
Integral Role of the Mitochondrial Ribosome in Supporting Ovarian Function: MRPS7 Variants in Syndromic Premature Ovarian Insufficiency
 , , , ,
, , , ,
Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
| Gene | OMIM | Inheritance | Clinical Presentation |
|---|---|---|---|
| Large 39S-LSU | |||
| MRPL3 | 607118 | AR/AD | COXPD and cardiomyopathy. Suggestive implication in Tourette’s syndrome/chronic tick disorder [9,10] |
| MRPL12 | 602375 | AR | Growth retardation, neurological defects and OXPHOS deficiency [11] |
| MRPL24 | 611986 | AR | Cerebellar atrophy, combined defect of complexes I and IV and choreoathetosis [13] |
| MRPL44 | 611849 | AR | Infantile cardiomyopathy [12] |
| Small 28S-SSU | |||
| MRPS2 | 611971 | AR | Hypoglycaemia and lactic acidosis [8,14] |
| MRPS7 | 611974 | AR | Sensorineural hearing loss, hepatic and renal failure delayed pubertal onset, primary hypogonadism [24]. |
| MRPS14 | 611978 | AR | Hypertrophic cardiomyopathy, lactic acidosis, developmental delay and muscle hypotonia [19] |
| MRPS16 | 609204 | AR | Agenesis of corpus callosum, dysmorphism and fatal neonatal lactic acidosis [15] |
| MRPS22 | 605810 | AR | Cornelia de Lange-like dysmorphic features, brain abnormalities, hypertrophic cardiomyopathy and isolated POI [22,23] |
| MRPS23 | 611985 | AR | Hepatic disease [16] |
| MRPS25 | 611987 | AR | Cerebral palsy, partial corpus callosum agenesis and mitochondrial myopathy [21] |
| MRPS34 | 611994 | AR | Neurological dysfunction, Leigh syndrome [14,17] |
| MRPS39 | 614918 | AR | Leigh syndrome [20] |
2. Materials and Methods
2.1. Ethics
2.2. Participants
2.3. General Molecular Techniques
2.4. Whole-Exome Sequencing (WES)
2.5. Variant Phasing
3. Results
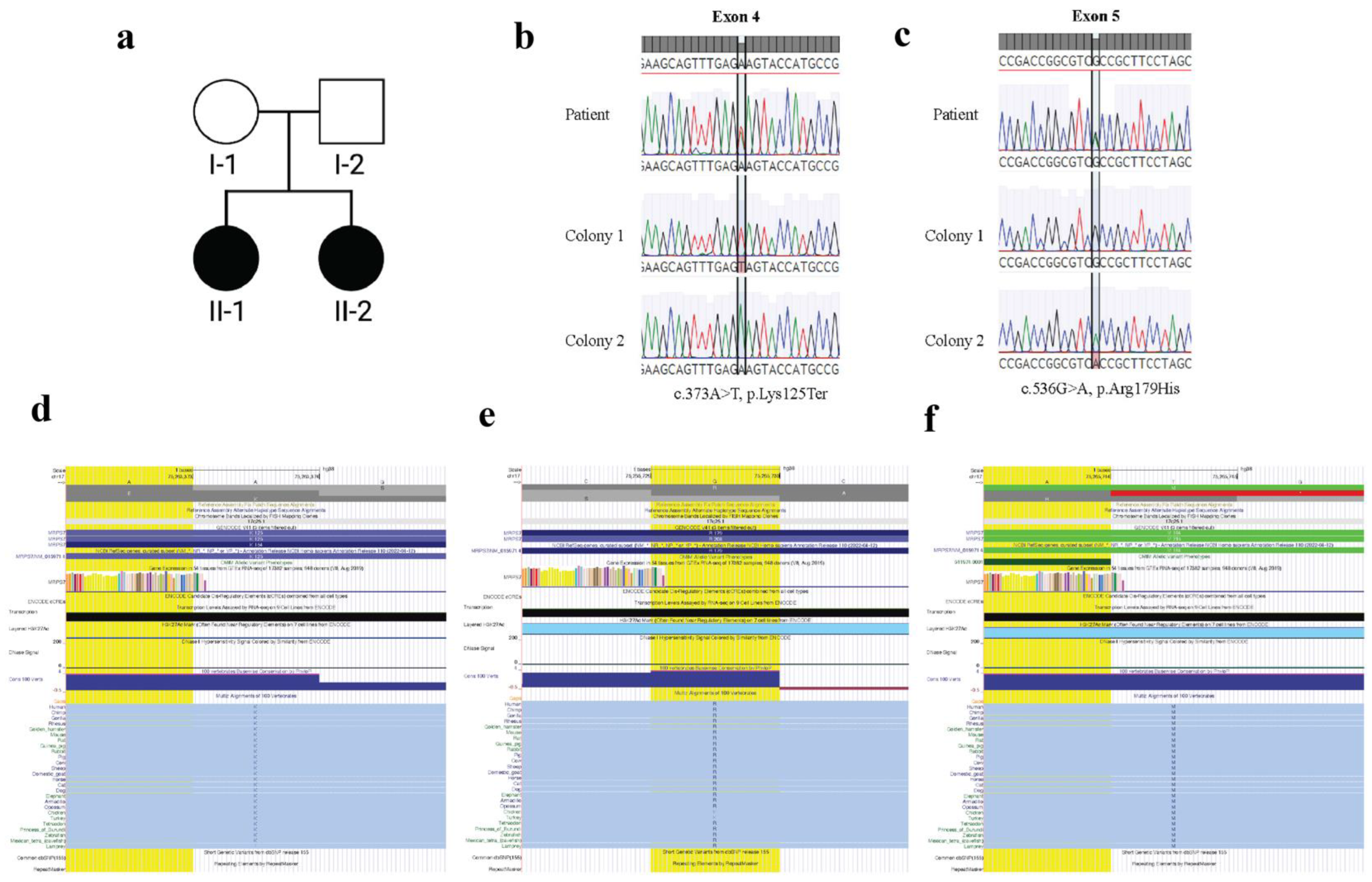
3.1. Diagnosis of Syndromic Premature Ovarian Insufficiency
3.2. Whole Exome Sequencing Identifies MRPS7 Variants
3.3. MRPS7 Variants Are Inherited Bi-Allelically
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tucker, E.J.; Grover, S.R.; Bachelot, A.; Touraine, P.; Sinclair, A.H. Premature Ovarian Insufficiency: New Perspectives on Genetic Cause and Phenotypic Spectrum. Endocr. Rev. 2016, 37, 609–635. [Google Scholar] [CrossRef] [PubMed]
- Newman, W.G.; Friedman, T.B.; Conway, G.S.; Demain, L.A.M. Perrault Syndrome. In GeneReviews((R)); Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1116/ (accessed on 9 October 2022).
- Tucker, E.J.; Rius, R.; Jaillard, S.; Bell, K.; Lamont, P.J.; Travessa, A.; Dupont, J.; Sampaio, L.; Dulon, J.; Vuillaumier-Barrot, S.; et al. Genomic sequencing highlights the diverse molecular causes of Perrault syndrome: A peroxisomal disorder (PEX6), metabolic disorders (CLPP, GGPS1), and mtDNA maintenance/translation disorders (LARS2, TFAM). Hum. Genet. 2020, 139, 1325–1343. [Google Scholar] [CrossRef] [PubMed]
- Foley, A.R.; Zou, Y.; Dunford, J.E.; Rooney, J.; Chandra, G.; Xiong, H.; Straub, V.; Voit, T.; Romero, N.; Donkervoort, S.; et al. GGPS1 Mutations Cause Muscular Dystrophy/Hearing Loss/Ovarian Insufficiency Syndrome. Ann. Neurol. 2020, 88, 332–347. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Ruiz, M.; Garcia-Martinez, A.; Corral-Juan, M.; Perez-Alvarez, A.I.; Plasencia, A.M.; Villamar, M.; Moreno-Pelayo, M.A.; Matilla-Duenas, A.; Menendez-Gonzalez, M.; Del Castillo, I. Perrault syndrome with neurological features in a compound heterozygote for two TWNK mutations: Overlap of TWNK-related recessive disorders. J. Transl. Med. 2019, 17, 290. [Google Scholar] [CrossRef] [PubMed]
- Hochberg, I.; Demain, L.A.M.; Richer, J.; Thompson, K.; Urquhart, J.E.; Rea, A.; Pagarkar, W.; Rodriguez-Palmero, A.; Schluter, A.; Verdura, E.; et al. Bi-allelic variants in the mitochondrial RNase P subunit PRORP cause mitochondrial tRNA processing defects and pleiotropic multisystem presentations. Am. J. Hum. Genet. 2021, 108, 2195–2204. [Google Scholar] [CrossRef]
- Amunts, A.; Brown, A.; Toots, J.; Scheres, S.H.W.; Ramakrishnan, V. The structure of the human mitochondrial ribosome. Science 2015, 348, 95–98. [Google Scholar] [CrossRef] [Green Version]
- Gardeitchik, T.; Mohamed, M.; Ruzzenente, B.; Karall, D.; Guerrero-Castillo, S.; Dalloyaux, D.; van den Brand, M.; van Kraaij, S.; van Asbeck, E.; Assouline, Z.; et al. Bi-allelic Mutations in the Mitochondrial Ribosomal Protein MRPS2 Cause Sensorineural Hearing Loss, Hypoglycemia, and Multiple OXPHOS Complex Deficiencies. Am. J. Hum. Genet. 2018, 102, 685–695. [Google Scholar] [CrossRef] [Green Version]
- Galmiche, L.; Serre, V.; Beinat, M.; Assouline, Z.; Lebre, A.S.; Chretien, D.; Nietschke, P.; Benes, V.; Boddaert, N.; Sidi, D.; et al. Exome sequencing identifies MRPL3 mutation in mitochondrial cardiomyopathy. Hum. Mutat. 2011, 32, 1225–1231. [Google Scholar] [CrossRef]
- Sundaram, S.K.; Huq, A.M.; Sun, Z.; Yu, W.; Bennett, L.; Wilson, B.J.; Behen, M.E.; Chugani, H.T. Exome sequencing of a pedigree with Tourette syndrome or chronic tic disorder. Ann. Neurol. 2011, 69, 901–904. [Google Scholar] [CrossRef]
- Serre, V.; Rozanska, A.; Beinat, M.; Chretien, D.; Boddaert, N.; Munnich, A.; Rotig, A.; Chrzanowska-Lightowlers, Z.M. Mutations in mitochondrial ribosomal protein MRPL12 leads to growth retardation, neurological deterioration and mitochondrial translation deficiency. Biochim. Biophys. Acta 2013, 1832, 1304–1312. [Google Scholar] [CrossRef]
- Carroll, C.J.; Isohanni, P.; Poyhonen, R.; Euro, L.; Richter, U.; Brilhante, V.; Gotz, A.; Lahtinen, T.; Paetau, A.; Pihko, H.; et al. Whole-exome sequencing identifies a mutation in the mitochondrial ribosome protein MRPL44 to underlie mitochondrial infantile cardiomyopathy. J. Med. Genet. 2013, 50, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Di Nottia, M.; Marchese, M.; Verrigni, D.; Mutti, C.D.; Torraco, A.; Oliva, R.; Fernandez-Vizarra, E.; Morani, F.; Trani, G.; Rizza, T.; et al. A homozygous MRPL24 mutation causes a complex movement disorder and affects the mitoribosome assembly. Neurobiol. Dis. 2020, 141, 104880. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhou, W.; Liu, Q.; Peng, Z. Hypoglycemia with lactic acidosis caused by a new MRPS2 gene mutation in a Chinese girl: A case report. BMC Endocr. Disord. 2022, 22, 15. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.; Saada, A.; Shaul, N.; Shabtai, N.; Ben-Shalom, E.; Shaag, A.; Hershkovitz, E.; Elpeleg, O. Defective mitochondrial translation caused by a ribosomal protein (MRPS16) mutation. Ann. Neurol. 2004, 56, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Kohda, M.; Tokuzawa, Y.; Kishita, Y.; Nyuzuki, H.; Moriyama, Y.; Mizuno, Y.; Hirata, T.; Yatsuka, Y.; Yamashita-Sugahara, Y.; Nakachi, Y.; et al. A Comprehensive Genomic Analysis Reveals the Genetic Landscape of Mitochondrial Respiratory Chain Complex Deficiencies. PLoS Genet. 2016, 12, e1005679. [Google Scholar] [CrossRef]
- Lake, N.J.; Webb, B.D.; Stroud, D.A.; Richman, T.R.; Ruzzenente, B.; Compton, A.G.; Mountford, H.S.; Pulman, J.; Zangarelli, C.; Rio, M.; et al. Biallelic Mutations in MRPS34 Lead to Instability of the Small Mitoribosomal Subunit and Leigh Syndrome. Am. J. Hum. Genet. 2018, 102, 713. [Google Scholar] [CrossRef] [Green Version]
- Pulman, J.; Ruzzenente, B.; Bianchi, L.; Rio, M.; Boddaert, N.; Munnich, A.; Rotig, A.; Metodiev, M.D. Mutations in the MRPS28 gene encoding the small mitoribosomal subunit protein bS1m in a patient with intrauterine growth retardation, craniofacial dysmorphism and multisystemic involvement. Hum. Mol. Genet. 2019, 28, 1445–1462. [Google Scholar] [CrossRef]
- Jackson, C.B.; Huemer, M.; Bolognini, R.; Martin, F.; Szinnai, G.; Donner, B.C.; Richter, U.; Battersby, B.J.; Nuoffer, J.M.; Suomalainen, A.; et al. A variant in MRPS14 (uS14m) causes perinatal hypertrophic cardiomyopathy with neonatal lactic acidosis, growth retardation, dysmorphic features and neurological involvement. Hum. Mol. Genet. 2019, 28, 639–649. [Google Scholar] [CrossRef]
- Borna, N.N.; Kishita, Y.; Kohda, M.; Lim, S.C.; Shimura, M.; Wu, Y.; Mogushi, K.; Yatsuka, Y.; Harashima, H.; Hisatomi, Y.; et al. Mitochondrial ribosomal protein PTCD3 mutations cause oxidative phosphorylation defects with Leigh syndrome. Neurogenetics 2019, 20, 9–25. [Google Scholar] [CrossRef]
- Bugiardini, E.; Mitchell, A.L.; Rosa, I.D.; Horning-Do, H.T.; Pitmann, A.M.; Poole, O.V.; Holton, J.L.; Shah, S.; Woodward, C.; Hargreaves, I.; et al. MRPS25 mutations impair mitochondrial translation and cause encephalomyopathy. Hum. Mol. Genet. 2019, 28, 2711–2719. [Google Scholar] [CrossRef]
- Smits, P.; Saada, A.; Wortmann, S.B.; Heister, A.J.; Brink, M.; Pfundt, R.; Miller, C.; Haas, D.; Hantschmann, R.; Rodenburg, R.J.; et al. Mutation in mitochondrial ribosomal protein MRPS22 leads to Cornelia de Lange-like phenotype, brain abnormalities and hypertrophic cardiomyopathy. Eur. J. Hum. Genet. 2011, 19, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Tiosano, D.; Guran, T.; Baris, H.N.; Bayram, Y.; Mory, A.; Shapiro-Kulnane, L.; Hodges, C.A.; Akdemir, Z.C.; Turan, S.; et al. Mutations in the mitochondrial ribosomal protein MRPS22 lead to primary ovarian insufficiency. Hum. Mol. Genet. 2018, 27, 1913–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menezes, M.J.; Guo, Y.; Zhang, J.; Riley, L.G.; Cooper, S.T.; Thorburn, D.R.; Li, J.; Dong, D.; Li, Z.; Glessner, J.; et al. Mutation in mitochondrial ribosomal protein S7 (MRPS7) causes congenital sensorineural deafness, progressive hepatic and renal failure and lactic acidemia. Hum. Mol. Genet. 2015, 24, 2297–2307. [Google Scholar] [CrossRef] [Green Version]
- The ESHRE Guideline Group on POI; Webber, L.; Davies, M.; Anderson, R.; Bartlett, J.; Braat, D.; Cartwright, B.; Cifkova, R.; de Muinck Keizer-Schrama, S.; Hogervorst, E.; et al. ESHRE Guideline: Management of women with premature ovarian insufficiency. Hum. Reprod. 2016, 31, 926–937. [Google Scholar] [CrossRef] [PubMed]
- Sadedin, S.P.; Dashnow, H.; James, P.A.; Bahlo, M.; Bauer, D.C.; Lonie, A.; Lunke, S.; Macciocca, I.; Ross, J.P.; Siemering, K.R.; et al. Cpipe: A shared variant detection pipeline designed for diagnostic settings. Genome Med. 2015, 7, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venselaar, H.; Te Beek, T.A.; Kuipers, R.K.; Hekkelman, M.L.; Vriend, G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinform. 2010, 11, 548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- UniProt, C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Cavdar Koc, E.; Burkhart, W.; Blackburn, K.; Moseley, A.; Spremulli, L.L. The small subunit of the mammalian mitochondrial ribosome. Identification of the full complement of ribosomal proteins present. J. Biol. Chem. 2001, 276, 19363–19374. [Google Scholar] [CrossRef] [Green Version]
- Prezant, T.R.; Agapian, J.V.; Bohlman, M.C.; Bu, X.; Oztas, S.; Qiu, W.Q.; Arnos, K.S.; Cortopassi, G.A.; Jaber, L.; Rotter, J.I.; et al. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nat. Genet. 1993, 4, 289–294. [Google Scholar] [CrossRef]
- Sorrentino, V.; Menzies, K.J.; Auwerx, J. Repairing Mitochondrial Dysfunction in Disease. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 353–389. [Google Scholar] [CrossRef]
- Wai, T.; Ao, A.; Zhang, X.; Cyr, D.; Dufort, D.; Shoubridge, E.A. The role of mitochondrial DNA copy number in mammalian fertility. Biol. Reprod. 2010, 83, 52–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiaratti, M.R. Uncovering the important role of mitochondrial dynamics in oogenesis: Impact on fertility and metabolic disorder transmission. Biophys. Rev. 2021, 13, 967–981. [Google Scholar] [CrossRef] [PubMed]
- Kirillova, A.; Smitz, J.E.J.; Sukhikh, G.T.; Mazunin, I. The Role of Mitochondria in Oocyte Maturation. Cells 2021, 10, 2484. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, G.R.; Yadav, P.K.; Yadav, A.K.; Tiwari, M.; Gupta, A.; Sharma, A.; Pandey, A.N.; Pandey, A.K.; Chaube, S.K. Necroptosis in stressed ovary. J. Biomed. Sci. 2019, 26, 11. [Google Scholar] [CrossRef] [Green Version]
- Tiosano, D.; Mears, J.A.; Buchner, D.A. Mitochondrial Dysfunction in Primary Ovarian Insufficiency. Endocrinology 2019, 160, 2353–2366. [Google Scholar] [CrossRef]
- Dallabona, C.; Diodato, D.; Kevelam, S.H.; Haack, T.B.; Wong, L.J.; Salomons, G.S.; Baruffini, E.; Melchionda, L.; Mariotti, C.; Strom, T.M.; et al. Novel (ovario) leukodystrophy related to AARS2 mutations. Neurology 2014, 82, 2063–2071. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Yang, H.J.; Kwon, J.H.; Kim, W.J.; Kim, S.Y.; Lee, E.M.; Park, J.Y.; Weon, Y.C.; Park, S.H.; Gwon, B.J.; et al. Two Korean siblings with recently described ovarioleukodystrophy related to AARS2 mutations. Eur. J. Neurol. 2017, 24, e21–e22. [Google Scholar] [CrossRef]
- Tucker, E.J.; Baker, M.J.; Hock, D.H.; Warren, J.T.; Jaillard, S.; Bell, K.M.; Sreenivasan, R.; Bakhshalizadeh, S.; Hanna, C.A.; Caruana, N.J.; et al. Premature ovarian insufficiency in CLPB deficiency: Transcriptomic, proteomic and phenotypic insights. J. Clin. Endocrinol. Metab. 2022, in press. [CrossRef]
- Jenkinson, E.M.; Rehman, A.U.; Walsh, T.; Clayton-Smith, J.; Lee, K.; Morell, R.J.; Drummond, M.C.; Khan, S.N.; Naeem, M.A.; Rauf, B.; et al. Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP-dependent chambered protease. Am. J. Hum. Genet. 2013, 92, 605–613. [Google Scholar] [CrossRef] [Green Version]
- Gispert, S.; Parganlija, D.; Klinkenberg, M.; Drose, S.; Wittig, I.; Mittelbronn, M.; Grzmil, P.; Koob, S.; Hamann, A.; Walter, M.; et al. Loss of mitochondrial peptidase Clpp leads to infertility, hearing loss plus growth retardation via accumulation of CLPX, mtDNA and inflammatory factors. Hum. Mol. Genet. 2013, 22, 4871–4887. [Google Scholar] [CrossRef]
- Pierce, S.B.; Chisholm, K.M.; Lynch, E.D.; Lee, M.K.; Walsh, T.; Opitz, J.M.; Li, W.; Klevit, R.E.; King, M.C. Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc. Natl. Acad. Sci. USA 2011, 108, 6543–6548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karstensen, H.G.; Rendtorff, N.D.; Hindbaek, L.S.; Colombo, R.; Stein, A.; Birkebaek, N.H.; Hartmann-Petersen, R.; Lindorff-Larsen, K.; Hojland, A.T.; Petersen, M.B.; et al. Novel HARS2 missense variants identified in individuals with sensorineural hearing impairment and Perrault syndrome. Eur. J. Med. Genet. 2020, 63, 103733. [Google Scholar] [CrossRef] [PubMed]
- Pierce, S.B.; Gersak, K.; Michaelson-Cohen, R.; Walsh, T.; Lee, M.K.; Malach, D.; Klevit, R.E.; King, M.C.; Levy-Lahad, E. Mutations in LARS2, encoding mitochondrial leucyl-tRNA synthetase, lead to premature ovarian failure and hearing loss in Perrault syndrome. Am. J. Hum. Genet. 2013, 92, 614–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosaki, R.; Horikawa, R.; Fujii, E.; Kosaki, K. Biallelic mutations in LARS2 can cause Perrault syndrome type 2 with neurologic symptoms. Am. J. Med. Genet. A 2018, 176, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Luoma, P.; Melberg, A.; Rinne, J.O.; Kaukonen, J.A.; Nupponen, N.N.; Chalmers, R.M.; Oldfors, A.; Rautakorpi, I.; Peltonen, L.; Majamaa, K.; et al. Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: Clinical and molecular genetic study. Lancet 2004, 364, 875–882. [Google Scholar] [CrossRef]
- Pagnamenta, A.T.; Taanman, J.W.; Wilson, C.J.; Anderson, N.E.; Marotta, R.; Duncan, A.J.; Bitner-Glindzicz, M.; Taylor, R.W.; Laskowski, A.; Thorburn, D.R.; et al. Dominant inheritance of premature ovarian failure associated with mutant mitochondrial DNA polymerase gamma. Hum. Reprod. 2006, 21, 2467–2473. [Google Scholar] [CrossRef] [Green Version]
- Morino, H.; Pierce, S.B.; Matsuda, Y.; Walsh, T.; Ohsawa, R.; Newby, M.; Hiraki-Kamon, K.; Kuramochi, M.; Lee, M.K.; Klevit, R.E.; et al. Mutations in Twinkle primase-helicase cause Perrault syndrome with neurologic features. Neurology 2014, 83, 2054–2061. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Mumusoglu, S.; Qin, Y.; Sun, Y.; Hsueh, A.J. A kaleidoscopic view of ovarian genes associated with premature ovarian insufficiency and senescence. FASEB J. 2021, 35, e21753. [Google Scholar] [CrossRef]
- Ghaddhab, C.; Morin, C.; Brunel-Guitton, C.; Mitchell, G.A.; Van Vliet, G.; Huot, C. Premature Ovarian Failure in French Canadian Leigh Syndrome. J. Pediatr. 2017, 184, 227–229 e221. [Google Scholar] [CrossRef]
- Demain, L.A.M.; Antunes, D.; O’Sullivan, J.; Bhaskhar, S.S.; O’Keefe, R.T.; Newman, W.G. A known pathogenic variant in the essential mitochondrial translation gene RMND1 causes a Perrault-like syndrome with renal defects. Clin. Genet. 2018, 94, 276–277. [Google Scholar] [CrossRef]
- Schlieben, L.D.; Prokisch, H. The Dimensions of Primary Mitochondrial Disorders. Front. Cell Dev. Biol. 2020, 8, 600079. [Google Scholar] [CrossRef] [PubMed]



| Patient | Age at Diagnosis | Karyotype | Amenorrhea | Secondary Sex Characteristics | Ultrasound | Auditory Phenotype | FSH (IU/L) | LH (IU/L) | Estradiol (pg/mL) | AMH (ng/mL) | TSH (mIU/L) | Anti-TPO (IU/mL) | Other |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Proband | 25 | XX | Secondary | Normal puberty | Small ovaries, two microfollicles in right ovary absent in left | Sensorineural hearing loss | 102 | 34 | 29 | 0.15 | 12.89 | 102 | Hashimoto’s disease |
| Sister | 21 | XX | NR | NR | NR | Congenital hearing loss | NR | NR | NR | NR | NR | NR | NR |
| Menezes et al. P1 | - | XX | NR | NR | NR | Congenital sensorineural hearing loss | NR | NR | NR | NR | NR | NR | Lactic acidemia, progressive hepatic and renal failure |
| Menezes et al. P2 | 16 | XX | Primary | Failed puberty | Primary hypogonadism | Congenital sensorineural hearing loss | NR | NR | NR | NR | 6.75 | NR | Mild learning difficulties Renal failure Liver failure Encephalopathy secondary to liver failure. Liver and renal transplant with positive outcome |
| Median Depth | 93 | ||
|---|---|---|---|
| % Bases > ×10 | 98.7 | ||
| Criteria | Number of Genes of Interest | ||
| Gene-centric | Moderate-high priority (POI) data | 3 | DNAH5, GAB2, YBX2 |
| Variant centric | Bi-allelic (all) | 11 | MAP3K6, MMACHC, NBPF14, MAGI1, ALB, PDCD11, SRPR, SOX21, DMXL2, IFT140, MRPS7* |
| High priority (all) | 17 | MAP3K6, RGPD2, HEG1, GYG1, CPEB2, TTC37, GRIFIN, SRPR, SLC38A6, TTC8, BLOC1S6, GOLGA6B, KCNG4, PRA1, YBX2, ATP8B3, MRPS7* |
| Patient | gDNA Variant (GrCH38) | cDNA Variant | Protein Variant | Polyphen | Mutation Taster | CADD | SIFT | Provean | ACMG Classification |
|---|---|---|---|---|---|---|---|---|---|
| Proband | chr17: 75263373 | c.373A>T | p.(Lys125*) | NA | Disease causing (score 1.000) | Harmful (score 44) | NA | NA | Likely pathogenic |
| chr17: 75265730 | c.536G>A | p.(Arg179His) | Probably Damaging (score 0.989) | Disease causing (score 1.000) | Harmful (score 23.3) | Damaging (score 0.041) | Neutral (score −2.35) | Likely pathogenic | |
| Menezes et al. [24] | chr17: 75265744 | c.550A>G | p.(Met184Val) | Probably Damaging (score 1.000) | Disease causing (score 1.000) | Harmful (score 25.4) | Damaging (score 0.048 | Deleterious (score −3.06) | Pathogenic [24] |
| Gene | Inheritance | Clinical Presentation | Gene Function | Molecular Function in Mitochondria |
|---|---|---|---|---|
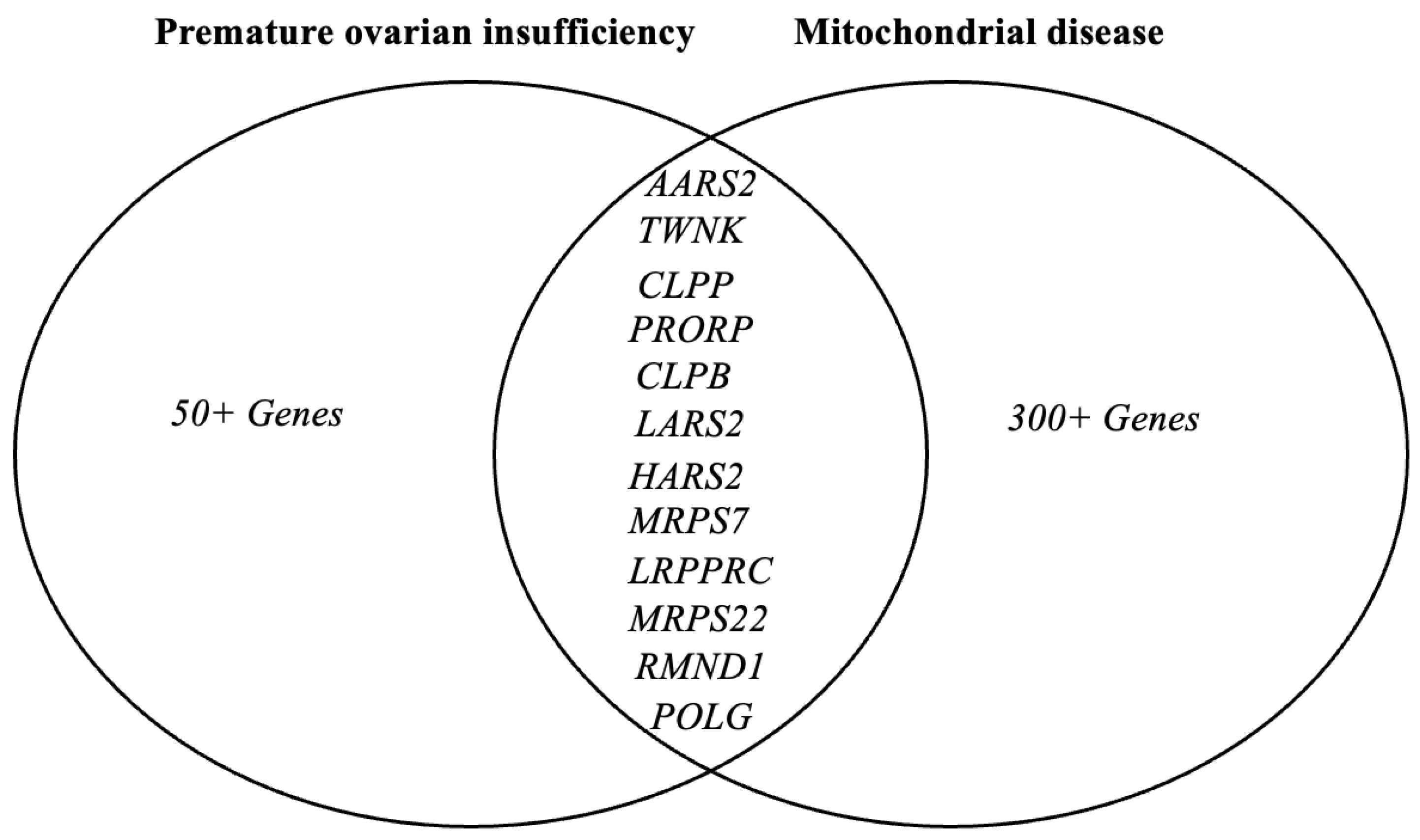
| AARS2 | AR | Ovarioleukodystrophy [37,38] | Aminoacylates alanyl-tRNA | mRNA translation |
| CLPB | AR | Progressive encephalopathy, intellectual disability, epilepsy, congenital neutropenia, cataracts, POI [39] | Caseinolytic peptidase | Mitochondrial matrix Peptidase Chaperone |
| CLPP | AR | Perrault syndrome [40,41] | Mitochondrial matrix protease | Protein degradation |
| HARS2 | AR | Perrault syndrome [42,43] | Histidine tRNA | mRNA translation |
| LARS2 | AR | Perrault syndrome [44,45] | Leucine tRNA | mRNA translation |
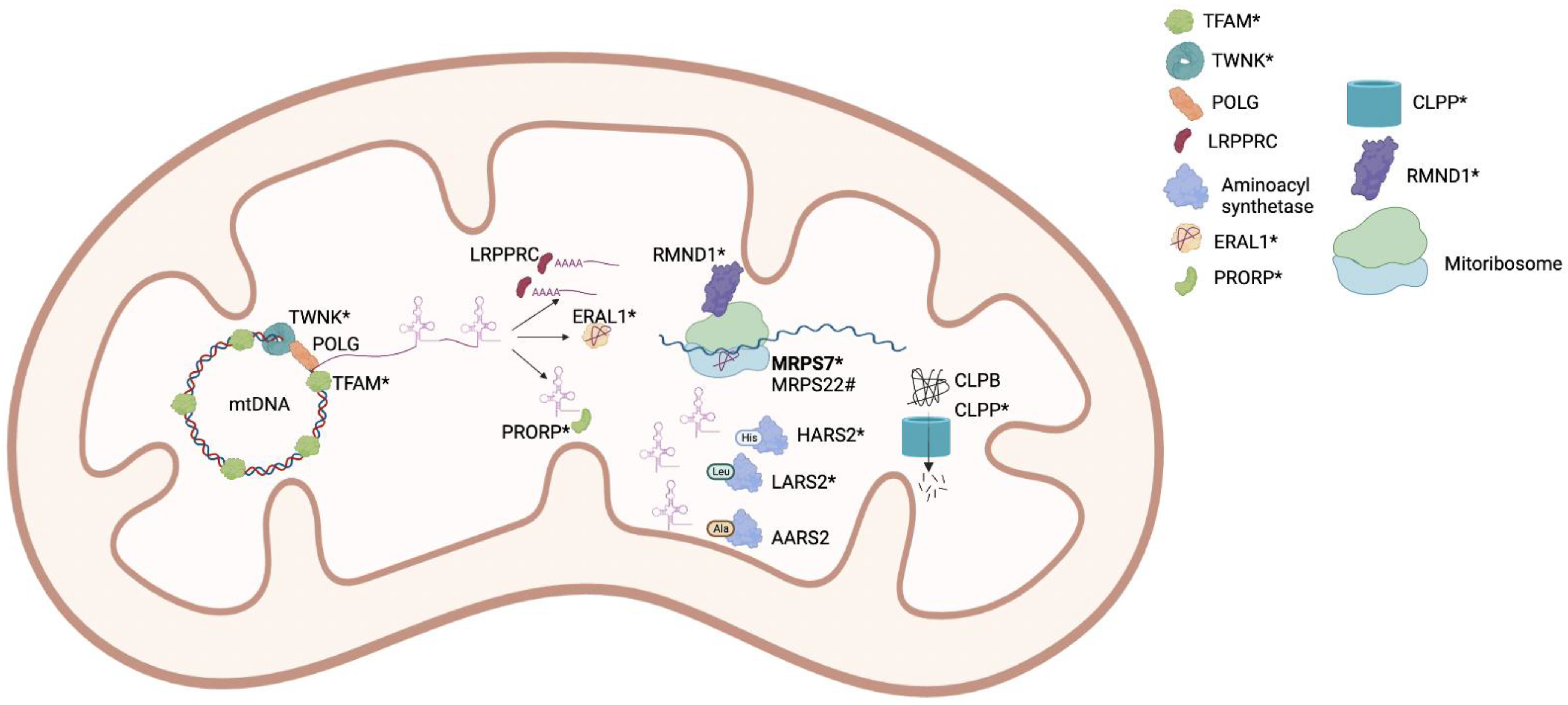
| MRPS7 | AR | Perrault syndrome, hepatic and renal failure [24] | Mitochondrial ribosome subunit | Mitochondrial protein synthesis |
| MRPS22 | AR | Isolated POI [23] | Mitochondrial ribosome subunit | mRNA translation |
| POLG | AD/AR | Premature ovarian insufficiency, mitochondrial recessive ataxia [46] Premature ovarian insufficiency, chronic progressive external ophthalmoplegia (CPEO) [47] | DNA polymerase γ | mtDNA replication |
| TWNK | AR | Perrault syndrome [48,49] | mtDNA Helicase | mtDNA replication and proofreading |
| LRPPRC | AR | Leigh Syndrome [50] | RNA binding protein | Mitochondrial gene expression |
| RMND1 | AR | Perrault syndrome, renal, neural, muscular defects [36,51] | Integral membrane protein | Mitochondrial mRNA translation |
| PRORP | AR | Perrault syndrome, developmental delay [6] | Mitochondrial-tRNA processing | Mitochondrial RNA maturation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kline, B.L.; Jaillard, S.; Bell, K.M.; Bakhshalizadeh, S.; Robevska, G.; van den Bergen, J.; Dulon, J.; Ayers, K.L.; Christodoulou, J.; Tchan, M.C.; et al. Integral Role of the Mitochondrial Ribosome in Supporting Ovarian Function: MRPS7 Variants in Syndromic Premature Ovarian Insufficiency. Genes 2022, 13, 2113. https://doi.org/10.3390/genes13112113
Kline BL, Jaillard S, Bell KM, Bakhshalizadeh S, Robevska G, van den Bergen J, Dulon J, Ayers KL, Christodoulou J, Tchan MC, et al. Integral Role of the Mitochondrial Ribosome in Supporting Ovarian Function: MRPS7 Variants in Syndromic Premature Ovarian Insufficiency. Genes. 2022; 13(11):2113. https://doi.org/10.3390/genes13112113
Chicago/Turabian StyleKline, Brianna L., Sylvie Jaillard, Katrina M. Bell, Shabnam Bakhshalizadeh, Gorjana Robevska, Jocelyn van den Bergen, Jérôme Dulon, Katie L. Ayers, John Christodoulou, Michel C. Tchan, and et al. 2022. "Integral Role of the Mitochondrial Ribosome in Supporting Ovarian Function: MRPS7 Variants in Syndromic Premature Ovarian Insufficiency" Genes 13, no. 11: 2113. https://doi.org/10.3390/genes13112113