
Next-Generation Sequencing Identifies Novel PMPCA Variants in Patients with Late-Onset Dominant Optic Atrophy

 ,
,  , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Consent for Genetic Investigations
2.2. Genetic Analysis
2.3. Fibroblasts Study
2.4. Time Lapse and Deconvolution Microscopy
2.4.1. Immunofluorescence
2.4.2. STORM Acquisition
3. Results
3.1. Identification of PMPCA Variants in Individuals with Primary Dominant Optic Atrophy
3.2. Clinical Manifestations of PMPCA Patients
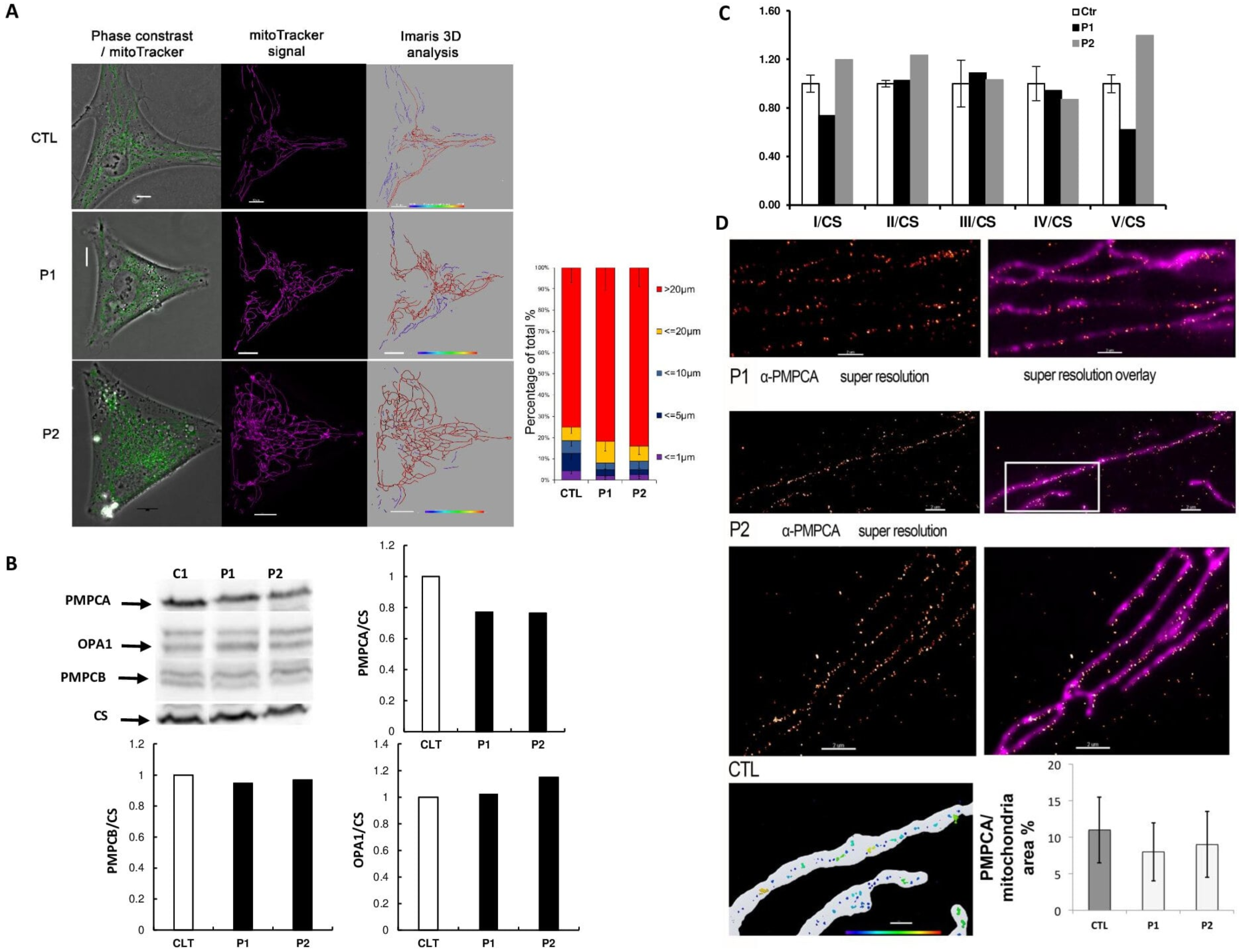
3.3. Functional Effects of PMPCA Variants
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Carelli, V.; Ross-Cisneros, F.N.; Sadun, A.A. Mitochondrial dysfunction as a cause of optic neuropathies. Prog. Retin. Eye Res. 2004, 23, 53–89. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Shankar, S.P.; Biousse, V.; Miller, N.R.; Bean, L.J.H.; Coffee, B.; Hegde, M.; Newman, N.J. Genetic screening for OPA1 and OPA3 mutations in patients with suspected inherited optic neuropathies. Ophthalmology 2011, 118, 558–563. [Google Scholar] [CrossRef] [Green Version]
- Lenaers, G.; Hamel, C.; Delettre, C.; Amati-Bonneau, P.; Procaccio, V.; Bonneau, D.; Reynier, P.; Milea, D. Dominant optic atrophy. Orphanet J. Rare Dis. 2012, 7, 46. [Google Scholar] [CrossRef] [Green Version]
- Newman, N.J.; Biousse, V. Hereditary optic neuropathies. Eye 2004, 18, 1144–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagli, E.; Zikou, A.K.; Agnantis, N.; Kitsos, G. Mitochondrial Membrane Dynamics and Inherited Optic Neuropathies. In Vivo 2017, 31, 511–525. [Google Scholar]
- Le Roux, B.; Lenaers, G.; Zanlonghi, X.; Amati-Bonneau, P.; Chabrun, F.; Foulonneau, T.; Caignard, A.; Leruez, S.; Gohier, P.; Procaccio, V.; et al. OPA1: 516 unique variants and 831 patients registered in an updated centralized Variome database. Orphanet J. Rare Dis. 2019, 14, 214. [Google Scholar] [CrossRef]
- Alexander, C.; Votruba, M.; Pesch, U.E.A.; Thiselton, D.L.; Mayer, S.; Rodriguez, M.; Kellner, U.; Leo-kottler, B.; Auburger, G.; Bhattacharya, S.S.; et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Delettre, C.; Lenaers, G.; Pelloquin, L.; Belenguer, P.; Hamel, C.P. OPA1 (Kjer type) dominant optic atrophy: A novel mitochondrial disease. Mol. Genet. Metab. 2002, 75, 97–107. [Google Scholar] [CrossRef]
- Jurkute, N.; Majander, A.; Bowman, R.; Votruba, M.; Abbs, S.; Acheson, J.; Lenaers, G.; Amati-Bonneau, P.; Moosajee, M.; Arno, G.; et al. Clinical utility gene card for: Inherited optic neuropathies including next-generation sequencing-based approaches. Eur. J. Hum. Genet. 2019, 27, 494–502. [Google Scholar] [CrossRef]
- Charif, M.; Chevrollier, A.; Gueguen, N.; Bris, C.; Goudenège, D.; Desquiret-Dumas, V.; Leruez, S.; Colin, E.; Meunier, A.; Vignal, C.; et al. Mutations in the m-AAA proteases AFG3L2 and SPG7 are causing isolated dominant optic atrophy. Neurol. Genet. 2020, 6, e428. [Google Scholar] [CrossRef]
- Charif, M.; Roubertie, A.; Salime, S.; Mamouni, S.; Goizet, C.; Hamel, C.P.; Lenaers, G. A novel mutation of AFG3L2 might cause dominant optic atrophy in patients with mild intellectual disability. Front. Genet. 2015, 6, 311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerber, S.; Charif, M.; Chevrollier, A.; Chaumette, T.; Angebault, C.; Kane, M.S.; Paris, A.; Alban, J.; Quiles, M.; Delettre, C.; et al. Mutations in DNM1L, as in OPA1, result indominant optic atrophy despite opposite effectson mitochondrial fusion and fission. Brain 2017, 140, 2586–2596. [Google Scholar] [CrossRef] [PubMed]
- Klebe, S.; Depienne, C.; Gerber, S.; Challe, G.; Anheim, M.; Charles, P.; Fedirko, E.; Lejeune, E.; Cottineau, J.; Brusco, A.; et al. Spastic paraplegia gene 7 in patients with spasticity and/or optic neuropathy. Brain 2012, 135, 2980–2993. [Google Scholar] [CrossRef]
- Piro-Mégy, C.; Sarzi, E.; Tarrés-Solé, A.; Péquignot, M.; Hensen, F.; Quilès, M.; Manes, G.; Chakraborty, A.; Sénéchal, A.; Bocquet, B.; et al. Dominant mutations in mtDNA maintenance gene SSBP1 cause optic atrophy and foveopathy. J. Clin. Investig. 2020, 130, 143–156. [Google Scholar] [CrossRef]
- Reynier, P.; Amati-Bonneau, P.; Verny, C.; Olichon, A.; Simard, G.; Guichet, A.; Bonnemains, C.; Malecaze, F.; Malinge, M.C.; Pelletier, J.B.; et al. OPA3 gene mutations responsible for autosomal dominant optic atrophy and cataract. J. Med. Genet. 2004, 41, e110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouzier, C.; Bannwarth, S.; Chaussenot, A.; Chevrollier, A.; Verschueren, A.; Bonello-Palot, N.; Fragaki, K.; Cano, A.; Pouget, J.; Pellissier, J.F.; et al. The MFN2 gene is responsible for mitochondrial DNA instability and optic atrophy “plus” phenotype. Brain 2012, 135, 23–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenaers, G.; Neutzner, A.; Le Dantec, Y.; Jüschke, C.; Xiao, T.; Decembrini, S.; Swirski, S.; Kieninger, S.; Agca, C.; Kim, U.S.; et al. Dominant optic atrophy: Culprit mitochondria in the optic nerve. Prog. Retin. Eye Res. 2021, 83, 100935. [Google Scholar] [CrossRef] [PubMed]
- Rendtorff, N.D.; Lodahl, M.; Boulahbel, H.; Johansen, I.R.; Pandya, A.; Welch, K.O.; Norris, V.W.; Arnos, K.S.; Bitner-Glindzicz, M.; Emery, S.B.; et al. Identification of p.A684V missense mutation in the WFS1 gene as a frequent cause of autosomal dominant optic atrophy and hearing impairment. Am. J. Med. Genet. Part A 2011, 155, 1298–1313. [Google Scholar] [CrossRef] [Green Version]
- Grenier, J.; Meunier, I.; Daien, V.; Baudoin, C.; Halloy, F.; Bocquet, B.; Blanchet, C.; Delettre, C.; Esmenjaud, E.; Roubertie, A.; et al. WFS1 in Optic Neuropathies: Mutation Findings in Nonsyndromic Optic Atrophy and Assessment of Clinical Severity. Ophthalmology 2016, 123, 1989–1998. [Google Scholar] [CrossRef]
- Gakh, O.; Cavadini, P.; Isaya, G. Mitochondrial processing peptidases. Biochim. Biophys. Acta—Mol. Cell Res. 2002, 1592, 63–77. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, P.F.; Glaser, E. Processing peptidases in mitochondria and chloroplasts. Biochim. Biophys. Acta—Mol. Cell Res. 2013, 1833, 360–370. [Google Scholar] [CrossRef] [Green Version]
- Choquet, K.; Zurita-Rendón, O.; La Piana, R.; Yang, S.; Dicaire, M.J.; Boycott, K.M.; Majewski, J.; Shoubridge, E.A.; Brais, B.; Tétreault, M. Autosomal recessive cerebellar ataxia caused by a homozygous mutation in PMPCA. Brain 2016, 139, e19. [Google Scholar] [CrossRef] [Green Version]
- Joshi, M.; Anselm, I.; Shi, J.; Bale, T.A.; Towne, M.; Schmitz-Abe, K.; Crowley, L.; Giani, F.C.; Kazerounian, S.; Markianos, K.; et al. Mutations in the substrate binding glycine-rich loop of the mitochondrial processing peptidase-α protein (PMPCA) cause a severe mitochondrial disease. Mol. Case Stud. 2016, 2, a000786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, Y.; Kubota, M.; Kosaki, R.; Kosaki, K.; Ishiguro, A. A severe form of autosomal recessive spinocerebellar ataxia associated with novel PMPCA variants. Brain Dev. 2020, 43, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Jobling, R.K.; Assoum, M.; Gakh, O.; Blaser, S.; Raiman, J.A.; Mignot, C.; Roze, E.; Dürr, A.; Brice, A.; Lévy, N.; et al. PMPCA mutations cause abnormal mitochondrial protein processing in patients with non-progressive cerebellar ataxia. Brain 2015, 138, 1505–1517. [Google Scholar] [CrossRef] [Green Version]
- Codron, P.; Cassereau, J.; Vourc’h, P.; Veyrat-Durebex, C.; Blasco, H.; Kane, S.; Procaccio, V.; Letournel, F.; Verny, C.; Lenaers, G.; et al. Primary fibroblasts derived from sporadic amyotrophic lateral sclerosis patients do not show ALS cytological lesions. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 446–456. [Google Scholar] [CrossRef]
- Kechkar, A.; Nair, D.; Heilemann, M.; Choquet, D.; Sibarita, J.B. Real-Time Analysis and Visualization for Single-Molecule Based Super-Resolution Microscopy. PLoS ONE 2013, 8, e62918. [Google Scholar] [CrossRef]
- Amati-Bonneau, P.; Valentino, M.L.; Reynier, P.; Gallardo, M.E.; Bornstein, B.; Boissière, A.; Campos, Y.; Rivera, H.; De La Aleja, J.G.; Carroccia, R.; et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy “plus” phenotypes. Brain 2008, 131, 338–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amati-bonneau, P.; Odent, S.; Derrien, C.; Pasquier, L.; Malthiéry, Y.; Reynier, P.; Bonneau, D. The association of autosomal dominant optic atrophy and moderate deafness may be due to the R445H mutation in the OPA1 gene. Am. J. Ophthalmol. 2003, 136, 1170–1171. [Google Scholar] [CrossRef]
- Hudson, G.; Amati-bonneau, P.; Blakely, E.L.; Stewart, J.D.; He, L.; Schaefer, A.M.; Griffiths, P.G.; Ahlqvist, K.; Suomalainen, A.; Reynier, P.; et al. Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: A novel disorder of mtDNA maintenance. Brain 2008, 131, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Votruba, M.; Burté, F.; La Morgia, C.; Barboni, P.; Carelli, V. A neurodegenerative perspective on mitochondrial optic neuropathies. Acta Neuropathol. 2016, 132, 789–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priglinger, C.; Klopstock, T.; Rudolph, G.; Priglinger, S.G. Leber’s Hereditary Optic Neuropathy. Klin. Monbl. Augenheilkd. 2019, 236, 1271–1282. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
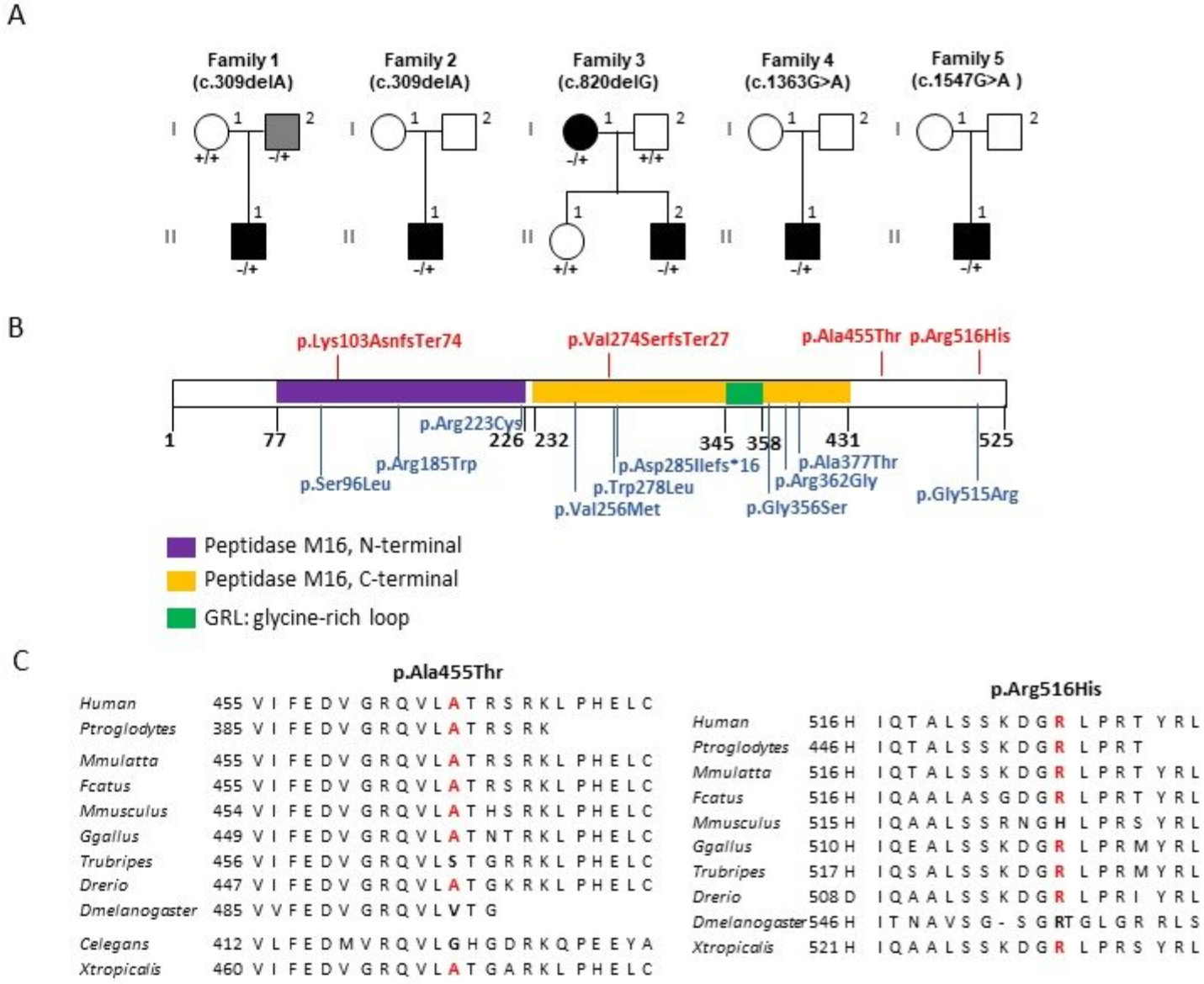
| Family | Patient | Sex | Age | VA | Other Symptoms | Brain MRI | ORF Change | Protein Change | rs # | Gnomad Freq. | ACMG Classification |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | (II.1) | M | 30 | counting fingers | - | normal | c.309delA | p.(Lys103AsnfsTer74) | unknown | - | PVS1 and PM2 Class 5 |
| 2 | (II.1) | M | 53 | peripheral neuropathy | normal | ||||||
| 3 | (II.2) | M | 45 | 4/10 | - | normal | c.820delG | p.(Val274SerfsTer27) | rs777445198 | 4.01 × 10−6 | PVS1 and PM2 Class 5 |
| 4 | (II.1) | M | 35 | 0.5/10 | multiple sclerosis | ND | c.1363G>A | p.(Ala455Thr) | unknown | - | PM2 and BP4 Class 3 |
| 5 | (II.1) | M | 69 | 6/10 | - | ND | c.1547G>A | p.(Arg516His) | rs768196711 | 8.01 × 10−6 | PM2 Class 3 |
| Variant | Polyphen | SIFT | MutationTaster | FATHMM-MKL | LRT | PROVEAN | DANN |
|---|---|---|---|---|---|---|---|
| c.1363G>A (p.Ala455Thr) | 0.568 possibly damaging | 0.492 tolerated | 0.9999 disease-causing | 0.9334 damaging | 0 deleterious | −2.46, −2.42, −2.78 damaging | 0.9956 damaging |
| c.1547G>A (p.Arg516His) | 0.017 benign | 0.025 damaging | 0.9999 disease-causing | 0.9669 damaging | 9.9999 × 10−7 neutral | −2.83, −2.53 damaging | 0.9982 damaging |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Charif, M.; Chevrollier, A.; Gueguen, N.; Kane, S.; Bris, C.; Goudenège, D.; Desquiret-Dumas, V.; Meunier, I.; Mochel, F.; Jeanjean, L.; et al. Next-Generation Sequencing Identifies Novel PMPCA Variants in Patients with Late-Onset Dominant Optic Atrophy. Genes 2022, 13, 1202. https://doi.org/10.3390/genes13071202
Charif M, Chevrollier A, Gueguen N, Kane S, Bris C, Goudenège D, Desquiret-Dumas V, Meunier I, Mochel F, Jeanjean L, et al. Next-Generation Sequencing Identifies Novel PMPCA Variants in Patients with Late-Onset Dominant Optic Atrophy. Genes. 2022; 13(7):1202. https://doi.org/10.3390/genes13071202
Chicago/Turabian StyleCharif, Majida, Arnaud Chevrollier, Naïg Gueguen, Selma Kane, Céline Bris, David Goudenège, Valerie Desquiret-Dumas, Isabelle Meunier, Fanny Mochel, Luc Jeanjean, and et al. 2022. "Next-Generation Sequencing Identifies Novel PMPCA Variants in Patients with Late-Onset Dominant Optic Atrophy" Genes 13, no. 7: 1202. https://doi.org/10.3390/genes13071202