 28 M in P8; <1 M in P9 | P8-none; P9-none | yes | P8-yes | P8-normal; P9-hemi-lobar HPE |
| 28 | c.2683C>G | Arg895Gly | Coil | Missense | Skewed-P1; n/a-P2, P9 from Kruszka | Oguni, 2019 [17]; Kruszka, 2019 [23] (P9) | 25 M-P1 Oguni, P2 (mother of P1)-Oguni- 12y; n/a- P8-Kruszka | Oguin-none; Kruszka-n/a | P1-yes; P2-normal until sz onset | Oguni-P1-no, P2-yes, Kruszka-n/a | P2: cerebellar atrophy; P9-Kruszka-semi-lobar HPE |
| 29 | c.2769dupC | Ser924Glnfs*2 | Coil | Frameshift | n/a | Barañano, 2022 [19] (P10) | 24 M | no | yes | n/a | n/a |
| 30 | c.2834delG | Gly945Alafs*19 | Coil | Frameshift | n/a | Kruszka, 2019 [23] (P11) | n/a | n/a | yes | n/a | semi-lobar HPE |
| 31 | c.2853_2856delTCAG | Ser951Argfs*12 | Coil | Frameshift | Skewed | Goldstein, 2015 [12] (P1) | 4 M | n/a | severe | no | mild ventriculomegaly, mildly rotated hippocampal heads |
| 32 | c.2873delA | Gln958Argfs*6 | Coil | Frameshift | Random | Barañano, 2022 [19] (P7) | 3 M | none | Severe | no | n/a |
| 33 | c.2923C>T | Arg975Ter | Coil | Nonsense | N/A | Symonds, 2017 [15] (P6) | 5 M | Lost speech after status | Moderate to severe | yes | normal |
| 34 | c.3046_3048delGTGinsG | Val1016Alafs*28 | Coil | Frameshift | Random | Barañano, 2022 [19] (P4) | Neonate | n/a | yes | no | n/a |
| 35 | c.3115C>T | Gln1039Ter | Coil | Nonsense | Random 76:24 | Symonds, 2017 [15] (P10) | 2 M | none | yes | no | thin abnormally shaped CC and minimal cerebral atrophy |
| 36 | c.3145C>T | Arg1049Ter | Coil | Nonsense | n/a | Symonds, 2017 [15] (P2) | 5–6 weeks | none | yes | no | cerebral volume loss |
| 37 | c.3241A>T | Ile1081Phe | Close to P2 head | Missense | n/a | Unreported | 4 yr | none | yes | yes | mildly prominent lateral ventricles |
| 38 | c.3285+1G>C | p1095 | Close to P2 head | Splice-site | n/a | Kruszka, 2019 [23] (P7) | not reported | n/a | yes | n/a | middle interhemispheric variant, HPE |
| 39 | c.3312C>A | Tyr1107Ter | Close to P2 head | Nonsense | n/a | Elwan, 2022 [20] | 12 yr | normal | normal before SE | yes | normal |
| 40 | c.3326_3330delATGGCinsC | Asp1109Alafs*102 | Close to P2 head | Frameshift | n/a | Symonds, 2017 [15] (P5) | 6 M | none | yes | no | small cavum septum vergae |
| 41 | c.3549_3552dupGGCC | Ile1185glyfs*23 | P2 head | Frameshift | Random in patient 1-Goldstein, 2015; n/a-P3- Barañano, 2022 | Goldstein, 2015 [12] (P2); Barañano, 2022 [19] (P13) | 17 M- P2- Goldstein 2015 [12]; 16 M- P13-Barañano 2022 [19] | n/a-Goldstein; P13-Barañano-limited | yes | n/a-Goldstein; P13Barababi-yes | P2 Goldstein-mild enlarged extra-axial spaces and slight thinning of CC |
CC = corpus callosum; HPE = holoprosencephaly; P = patient; SE = status epilepticus; sz = seizure.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bozarth, X.L.; Lopez, J.; Fang, H.; Lee-Eng, J.; Duan, Z.; Deng, X. Phenotypes and Genotypes in Patients with SMC1A-Related Developmental and Epileptic Encephalopathy. Genes 2023, 14, 852. https://doi.org/10.3390/genes14040852
AMA Style
Bozarth XL, Lopez J, Fang H, Lee-Eng J, Duan Z, Deng X. Phenotypes and Genotypes in Patients with SMC1A-Related Developmental and Epileptic Encephalopathy. Genes. 2023; 14(4):852. https://doi.org/10.3390/genes14040852
Chicago/Turabian StyleBozarth, Xiuhua L., Jonathan Lopez, He Fang, Jacqueline Lee-Eng, Zhijun Duan, and Xinxian Deng. 2023. "Phenotypes and Genotypes in Patients with SMC1A-Related Developmental and Epileptic Encephalopathy" Genes 14, no. 4: 852. https://doi.org/10.3390/genes14040852
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.