

Profiling Genome-Wide DNA Methylation in Children with Autism Spectrum Disorder and in Children with Fragile X Syndrome
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Participants and Samples
2.2. CGG Allele Sizing and Methylation Status
2.3. Data Processing
2.4. Principal Component Analysis
2.5. Cell Type Composition Estimates
2.6. Differentially Methylated Sites (DMS)
2.7. Differential Methylated Regions (DMR)
2.8. Gene Set Enrichment Analysis
2.9. Measurements of mRNA Expression Levels
2.10. Measurement of Protein Expression Levels
3. Results
3.1. Subjects
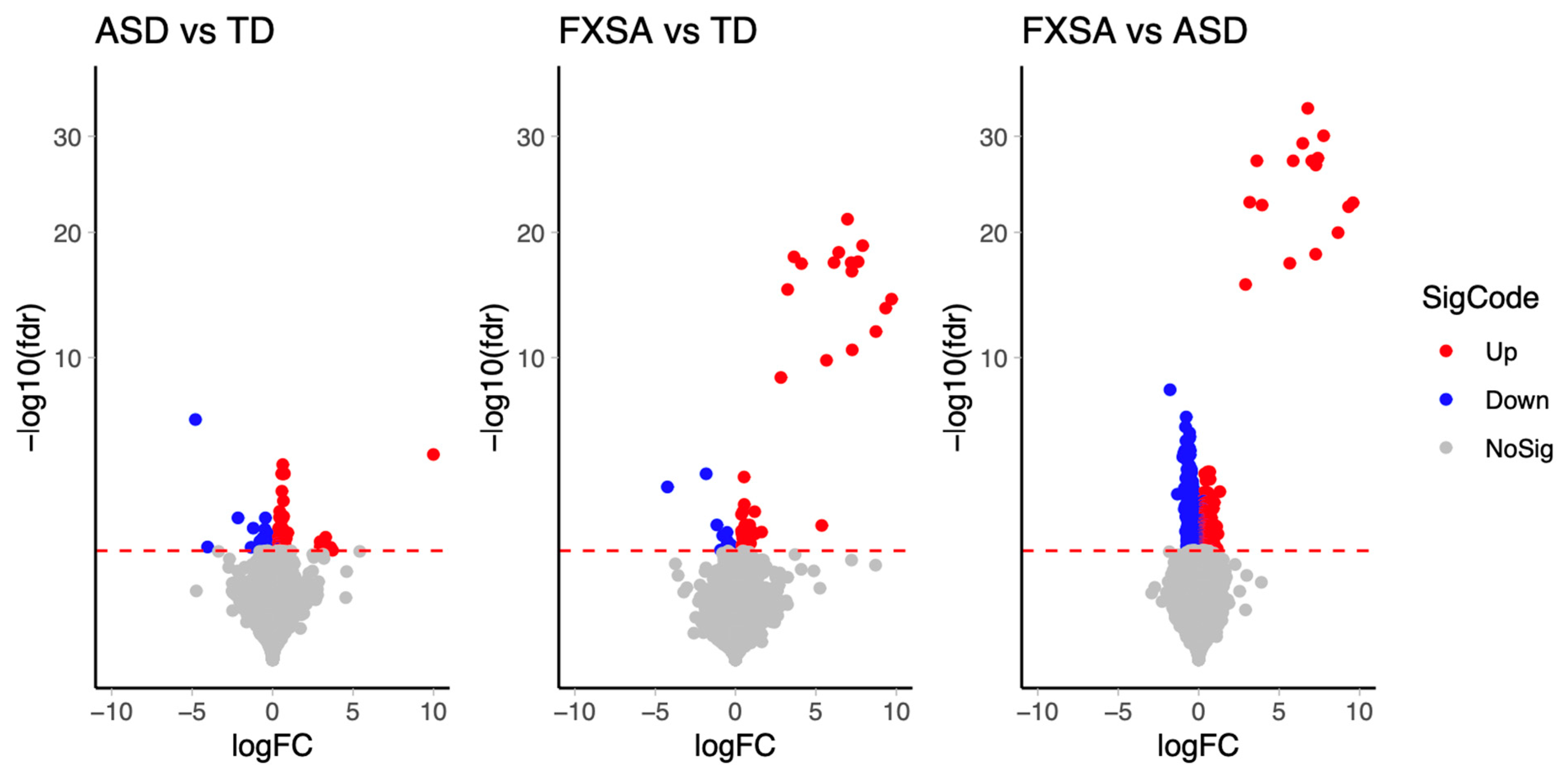
3.2. Differentially Methylated Sites (DMS)
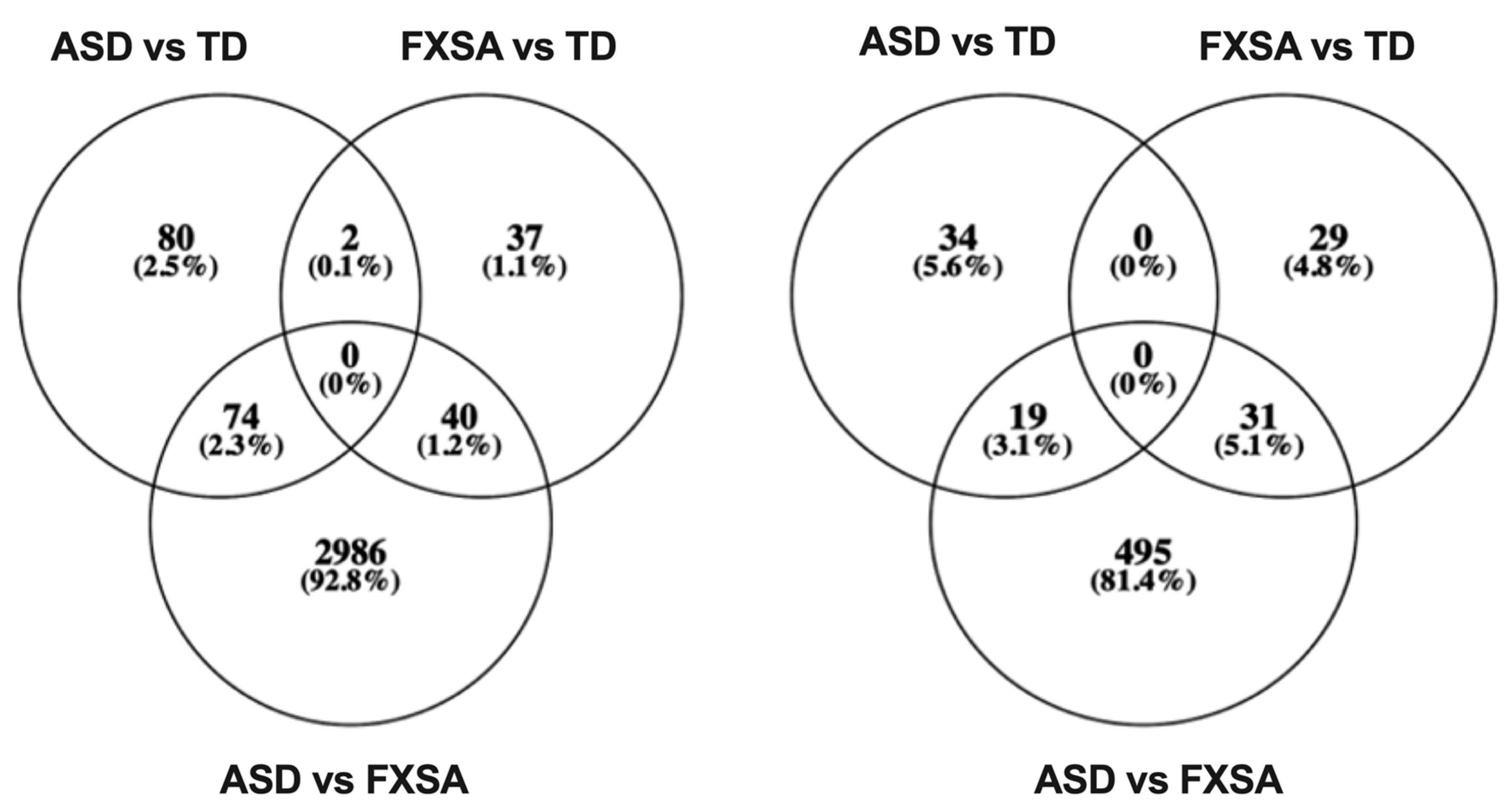
3.3. Differentially Methylated Regions (DMR)
3.4. Risk Genes for Autism Are Differentially Methylated and among Those Identified in the Three Groups
3.5. Functional Implications of Differentially Methylated Sites
3.6. Validation of Gene Expression of a Subset of Differentially Methylated Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Widiger, T. Diagnostic and Statistical Manual of Mental Disorders (DSM). Psychology 2011, 12, 347. [Google Scholar]
- Baio, J.; Wiggins, L.; Christensen, D.L.; Maenner, M.J.; Daniels, J.; Warren, Z.; Kurzius-Spencer, M.; Zahorodny, W.; Robinson, C.; Rosenberg; et al. Prevalence of Autism Spectrum Disorder among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2014. MMWR. Surveill. Summ. 2018, 67, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Jacquemont, S.; Coe, B.P.; Hersch, M.; Duyzend, M.H.; Krumm, N.; Bergmann, S.; Beckmann, J.S.; Rosenfeld, J.A.; Eichler, E.E. A Higher Mutational Burden in Females Supports a “Female Protective Model” in Neurodevelopmental Disorders. Am. J. Hum. Genet. 2014, 94, 415–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, E.B.; Lichtenstein, P.; Anckarsäter, H.; Happé, F.; Ronald, A. Examining and Interpreting the Female Protective Effect against Autistic Behavior. Proc. Natl. Acad. Sci. USA 2013, 110, 5258–5262. [Google Scholar] [CrossRef] [Green Version]
- Doan, R.N.; Lim, E.T.; De Rubeis, S.; Betancur, C.; Cutler, D.J.; Chiocchetti, A.G.; Overman, L.M.; Soucy, A.; Goetze, S.; Autism Sequencing Consortium; et al. Recessive Gene Disruptions in Autism Spectrum Disorder. Nat. Genet. 2019, 51, 1092–1098. [Google Scholar] [CrossRef]
- Quesnel-Vallières, M.; Weatheritt, R.J.; Cordes, S.P.; Blencowe, B.J. Autism Spectrum Disorder: Insights into Convergent Mechanisms from Transcriptomics. Nat. Rev. Genet. 2019, 20, 51–63. [Google Scholar] [CrossRef]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, Transcriptional and Chromatin Genes Disrupted in Autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The Contribution of de Novo Coding Mutations to Autism Spectrum Disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [Green Version]
- Gaugler, T.; Klei, L.; Sanders, S.J.; Bodea, C.A.; Goldberg, A.P.; Lee, A.B.; Mahajan, M.; Manaa, D.; Pawitan, Y.; Reichert, J.; et al. Most Genetic Risk for Autism Resides with Common Variation. Nat. Genet. 2014, 46, 881–885. [Google Scholar] [CrossRef] [Green Version]
- Sanders, S.J.; He, X.; Willsey, A.J.; Ercan-Sencicek, A.G.; Samocha, K.E.; Cicek, A.E.; Murtha, M.T.; Bal, V.H.; Bishop, S.L.; Dong, S.; et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 2015, 87, 1215–1233. [Google Scholar] [CrossRef] [Green Version]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; et al. Convergence of Genes and Cellular Pathways Dysregulated in Autism Spectrum Disorders. Am. J. Hum. Genet. 2014, 94, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.W.; Chahrour, M.H.; Coulter, M.E.; Jiralerspong, S.; Okamura-Ikeda, K.; Ataman, B.; Schmitz-Abe, K.; Harmin, D.A.; Adli, M.; Malik, A.N.; et al. Using Whole-Exome Sequencing to Identify Inherited Causes of Autism. Neuron 2013, 77, 259–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, E.T.; Uddin, M.; De Rubeis, S.; Chan, Y.; Kamumbu, A.S.; Zhang, X.; D’Gama, A.M.; Kim, S.N.; Hill, R.S.; Goldberg, A.P.; et al. Rates, Distribution and Implications of Postzygotic Mosaic Mutations in Autism Spectrum Disorder. Nat. Neurosci. 2017, 20, 1217–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bird, A. DNA Methylation Patterns and Epigenetic Memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffney, L.J.; Valdez, P.; Tremblay, M.W.; Cao, X.; Montgomery, S.; McConkie-Rosell, A.; Jiang, Y.-H. Epigenetics and Autism Spectrum Disorder: A Report of an Autism Case with Mutation in H1 Linker Histone HIST1H1E and Literature Review. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2018, 177, 426–433. [Google Scholar] [CrossRef]
- Hall, L.; Kelley, E. The Contribution of Epigenetics to Understanding Genetic Factors in Autism. Autism 2014, 18, 872–881. [Google Scholar] [CrossRef]
- Loke, Y.J.; Hannan, A.J.; Craig, J.M. The Role of Epigenetic Change in Autism Spectrum Disorders. Front. Neurology 2015, 6, 107. [Google Scholar] [CrossRef] [Green Version]
- Rylaarsdam, L.; Guemez-Gamboa, A. Genetic Causes and Modifiers of Autism Spectrum Disorder. Front. Cell. Neurosci. 2019, 13, 385. [Google Scholar] [CrossRef]
- Tran, S.S.; Jun, H.-I.; Bahn, J.H.; Azghadi, A.; Ramaswami, G.; Van Nostrand, E.L.; Nguyen, T.B.; Hsiao, Y.-H.E.; Lee, C.; Pratt, G.A.; et al. Widespread RNA Editing Dysregulation in Brains from Autistic Individuals. Nat. Neurosci. 2019, 22, 25–36. [Google Scholar] [CrossRef]
- Zhubi, A.; Cook, E.H.; Guidotti, A.; Grayson, D.R. Epigenetic Mechanisms in Autism Spectrum Disorder. Int. Rev. Neurobiol. 2014, 115, 203–244. [Google Scholar]
- Harris, S.W.; Hessl, D.; Goodlin-Jones, B.; Ferranti, J.; Bacalman, S.; Barbato, I.; Tassone, F.; Hagerman, P.J.; Herman, K.; Hagerman, R.J. Autism Profiles of Males with Fragile X Syndrome. Am. J. Ment. Retard. 2008, 113, 427–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, A.C.; Mussey, J.; Villagomez, A.; Bishop, E.; Raspa, M.; Edwards, A.; Bodfish, J.; Bann, C.; Bailey, D.B., Jr. DSM-5 Changes and the Prevalence of Parent-Reported Autism Spectrum Symptoms in Fragile X Syndrome. J. Autism Dev. Disord. 2015, 45, 816–829. [Google Scholar] [CrossRef] [PubMed]
- Verheij, C.; Bakker, C.E.; de Graaff, E.; Keulemans, J.; Willemsen, R.; Verkerk, A.J.M.; Galjaard, H.; Reuser, A.J.J.; Hoogeveen, A.T.; Oostra, B.A. Characterization and Localization of the FMR-1 Gene Product Associated with Fragile X Syndrome. Nature 1993, 363, 722–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doll, C.A.; Broadie, K. Neuron Class-Specific Requirements for Fragile X Mental Retardation Protein in Critical Period Development of Calcium Signaling in Learning and Memory Circuitry. Neurobiol. Dis. 2016, 89, 76–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, J.K.; Broadie, K. Multifarious Functions of the Fragile X Mental Retardation Protein. Trends Genet. 2017, 33, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; Ronemus, M.; Levy, D.; Wang, Z.; Hakker, I.; Rosenbaum, J.; Yamrom, B.; Lee, Y.-H.; Narzisi, G.; Leotta, A.; et al. De Novo Gene Disruptions in Children on the Autistic Spectrum. Neuron 2012, 74, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Parikshak, N.N.; Luo, R.; Zhang, A.; Won, H.; Lowe, J.K.; Chandran, V.; Horvath, S.; Geschwind, D.H. Integrative Functional Genomic Analyses Implicate Specific Molecular Pathways and Circuits in Autism. Cell 2013, 155, 1008–1021. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, J.; Webber, C. The Roles of FMRP-Regulated Genes in Autism Spectrum Disorder: Single- and Multiple-Hit Genetic Etiologies. Am. J. Hum. Genet. 2013, 93, 825–839. [Google Scholar] [CrossRef] [Green Version]
- Fatemi, S.H.; Folsom, T.D. Dysregulation of Fragile × Mental Retardation Protein and Metabotropic Glutamate Receptor 5 in Superior Frontal Cortex of Individuals with Autism: A Postmortem Brain Study. Mol. Autism 2011, 2, 6. [Google Scholar] [CrossRef] [Green Version]
- Budimirovic, D.B.; Schlageter, A.; Sadic-Filipovic, S.; Protic, D.; Bram, E.; Mark Mahone, E.; Nicholson, K.; Culp, K.; Javanmardi, K.; Hadd, A.; et al. A Genotype-Phenotype Study of High-Resolution FMR1 Nucleic Acid and Protein Analyses in Fragile X Patients with Neurobehavioral Assessments. Brain Sci. 2020, 10, 694. [Google Scholar] [CrossRef]
- Folsom, T.D.; Thuras, P.D.; Fatemi, S.H. Protein Expression of Targets of the FMRP Regulon Is Altered in Brains of Subjects with Schizophrenia and Mood Disorders. Schizophr. Res. 2015, 165, 201–211. [Google Scholar] [CrossRef] [Green Version]
- Castle, J.C.; Zhang, C.; Shah, J.K.; Kulkarni, A.V.; Kalsotra, A.; Cooper, T.A.; Johnson, J.M. Expression of 24,426 Human Alternative Splicing Events and Predicted Cis Regulation in 48 Tissues and Cell Lines. Nat. Genet. 2008, 40, 1416–1425. [Google Scholar] [CrossRef] [PubMed]
- Hu, V.W. The Expanding Genomic Landscape of Autism: Discovering the “forest” beyond the “trees. ” Future Neurol. 2013, 8, 29–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraan, C.M.; Godler, D.E.; Amor, D.J. Epigenetics of Fragile X Syndrome and Fragile X-Related Disorders. Dev. Med. Child Neurol. 2019, 61, 121–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotham, K.; Pickles, A.; Lord, C. Standardizing ADOS Scores for a Measure of Severity in Autism Spectrum Disorders. J. Autism Dev. Disord. 2009, 39, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Regier, D.A.; Narrow, W.E.; Kuhl, E.A.; Kupfer, D.J. The Conceptual Development of DSM-V. Am. J. Psychiatry 2009, 166, 645–650. [Google Scholar] [CrossRef] [Green Version]
- Tassone, F.; Pan, R.; Amiri, K.; Taylor, A.K.; Hagerman, P.J. A Rapid Polymerase Chain Reaction-Based Screening Method for Identification of All Expanded Alleles of the Fragile X (FMR1) Gene in Newborn and High-Risk Populations. J. Mol. Diagn. 2008, 10, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Tassone, F.; Hagerman, R.J.; Iklé, D.N.; Dyer, P.N.; Lampe, M.; Willemsen, R.; Oostra, B.A.; Taylor, A.K. FMRP Expression as a Potential Prognostic Indicator in Fragile X Syndrome. Am. J. Med. Genet. 1999, 84, 250–261. [Google Scholar] [CrossRef]
- Filipovic-Sadic, S.; Sah, S.; Chen, L.; Krosting, J.; Sekinger, E.; Zhang, W.; Hagerman, P.J.; Stenzel, T.T.; Hadd, A.G.; Latham, G.J.; et al. A Novel FMR1 PCR Method for the Routine Detection of Low Abundance Expanded Alleles and Full Mutations in Fragile X Syndrome. Clin. Chem. 2010, 56, 399–408. [Google Scholar] [CrossRef] [Green Version]
- Nordlund, J.; Bäcklin, C.L.; Wahlberg, P.; Busche, S.; Berglund, E.C.; Eloranta, M.-L.; Flaegstad, T.; Forestier, E.; Frost, B.-M.; Harila-Saari, A.; et al. Genome-Wide Signatures of Differential DNA Methylation in Pediatric Acute Lymphoblastic Leukemia. Genome Biol. 2013, 14, r105. [Google Scholar] [CrossRef] [Green Version]
- Hicks, S.C.; Irizarry, R.A. methylCC: Technology-Independent Estimation of Cell Type Composition Using Differentially Methylated Regions. Genome Biol. 2019, 20, 261. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Zhang, X.; Huang, C.-C.; Jafari, N.; Kibbe, W.A.; Hou, L.; Lin, S.M. Comparison of beta-Value and M-Value Methods for Quantifying Methylation Levels by Microarray Analysis. BMC Bioinform. 2010, 11, 587. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Kuan, P.F. methylGSA: A Bioconductor Package and Shiny App for DNA Methylation Data Length Bias Adjustment in Gene Set Testing. Bioinformatics 2019, 35, 1958–1959. [Google Scholar] [CrossRef]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A Flexible and Comprehensive Bioconductor Package for the Analysis of Infinium DNA Methylation Microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- Boncinelli, E. Homeobox Genes and Disease. Curr. Opin. Genet. Dev. 1997, 7, 331–337. [Google Scholar] [CrossRef]
- Joshi, R.L.; Torero Ibad, R.; Rheey, J.; Castagner, F.; Prochiantz, A.; Moya, K.L. Cell Non-Autonomous Functions of Homeoproteins in Neuroprotection in the Brain. FEBS Lett. 2011, 585, 1573–1578. [Google Scholar] [CrossRef] [Green Version]
- Eshraghi, A.A.; Liu, G.; Kay, S.-I.S.; Eshraghi, R.S.; Mittal, J.; Moshiree, B.; Mittal, R. Epigenetics and Autism Spectrum Disorder: Is There a Correlation? Front. Cell. Neurosci. 2018, 12, 78. [Google Scholar] [CrossRef] [Green Version]
- AlOlaby, R.R.; Zafarullah, M.; Barboza, M.; Peng, G.; Varian, B.J.; Erdman, S.E.; Lebrilla, C.; Tassone, F. Differential Methylation Profile in Fragile X Syndrome-Prone Offspring Mice after in Utero Exposure to Lactobacillus Reuteri. Genes 2022, 13, 1300. [Google Scholar] [CrossRef]
- Mikeska, T.; Craig, J. DNA Methylation Biomarkers: Cancer and Beyond. Genes 2014, 5, 821–864. [Google Scholar] [CrossRef] [Green Version]
- Bokoch, G.M. Biology of the p21-Activated Kinases. Annu. Rev. Biochem. 2003, 72, 743–781. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zeng, C.; Li, J.; Zhou, Z.; Ju, X.; Xia, S.; Li, Y.; Liu, A.; Teng, H.; Zhang, K.; et al. PAK2 Haploinsufficiency Results in Synaptic Cytoskeleton Impairment and Autism-Related Behavior. Cell Rep. 2018, 24, 2029–2041. [Google Scholar] [CrossRef]
- Dolan, B.M.; Duron, S.G.; Campbell, D.A.; Vollrath, B.; Shankaranarayana Rao, B.S.; Ko, H.-Y.; Lin, G.G.; Govindarajan, A.; Choi, S.-Y.; Tonegawa, S. Rescue of Fragile X Syndrome Phenotypes in Fmr1 KO Mice by the Small-Molecule PAK Inhibitor FRAX486. Proc. Natl. Acad. Sci. USA 2013, 110, 5671–5676. [Google Scholar] [CrossRef] [Green Version]
- Zhu, K.; Sun, S.; Guo, F.; Gao, L. Impaired Fanconi Anemia Pathway Causes DNA Hypomethylation in Human Angiosarcomas. Hum. Cell 2022, 35, 1602–1611. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Lindenberg, A.; Mervis, C.B.; Berman, K.F. Neural Mechanisms in Williams Syndrome: A Unique Window to Genetic Influences on Cognition and Behaviour. Nat. Rev. Neurosci. 2006, 7, 380–393. [Google Scholar] [CrossRef] [PubMed]
- Antonell, A.; Del Campo, M.; Magano, L.F.; Kaufmann, L.; de la Iglesia, J.M.; Gallastegui, F.; Flores, R.; Schweigmann, U.; Fauth, C.; Kotzot, D.; et al. Partial 7q11.23 Deletions Further Implicate GTF2I and GTF2IRD1 as the Main Genes Responsible for the Williams-Beuren Syndrome Neurocognitive Profile. J. Med. Genet. 2010, 47, 312–320. [Google Scholar] [CrossRef]
- Attree, E.A.; Turner, M.J.; Cowell, N. A Virtual Reality Test Identifies the Visuospatial Strengths of Adolescents with Dyslexia. Cyberpsychol. Behav. 2009, 12, 163–168. [Google Scholar] [CrossRef]
- Kim, D.W.; Cochran, B.H. JAK2 Activates TFII-I and Regulates Its Interaction with Extracellular Signal-Regulated Kinase. Mol. Cell. Biol. 2001, 21, 3387–3397. [Google Scholar] [CrossRef] [Green Version]
- Massinen, S.; Tammimies, K.; Tapia-Páez, I.; Matsson, H.; Hokkanen, M.-E.; Söderberg, O.; Landegren, U.; Castrén, E.; Gustafsson, J.-A.; Treuter, E.; et al. Functional Interaction of DYX1C1 with Estrogen Receptors Suggests Involvement of Hormonal Pathways in Dyslexia. Hum. Mol. Genet. 2009, 18, 2802–2812. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.; Sétáló, G., Jr.; Guan, X.; Warren, M.; Toran-Allerand, C.D. Estrogen-Induced Activation of Mitogen-Activated Protein Kinase in Cerebral Cortical Explants: Convergence of Estrogen and Neurotrophin Signaling Pathways. J. Neurosci. 1999, 19, 1179–1188. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Snape, M.; Klann, E.; Stone, J.G.; Singh, A.; Petersen, R.B.; Castellani, R.J.; Casadesus, G.; Smith, M.A.; Zhu, X. Activation of the Extracellular Signal-regulated Kinase Pathway Contributes to the Behavioral Deficit of Fragile X-syndrome. J. Neurochem. 2012, 121, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.; Tessier-Lavigne, M. Hierarchical Organization of Guidance Receptors: Silencing of Netrin Attraction by Slit through a Robo/DCC Receptor Complex. Science 2001, 291, 1928–1938. [Google Scholar] [CrossRef] [PubMed]
- Tanimura, S.; Takeda, K. ERK Signalling as a Regulator of Cell Motility. J. Biochem. 2017, 162, 145–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozano, R.; Gbekie, C.; Siper, P.M.; Srivastava, S.; Saland, J.M.; Sethuram, S.; Tang, L.; Drapeau, E.; Frank, Y.; Buxbaum, J.D.; et al. FOXP1 Syndrome: A Review of the Literature and Practice Parameters for Medical Assessment and Monitoring. J. Neurodev. Disord. 2021, 13, 18. [Google Scholar] [CrossRef]
- Chen, L.; Johnson, R.C.; Milgram, S.L. P-CIP1, a Novel Protein That Interacts with the Cytosolic Domain of Peptidylglycine Alpha-Amidating Monooxygenase, Is Associated with Endosomes. J. Biol. Chem. 1998, 273, 33524–33532. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Feng, C.; Zhang, K.; Chen, Y.; Gao, Y.; Ke, J.; Chen, X.; Lin, J.; Li, C.; Iqbal, J.; et al. Proteomics Study of Peripheral Blood Mononuclear Cells (PBMCs) in Autistic Children. Front. Cell. Neurosci. 2019, 13, 105. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Bhatti, D.L.; Lee, K.-W.; Medrihan, L.; Cheng, J.; Wei, J.; Zhong, P.; Yan, Z.; Kooiker, C.; Song, C.; et al. Ahnak Scaffolds p11/Anxa2 Complex and L-Type Voltage-Gated Calcium Channel and Modulates Depressive Behavior. Mol. Psychiatry 2020, 25, 1035–1049. [Google Scholar] [CrossRef] [Green Version]
- Lavado, A.; He, Y.; Paré, J.; Neale, G.; Olson, E.N.; Giovannini, M.; Cao, X. Tumor Suppressor Nf2 Limits Expansion of the Neural Progenitor Pool by Inhibiting Yap/Taz Transcriptional Coactivators. Development 2013, 140, 3323–3334. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.S.; Lee, Y.-S.; Narasimhan, P.; Chan, P.H. Reduced Oxidative Stress Promotes NF-κB-Mediated Neuroprotective Gene Expression after Transient Focal Cerebral Ischemia: Lymphocytotrophic Cytokines and Antiapoptotic Factors. J. Cereb. Blood Flow Metab. 2007, 27, 764–775. [Google Scholar] [CrossRef]
- Cox, A.G.; Tsomides, A.; Kim, A.J.; Saunders, D.; Hwang, K.L.; Evason, K.J.; Heidel, J.; Brown, K.K.; Yuan, M.; Lien, E.C.; et al. Selenoprotein H Is an Essential Regulator of Redox Homeostasis That Cooperates with p53 in Development and Tumorigenesis. Proc. Natl. Acad. Sci. USA 2016, 113, E5562–E5571. [Google Scholar] [CrossRef] [Green Version]
- Lucá, R.; Averna, M.; Zalfa, F.; Vecchi, M.; Bianchi, F.; La Fata, G.; Del Nonno, F.; Nardacci, R.; Bianchi, M.; Nuciforo, P.; et al. The Fragile X Protein Binds mRNAs Involved in Cancer Progression and Modulates Metastasis Formation. EMBO Mol. Med. 2013, 5, 1523–1536. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| TD | ASD | FXSA | |
|---|---|---|---|
| (n = 11) | (n = 23) | (n = 23) | |
| Age (Years) | |||
| Mean (SD) | 3.8 (1.3) | 4.1 (1.1) | 3.6 (1.1) |
| Median (Range) | 4 (2–6) | 4 (2–6) | 4 (2–6) |
| CGG Repeats | |||
| Mean (SD) | 28.5 (3.1) | 28.7 (3.4) | (all > 200) |
| Median (Range) | 30 (21–31) | 30 (21–33) | |
| Mutation Category | |||
| Full | 0 | 0 | 12 (52.2%) |
| Mosaic (Size/Methylation) | 0 | 0 | 11 (47.8%) |
| No Mutation | 11 (100%) | 23 (100%) | 0 |
| TD vs. ASD | TD vs. FXSA | ASD vs. FXSA | |
|---|---|---|---|
| FDR ≤ 0.05 | 156 | 79 | 3100 |
| FDR ≤ 0.05 & BetaAbsDiff ≥ 0.05 | 53 | 60 | 545 |
| FDR ≤ 0.05 & BetaAbsDiff ≥ 0.1 | 14 | 38 | 42 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jasoliya, M.; Gu, J.; AlOlaby, R.R.; Durbin-Johnson, B.; Chedin, F.; Tassone, F. Profiling Genome-Wide DNA Methylation in Children with Autism Spectrum Disorder and in Children with Fragile X Syndrome. Genes 2022, 13, 1795. https://doi.org/10.3390/genes13101795
Jasoliya M, Gu J, AlOlaby RR, Durbin-Johnson B, Chedin F, Tassone F. Profiling Genome-Wide DNA Methylation in Children with Autism Spectrum Disorder and in Children with Fragile X Syndrome. Genes. 2022; 13(10):1795. https://doi.org/10.3390/genes13101795
Chicago/Turabian StyleJasoliya, Mittal, Jianlei Gu, Reem R. AlOlaby, Blythe Durbin-Johnson, Frederic Chedin, and Flora Tassone. 2022. "Profiling Genome-Wide DNA Methylation in Children with Autism Spectrum Disorder and in Children with Fragile X Syndrome" Genes 13, no. 10: 1795. https://doi.org/10.3390/genes13101795