
Genotype and Phenotype Analyses of a Novel WFS1 Variant (c.2512C>T p.(Pro838Ser)) Associated with DFNA6/14/38

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genetic Analyses
2.2. Clinical Evaluation
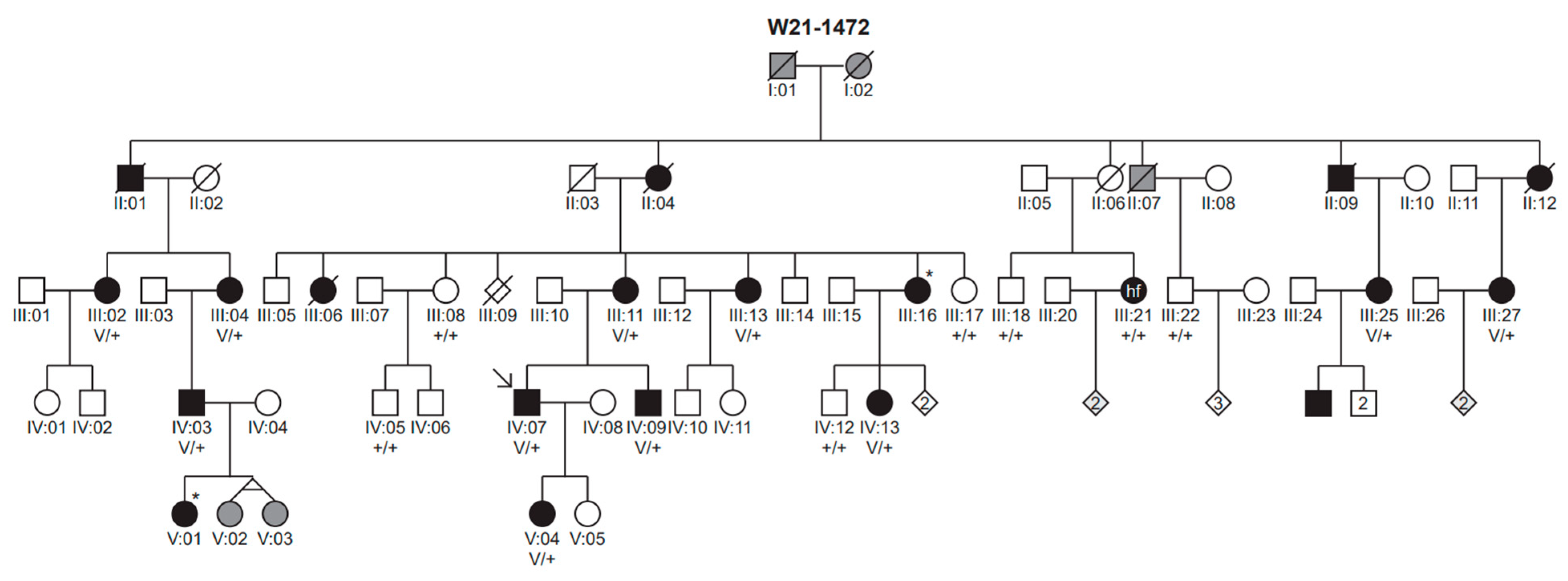
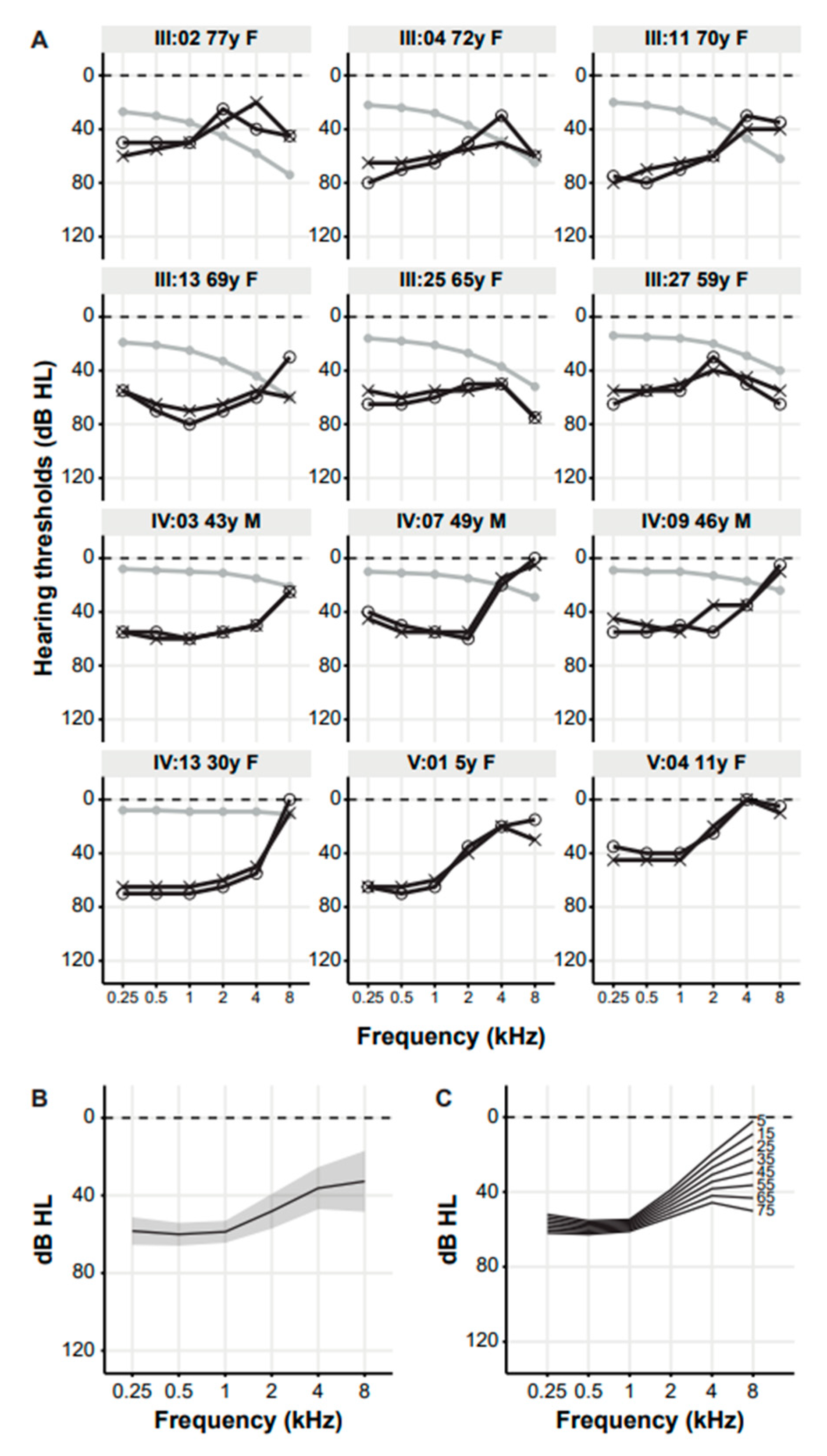
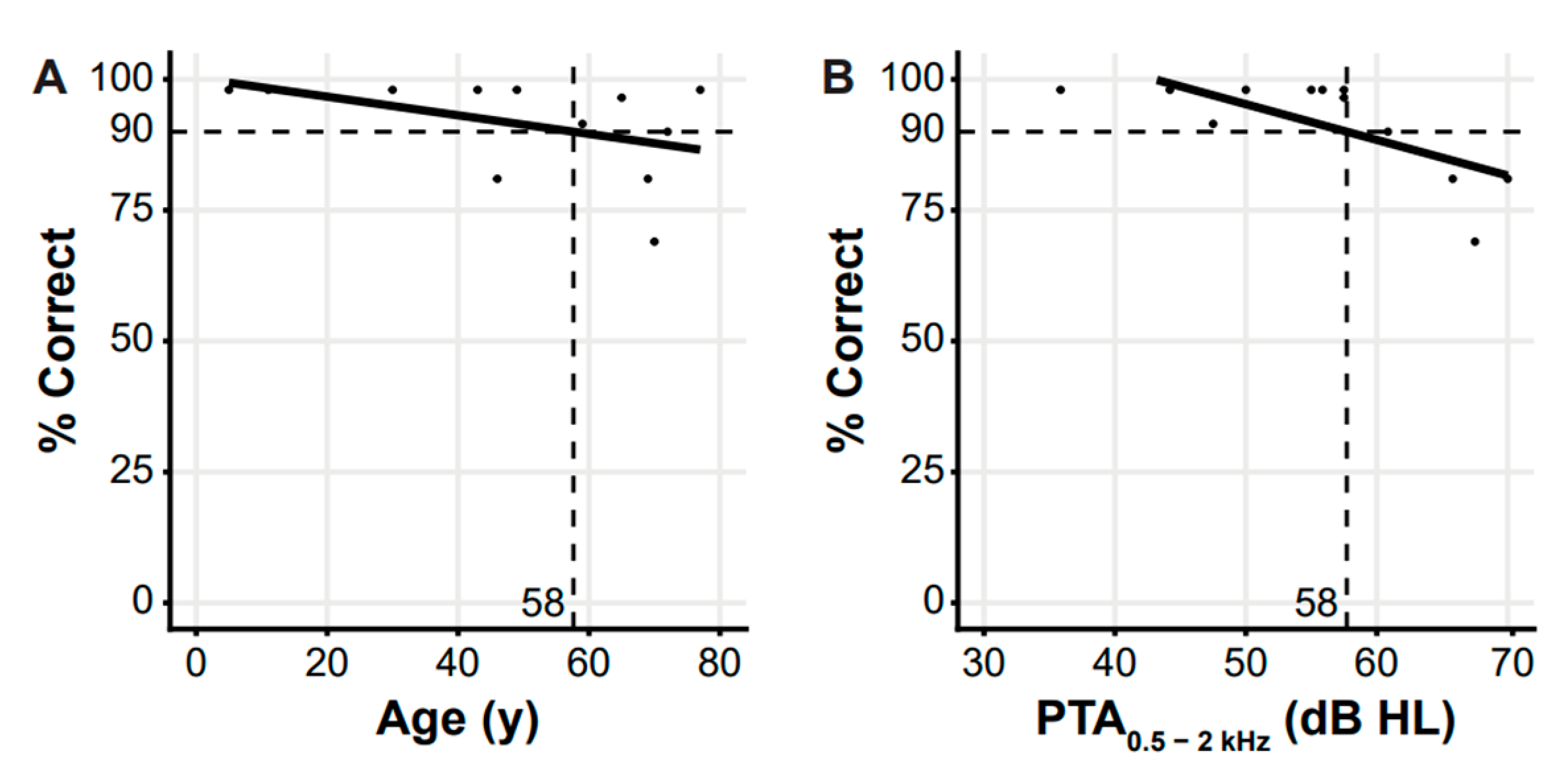
3. Results
3.1. Genetic Analyses
3.2. Clinical Evaluation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Camp, G.; Smith, R.J.H. Hereditary Hearing Loss Homepage. 2018. Available online: http://hereditaryhearingloss.org (accessed on 17 December 2021).
- Morton, N.E. Genetic Epidemiology of Hearing Impairment. Ann. N. Y. Acad. Sci. 1991, 630, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Lesperance, M.M.; Hall, J.W.; Bess, F.H.; Fukushima, K.; Jain, P.K.; Ploplis, B.; Agustin, T.B.; Skarka, H.; Smith, R.J.; Wills, M.; et al. A gene for autosomal dominant nonsyndromic hereditary hearing impairment maps to 4p16.3. Hum. Mol. Genet. 1995, 4, 1967–1972. [Google Scholar] [CrossRef] [PubMed]
- Van Camp, G.; Kunst, H.; Flothmann, K.; McGuirt, W.; Wauters, J.; Marres, H.; Verstreken, M.; Bespalova, I.N.; Burmeister, M.; Van de Heyning, P.H.; et al. A gene for autosomal dominant hearing impairment (DFNA14) maps to a region on chromosome 4p16.3 that does not overlap the DFNA6 locus. J. Med. Genet. 1999, 36, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Bespalova, I.N. Mutations in the Wolfram syndrome 1 gene (WFS1) are a common cause of low frequency sensorineural hearing loss. Hum. Mol. Genet. 2001, 10, 2501–2508. [Google Scholar] [CrossRef]
- Young, T.-L.; Ives, E.; Lynch, E.; Person, R.; Snook, S.; MacLaren, L.; Cator, T.; Griffin, A.; Fernandez, B.; Lee, M.K.; et al. Non-syndromic progressive hearing loss DFNA38 is caused by heterozygous missense mutation in the Wolfram syndrome gene WFS1. Hum. Mol. Genet. 2001, 10, 2509–2514. [Google Scholar] [CrossRef]
- Kobayashi, M.; Miyagawa, M.; Nishio, S.-Y.; Moteki, H.; Fujikawa, T.; Ohyama, K.; Sakaguchi, H.; Miyanohara, I.; Sugaya, A.; Naito, Y.; et al. WFS1 mutation screening in a large series of Japanese hearing loss patients: Massively parallel DNA sequencing-based analysis. PLoS ONE 2018, 13, e0193359. [Google Scholar] [CrossRef]
- Barrett, T.; Tranebjærg, L.; Gupta, R.; Rendtorff, N.D.; Williams, D.; Wright, B.; Dias, R. WFS1 Spectrum Disorder. In GeneReviews®; University of Washington: Seattle, WA, USA, 1993. Available online: https://www.ncbi.nlm.nih.gov/books/NBK4144/ (accessed on 16 December 2022).
- Seco, C.Z.; Wesdorp, M.; Feenstra, I.; Pfundt, R.; Hehir-Kwa, J.Y.; Lelieveld, S.H.; Castelein, S.; Gilissen, C.; de Wijs, I.J.; Admiraal, R.J.; et al. The diagnostic yield of whole-exome sequencing targeting a gene panel for hearing impairment in The Netherlands. Eur. J. Hum. Genet. 2016, 25, 308–314. [Google Scholar] [CrossRef]
- Strom, T.M.; Hörtnagel, K.; Hofmann, S.; Gekeler, F.; Scharfe, C.; Rabl, W.; Gerbitz, K.D.; Meitinger, T. Diabetes insipidus, diabetes mellitus, optic atrophy and deafness (DIDMOAD) caused by mutations in a novel gene (wolframin) coding for a predicted transmembrane protein. Hum. Mol. Genet. 1998, 7, 2021–2028. [Google Scholar] [CrossRef]
- Inoue, H.; Tanizawa, Y.; Wasson, J.; Behn, P.; Kalidas, K.; Bernal-Mizrachi, E.; Mueckler, M.; Marshall, H.; Donis-Keller, H.; Crock, P.; et al. A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome). Nat. Genet. 1998, 20, 143–148. [Google Scholar] [CrossRef]
- Plantinga, R.F.; Pennings, R.J.E.; Huygen, P.L.M.; Bruno, R.; Eller, P.; Barrett, T.; Vialettes, B.; Paquis-Fluklinger, V.; Lombardo, F.; Cremers, C.W.R.J. Hearing Impairment in Genotyped Wolfram Syndrome Patients. Ann. Otol. Rhinol. Laryngol. 2008, 117, 494–500. [Google Scholar] [CrossRef]
- Pennings, R.J.; Huygen, P.L.; van den Ouweland, J.M.; Cryns, K.; Dikkeschei, L.D.; Van Camp, G.; Cremers, C.W. Sex-Related Hearing Impairment in Wolfram Syndrome Patients Identified by Inactivating WFS1 Mutations. Audiol. Neurotol. 2004, 9, 51–62. [Google Scholar] [CrossRef]
- Barrett, T.G.; Bundey, S.E.; Macleod, A.F. Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet 1995, 346, 1458–1463. [Google Scholar] [CrossRef]
- Eiberg, H.; Hansen, L.; Kjer, B.; Hansen, T.; Pedersen, O.; Bille, M.; Rosenberg, T.; Tranebjærg, L. Autosomal dominant optic atrophy associated with hearing impairment and impaired glucose regulation caused by a missense mutation in the WFS1 gene. J. Med. Genet. 2005, 43, 435–440. [Google Scholar] [CrossRef]
- Zhu, M.; Li, Y.; Dong, G.; Chen, X.; Huang, K.; Wu, W.; Dai, Y.; Zhang, L.; Lin, H.; Wang, S.; et al. Prevalence and phenotypic features of diabetes due to recessive, non-syndromic WFS1 mutations. Eur. J. Endocrinol. 2022, 186, 163–170. [Google Scholar] [CrossRef]
- Bonnycastle, L.L.; Chines, P.S.; Hara, T.; Huyghe, J.R.; Swift, A.J.; Heikinheimo, P.; Mahadevan, J.; Peltonen, S.; Huopio, H.; Nuutila, P.; et al. Autosomal Dominant Diabetes Arising From a Wolfram Syndrome 1 Mutation. Diabetes 2013, 62, 3943–3950. [Google Scholar] [CrossRef]
- Berry, V.; Gregory-Evans, C.; Emmett, W.; Waseem, N.; Raby, J.; Prescott, D.; Moore, A.T.; Bhattacharya, S.S. Wolfram gene (WFS1) mutation causes autosomal dominant congenital nuclear cataract in humans. Eur. J. Hum. Genet. 2013, 21, 1356–1360. [Google Scholar] [CrossRef]
- De Franco, E.; Flanagan, S.E.; Yagi, T.; Abreu, D.; Mahadevan, J.; Johnson, M.B.; Jones, G.; Acosta, F.; Mulaudzi, M.; Lek, N.; et al. Dominant ER Stress–Inducing WFS1 Mutations Underlie a Genetic Syndrome of Neonatal/Infancy-Onset Diabetes, Congenital Sensorineural Deafness, and Congenital Cataracts. Diabetes 2017, 66, 2044–2053. [Google Scholar] [CrossRef]
- Majander, A.; Jurkute, N.; Burté, F.; Brock, K.; João, C.; Huang, H.; Neveu, M.M.; Chan, C.M.; Duncan, H.J.; Kelly, S.; et al. WFS1-Associated Optic Neuropathy: Genotype-Phenotype Correlations and Disease Progression. Am. J. Ophthalmol. 2022, 241, 9–27. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Hofmann, S.; Philbrook, C.; Gerbitz, K.-D.; Bauer, M.F. Wolfram syndrome: Structural and functional analyses of mutant and wild-type wolframin, the WFS1 gene product. Hum. Mol. Genet. 2003, 12, 2003–2012. [Google Scholar] [CrossRef] [Green Version]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2017, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed]
- Cryns, K.; Pfister, M.; Pennings, R.J.; Bom, S.J.; Flothmann, K.; Caethoven, G.; Kremer, H.; Schatteman, I.; Köln, K.A.; Tóth, T.; et al. Mutations in the WFS1 gene that cause low-frequency sensorineural hearing loss are small non-inactivating mutations. Hum. Genet. 2002, 110, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Fokkema, I.F.; Dunnen, J.T.D.; Taschner, P.E. LOVD: Easy creation of a locus-specific sequence variation database using an “LSDB-in-a-box” approach. Hum. Mutat. 2005, 26, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Qing, J.; Yan, D.; Zhou, Y.; Liu, Q.; Wu, W.; Xiao, Z.; Liu, Y.; Liu, J.; Du, L.; Xie, D.; et al. Whole-Exome Sequencing to Decipher the Genetic Heterogeneity of Hearing Loss in a Chinese Family with Deaf by Deaf Mating. PLoS ONE 2014, 9, e109178. [Google Scholar] [CrossRef]
- Gonçalves, A.; Matos, T.; Simões-Teixeira, H.; Machado, M.P.; Simão, M.; Dias, Ó.P.; Andrea, M.; Fialho, G.; Caria, H. WFS1 and non-syndromic low-frequency sensorineural hearing loss: A novel mutation in a Portuguese case. Gene 2014, 538, 288–291. [Google Scholar] [CrossRef]
- Fokkema, I.F.A.C.; Taschner, P.E.M.; Schaafsma, G.C.; Celli, J.; Laros, J.F.; den Dunnen, J.T. LOVD v.2.0: The next generation in gene variant databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef]
- Cryns, K.; Sivakumaran, T.A.; Ouweland, J.M.V.D.; Pennings, R.J.; Cremers, C.W.; Flothmann, K.; Young, T.-L.; Smith, R.J.; Lesperance, M.M.; Van Camp, G. Mutational spectrum of theWFS1 gene in Wolfram syndrome, nonsyndromic hearing impairment, diabetes mellitus, and psychiatric disease. Hum. Mutat. 2003, 22, 275–287. [Google Scholar] [CrossRef]
- Haer-Wigman, L.; Van Zelst-Stams, W.A.G.; Pfundt, R.; Born, L.I.V.D.; Klaver, C.; Verheij, J.B.G.M.; Hoyng, C.B.; Breuning, M.H.; Boon, C.; Kievit, A.J.; et al. Diagnostic exome sequencing in 266 Dutch patients with visual impairment. Eur. J. Hum. Genet. 2017, 25, 591–599. [Google Scholar] [CrossRef]
- Wallis, Y.; Payne, S.; McAnulty, C.; Bodmer, D.; Sistermans, E.; Robertson, K. Practice Guidelines for the Evaluation of Pathogenicity and the Reporting of Sequence Variants in Clinical Molecular Genetics; Association for Clinical Genetic Science: London, UK; Dutch Society of Clinical Genetic Laboratory Specialists: Nijmegen, The Netherlands, 2013; pp. 1–16. [Google Scholar]
- Rentzsch, P.; Schubach, M.; Shendure, J.; Kircher, M. CADD-Splice—Improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 2021, 13, 31. [Google Scholar] [CrossRef]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [Green Version]
- Jaganathan, K.; Panagiotopoulou, S.K.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Anesth. Analg. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Oza, A.M.; DiStefano, M.T.; Hemphill, S.E.; Cushman, B.J.; Grant, A.R.; Siegert, R.K.; Shen, J.; Chapin, A.; Boczek, N.J.; Schimmenti, L.A.; et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 2018, 39, 1593–1613. [Google Scholar] [CrossRef]
- Jacobson, G.P.; Newman, C.W. The Development of the Dizziness Handicap Inventory. Arch. Otolaryngol. Neck Surg. 1990, 116, 424–427. [Google Scholar] [CrossRef]
- Whitney, S.L.; Wrisley, D.M.; Brown, K.E.; Furman, J.M. Is Perception of Handicap Related to Functional Performance in Persons with Vestibular Dysfunction? Otol. Neurotol. 2004, 25, 139–143. [Google Scholar] [CrossRef]
- McCaslin, D.L.; Jacobson, G.P.; Lambert, W.; English, L.N.; Kemph, A.J. The development of the vanderbilt pediatric dizziness handicap inventory for patient caregivers (DHI-PC). Int. J. Pediatr. Otorhinolaryngol. 2015, 79, 1662–1666. [Google Scholar] [CrossRef]
- ISO 7029:2017; Acoustics—Statistical Distribution of Hearing Thresholds Related to Age and Gender. ISO: Geneva, Switzerland, 2017. Available online: https://www.iso.org/standard/42916.html (accessed on 16 December 2022).
- Mazzoli, M.G.; Van Camp, G.U.; Newton, V.; Giarbini, N.; Declau, F.; Parving, A. Recommendations for the description of genetic and audiological data for families with nonsyndromic hereditary hearing impairment. Audiol. Med. 2003, 1, 148–150. [Google Scholar] [CrossRef]
- Huygen, P.L.; Pennings, R.J.; Cremers, C.W. Characterizing and Distinguishing Progressive Phenotypes in Nonsyndromic Autosomal Dominant Hearing Impairment. Audiol. Med. 2003, 1, 37–46. [Google Scholar] [CrossRef]
- Bosmana, A.J.; Smoorenburg, G.F. Intelligibility of Dutch CVC Syllables and Sentences for Listeners with Normal Hearing and with Three Types of Hearing Impairment. Int. J. Audiol. 1995, 34, 260–284. [Google Scholar] [CrossRef]
- American Speech-Language-Hearing Association. Determining Threshold Level for Speech [Guidelines]; American Speech-Language-Hearing Association: Rockville, MD, USA, 1988; Available online: www.asha.org/policy (accessed on 16 December 2022).
- Koopmans, W.J.A.; Goverts, S.T.; Smits, C. Speech Recognition Abilities in Normal-Hearing Children 4 to 12 Years of Age in Stationary and Interrupted Noise. Ear Heart 2018, 39, 1091–1103. [Google Scholar] [CrossRef]
- Oonk, A.; Beynon, A.; Peters, T.; Kunst, H.; Admiraal, R.; Kremer, H.; Verbist, B.; Pennings, R. Vestibular function and temporal bone imaging in DFNB1. Heart Res. 2015, 327, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Lesperance, M.M.; Hall, J.W.; Agustin, T.B.S.; Leal, S.M. Mutations in the Wolfram Syndrome Type 1 Gene (WFS1) Define a Clinical Entity of Dominant Low-Frequency Sensorineural Hearing Loss. Arch. Otolaryngol. Neck Surg. 2003, 129, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Kytövuori, L.; Hannula, S.; Mäki-Torkko, E.; Sorri, M.; Majamaa, K. A nonsynonymous mutation in the WFS1 gene in a Finnish family with age-related hearing impairment. Heart Res. 2017, 355, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Li, M.M.; Tayoun, A.A.; DiStefano, M.; Pandya, A.; Rehm, H.L.; Robin, N.H.; Schaefer, A.M.; Yoshinaga-Itano, C. Clinical evaluation and etiologic diagnosis of hearing loss: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Anesth. Analg. 2022, 24, 1392–1406. [Google Scholar] [CrossRef]
- Ma, J.; Wang, R.; Zhang, L.; Wang, S.; Tong, S.; Bai, X.; Lu, Z. A Novel Missense WFS1 Variant: Expanding the Mutational Spectrum Associated with Nonsyndromic Low-Frequency Sensorineural Hearing Loss. BioMed Res. Int. 2022, 2022, 5068869. [Google Scholar] [CrossRef]
- Pennings, R.J.E.; Bom, S.J.H.; Cryns, K.; Flothmann, K.; Huygen, P.L.M.; Kremer, H.; Van Camp, G.; Cremers, C.W.R.J. Progression of Low-Frequency Sensorineural Hearing Loss (DFNA6/14-WFS1). Arch. Otolaryngol. Neck Surg. 2003, 129, 421–426. [Google Scholar] [CrossRef]
- Attitudes of deaf people and their families towards issues surrounding genetics. In Genes, Hearing, and Deafness; CRC Press: London, UK, 2007; pp. 173–182. [CrossRef]
- Walls, W.D.; Moteki, H.; Thomas, T.R.; Nishio, S.-Y.; Yoshimura, H.; Iwasa, Y.; Frees, K.L.; Nishimura, C.J.; Azaiez, H.; Booth, K.T.; et al. A comparative analysis of genetic hearing loss phenotypes in European/American and Japanese populations. Hum. Genet. 2020, 139, 1315–1323. [Google Scholar] [CrossRef]
- Collin, R.W.; Chellappa, R.; Pauw, R.-J.; Vriend, G.; Oostrik, J.; van Drunen, W.; Huygen, P.L.; Admiraal, R.; Hoefsloot, L.H.; Cremers, F.P.; et al. Missense mutations in POU4F3 cause autosomal dominant hearing impairment DFNA15 and affect subcellular localization and DNA binding. Hum. Mutat. 2008, 29, 545–554. [Google Scholar] [CrossRef]
- E de Bruijn, S.; Smits, J.J.; Liu, C.; Lanting, C.P.; Beynon, A.J.; Blankevoort, J.; Oostrik, J.; Koole, W.; de Vrieze, E.; Cremers, C.W.R.J.; et al. A RIPOR2 in-frame deletion is a frequent and highly penetrant cause of adult-onset hearing loss. J. Med Genet. 2020, 58, 96–104. [Google Scholar] [CrossRef]
- Bom, S.J.H.; Van Camp, G.; Cryns, K.; Admiraal, R.J.C.; Huygen, P.L.M.; Cremers, C.W.R.J. Autosomal Dominant Low-Frequency Hearing Impairment (DFNA6/14). Otol. Neurotol. 2002, 23, 876–884. [Google Scholar] [CrossRef]
- Thorpe, R.K.; Walls, W.D.; Corrigan, R.; Schaefer, A.; Wang, K.; Huygen, P.; Casavant, T.L.; Smith, R.J.H. AudioGene: Refining the natural history of KCNQ4, GSDME, WFS1, and COCH-associated hearing loss. Hum. Genet. 2022, 141, 877–887. [Google Scholar] [CrossRef]
- Sun, Y.; Cheng, J.; Lu, Y.; Li, J.; Lu, Y.; Jin, Z.; Dai, P.; Wang, R.; Yuan, H. Identification of two novel missense WFS1 mutations, H696Y and R703H, in patients with non-syndromic low-frequency sensorineural hearing loss. J. Genet. Genom. 2011, 38, 71–76. [Google Scholar] [CrossRef]
- Bramhall, N.F.; Kallman, J.C.; Verrall, A.M.; A Street, V. A novel WFS1 mutation in a family with dominant low frequency sensorineural hearing loss with normal VEMP and EcochG findings. BMC Med. Genet. 2008, 9, 48. [Google Scholar] [CrossRef]
- Cunningham, F.; E Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2021, 50, D988–D995. [Google Scholar] [CrossRef]
- Takeda, K.; Inoue, H.; Tanizawa, Y.; Matsuzaki, Y.; Oba, J.; Watanabe, Y.; Shinoda, K.; Oka, Y. WFS1 (Wolfram syndrome 1) gene product: Predominant subcellular localization to endoplasmic reticulum in cultured cells and neuronal expression in rat brain. Hum. Mol. Genet. 2001, 10, 477–484. [Google Scholar] [CrossRef]
- Osman, A.A.; Saito, M.; Makepeace, C.; Permutt, M.A.; Schlesinger, P.; Mueckler, M. Wolframin Expression Induces Novel Ion Channel Activity in Endoplasmic Reticulum Membranes and Increases Intracellular Calcium. J. Biol. Chem. 2003, 278, 52755–52762. [Google Scholar] [CrossRef]
- Takei, D.; Ishihara, H.; Yamaguchi, S.; Yamada, T.; Tamura, A.; Katagiri, H.; Maruyama, Y.; Oka, Y. WFS1 protein modulates the free Ca2+ concentration in the endoplasmic reticulum. FEBS Lett. 2006, 580, 5635–5640. [Google Scholar] [CrossRef]
- Zatyka, M.; Da Silva Xavier, G.; Bellomo, E.A.; Leadbeater, W.; Astuti, D.; Smith, J.; Michelangeli, F.; Rutter, G.A.; Barrett, T.G. Sarco(endo)plasmic reticulum ATPase is a molecular partner of Wolfram syndrome 1 protein, which negatively regulates its expression. Hum. Mol. Genet. 2014, 24, 814–827. [Google Scholar] [CrossRef]
- Nguyen, L.D.; Fischer, T.T.; Abreu, D.; Arroyo, A.; Urano, F.; Ehrlich, B.E. Calpain inhibitor and ibudilast rescue β cell functions in a cellular model of Wolfram syndrome. Proc. Natl. Acad. Sci. USA 2020, 117, 17389–17398. [Google Scholar] [CrossRef]
- Ishihara, H.; Takeda, S.; Tamura, A.; Takahashi, R.; Yamaguchi, S.; Takei, D.; Yamada, T.; Inoue, H.; Soga, H.; Katagiri, H.; et al. Disruption of the WFS1 gene in mice causes progressive -cell loss and impaired stimulus-secretion coupling in insulin secretion. Hum. Mol. Genet. 2004, 13, 1159–1170. [Google Scholar] [CrossRef]
- Fonseca, S.G.; Ishigaki, S.; Oslowski, C.M.; Lu, S.; Lipson, K.L.; Ghosh, R.; Hayashi, E.; Ishihara, H.; Oka, Y.; Permutt, M.A.; et al. Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. J. Clin. Investig. 2010, 120, 744–755. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, S.G.; Fukuma, M.; Lipson, K.L.; Nguyen, L.X.; Allen, J.R.; Oka, Y.; Urano, F. WFS1 Is a Novel Component of the Unfolded Protein Response and Maintains Homeostasis of the Endoplasmic Reticulum in Pancreatic β-Cells. J. Biol. Chem. 2005, 280, 39609–39615. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Ishihara, H.; Tamura, A.; Takahashi, R.; Yamaguchi, S.; Takei, D.; Tokita, A.; Satake, C.; Tashiro, F.; Katagiri, H.; et al. WFS1-deficiency increases endoplasmic reticulum stress, impairs cell cycle progression and triggers the apoptotic pathway specifically in pancreatic β-cells. Hum. Mol. Genet. 2006, 15, 1600–1609. [Google Scholar] [CrossRef] [PubMed]
- Cairns, G.; Burté, F.; Price, R.; O’Connor, E.; Toms, M.; Mishra, R.; Moosajee, M.; Pyle, A.; Sayer, J.A.; Yu-Wai-Man, P. A mutant wfs1 zebrafish model of Wolfram syndrome manifesting visual dysfunction and developmental delay. Sci. Rep. 2021, 11, 20491. [Google Scholar] [CrossRef] [PubMed]
- Cagalinec, M.; Liiv, M.; Hodurova, Z.; Hickey, M.A.; Vaarmann, A.; Mandel, M.; Zeb, A.; Choubey, V.; Kuum, M.; Safiulina, D.; et al. Role of Mitochondrial Dynamics in Neuronal Development: Mechanism for Wolfram Syndrome. PLOS Biol. 2016, 14, e1002511. [Google Scholar] [CrossRef]
- Zmyslowska, A.; Kuljanin, M.; Malachowska, B.; Stanczak, M.; Michalek, D.; Wlodarczyk, A.; Grot, D.; Taha, J.; Pawlik, B.; Lebiedzińska-Arciszewska, M.; et al. Multiomic analysis on human cell model of wolfram syndrome reveals changes in mitochondrial morphology and function. Cell Commun. Signal. 2021, 19, 116. [Google Scholar] [CrossRef]
- Wang, L.; Liu, H.; Zhang, X.; Song, E.; Wang, Y.; Xu, T.; Li, Z. WFS1 functions in ER export of vesicular cargo proteins in pancreatic β-cells. Nat. Commun. 2021, 12, 6996. [Google Scholar] [CrossRef]
- Angebault, C.; Fauconnier, J.; Patergnani, S.; Rieusset, J.; Danese, A.; Affortit, C.A.; Jagodzinska, J.; Mégy, C.; Quiles, M.; Cazevieille, C.; et al. ER-mitochondria cross-talk is regulated by the Ca2+ sensor NCS1 and is impaired in Wolfram syndrome. Sci. Signal. 2018, 11, eaaq1380. [Google Scholar] [CrossRef]
- Cryns, K.; Thys, S.; Van Laer, L.; Oka, Y.; Pfister, M.; Van Nassauw, L.; Smith, R.J.H.; Timmermans, J.-P.; Van Camp, G. The WFS1 gene, responsible for low frequency sensorineural hearing loss and Wolfram syndrome, is expressed in a variety of inner ear cells. Histochemistry 2003, 119, 247–256. [Google Scholar] [CrossRef]
- Teggi, R.; Carpini, S.D.; Zagato, L. Endolymphatic hydrops and ionic transporters: Genetic and biohumoral aspects. J. Neurol. 2019, 266, 47–51. [Google Scholar] [CrossRef]
- Lopez-Escamez, J.A.; Carey, J.; Chung, W.-H.; Goebel, J.A.; Magnusson, M.; Mandalà, M.; Newman-Toker, D.E.; Strupp, M.; Suzuki, M.; Trabalzini, F.; et al. Diagnostic criteria for Menière’s disease. J. Vestib. Res. 2015, 25, 1–7. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Subject | III:04 | III:11 | IV:07 | IV:09 |
|---|---|---|---|---|
| Age (y) | 72 | 70 | 49 | 46 |
| Vestibular complaints (DHI score) | Yes (NC) | Yes (34) | No (0) | No (0) |
| Caloric test a | ||||
| SPV b (R/L) | 44/36 | 26/32 | 18/14 | 45/14 |
| Rotating chair | ||||
| SCV b (R/L) | 38/61 | 52/NT | 31/29 | 41/27 |
| Tau b (R/L) | 25/17 | 25/NT | 47/55 | 21/29 |
| GA b (R/L) | 929/1015 | 1271/NT | 1421/1590 | 857/641 |
| vHIT (mean gain) | ||||
| Anterior (R/L) | N/N | N/N | N/N | N/N |
| Lateral (R/L) | N/N | N/N | N/N | N/N |
| Posterior (R/L) | N/N | N/N | N/N | N/N |
| VEMP | ||||
| Ocular (R/L) | N/A | A/A | N/N | A/A |
| Cervical (R/L) | A/N | A/N | N/N | N/N |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velde, H.M.; Huizenga, X.J.J.; Yntema, H.G.; Haer-Wigman, L.; Beynon, A.J.; Oostrik, J.; Pegge, S.A.H.; Kremer, H.; Lanting, C.P.; Pennings, R.J.E. Genotype and Phenotype Analyses of a Novel WFS1 Variant (c.2512C>T p.(Pro838Ser)) Associated with DFNA6/14/38. Genes 2023, 14, 457. https://doi.org/10.3390/genes14020457
Velde HM, Huizenga XJJ, Yntema HG, Haer-Wigman L, Beynon AJ, Oostrik J, Pegge SAH, Kremer H, Lanting CP, Pennings RJE. Genotype and Phenotype Analyses of a Novel WFS1 Variant (c.2512C>T p.(Pro838Ser)) Associated with DFNA6/14/38. Genes. 2023; 14(2):457. https://doi.org/10.3390/genes14020457
Chicago/Turabian StyleVelde, Hedwig M., Xanne J. J. Huizenga, Helger G. Yntema, Lonneke Haer-Wigman, Andy J. Beynon, Jaap Oostrik, Sjoert A. H. Pegge, Hannie Kremer, Cris P. Lanting, and Ronald J. E. Pennings. 2023. "Genotype and Phenotype Analyses of a Novel WFS1 Variant (c.2512C>T p.(Pro838Ser)) Associated with DFNA6/14/38" Genes 14, no. 2: 457. https://doi.org/10.3390/genes14020457