
Predominant Founder Effect among Recurrent Pathogenic Variants for an X-Linked Disorder

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Consent, Sample Collection, and Testing
2.2. Variant Classification
2.3. Minigene Assay
2.4. Haplotype Analysis
3. Results
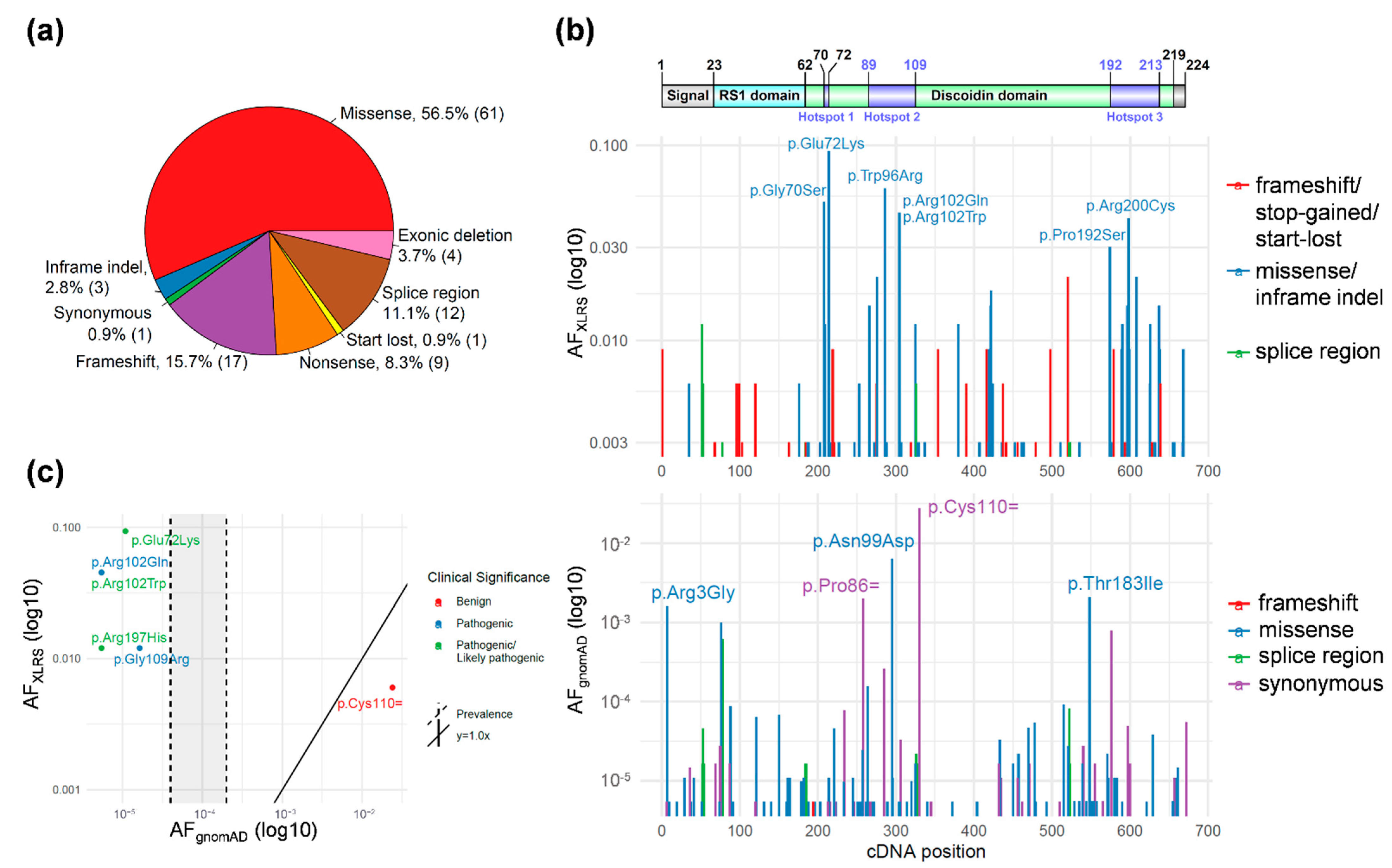
3.1. RS1 Gene-Level Variant Characterization
3.2. RS1 Population-Level Variant Analysis
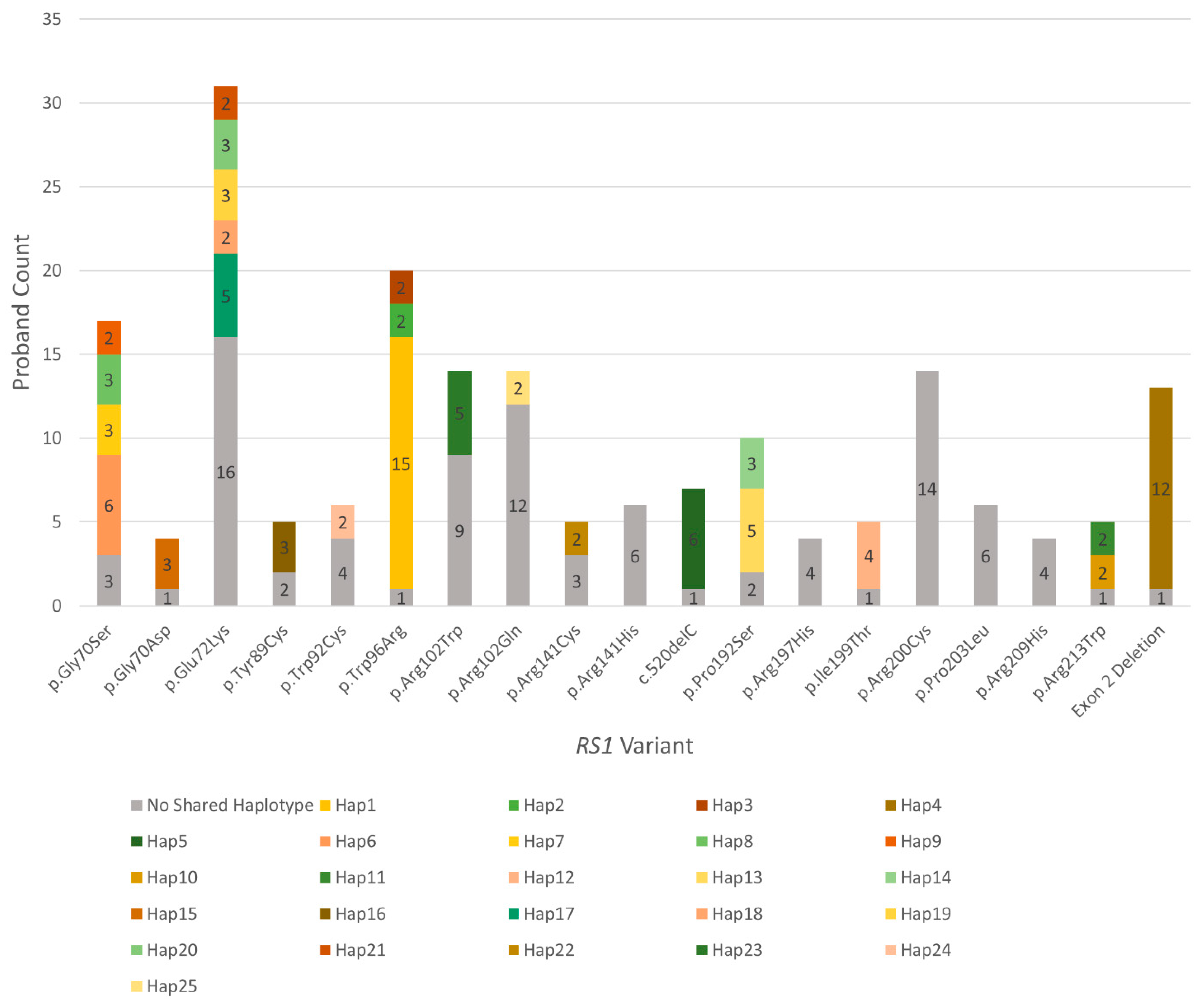
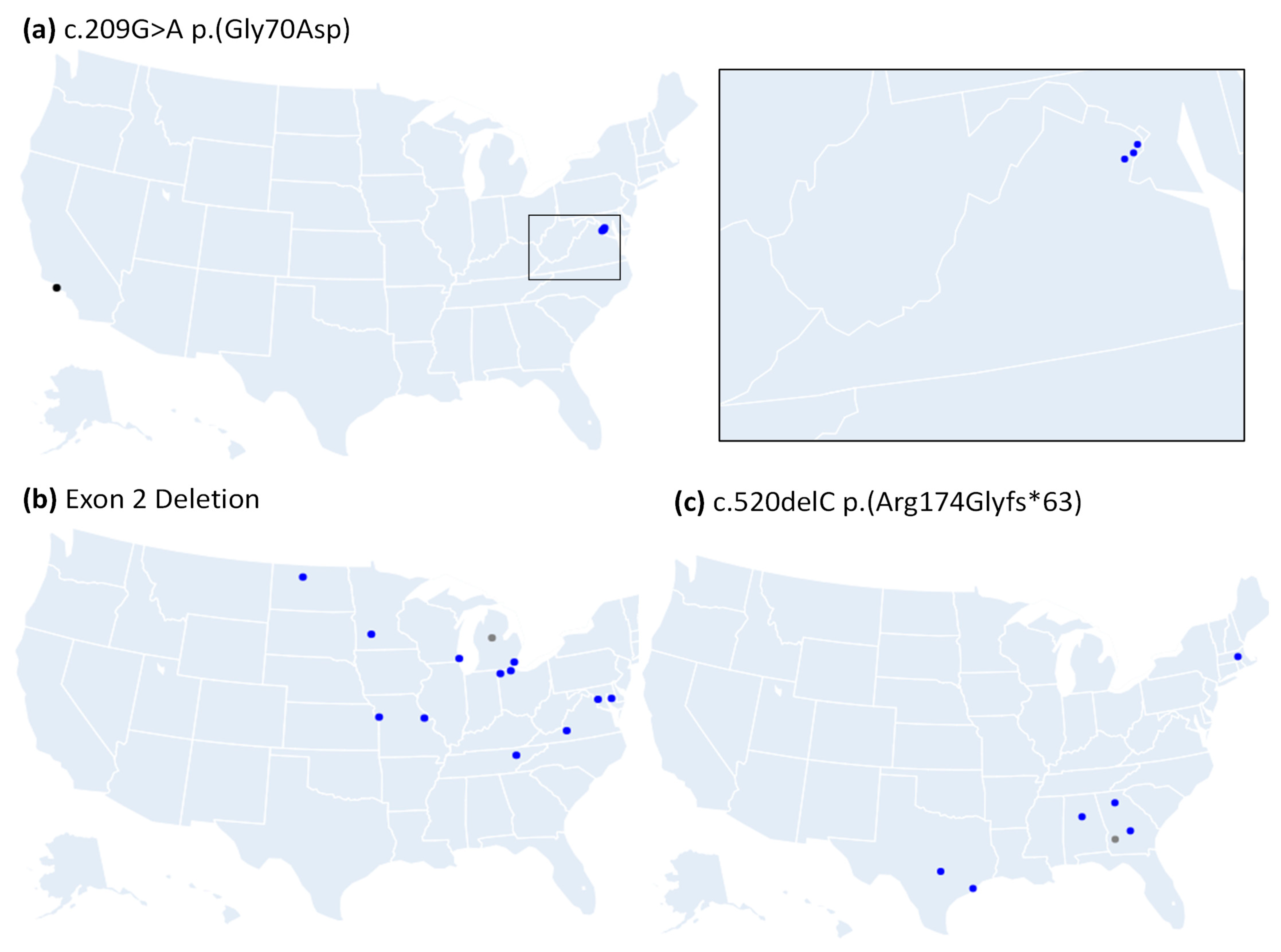
3.3. Many Recurrently Observed RS1 Variants Share Haplotypes
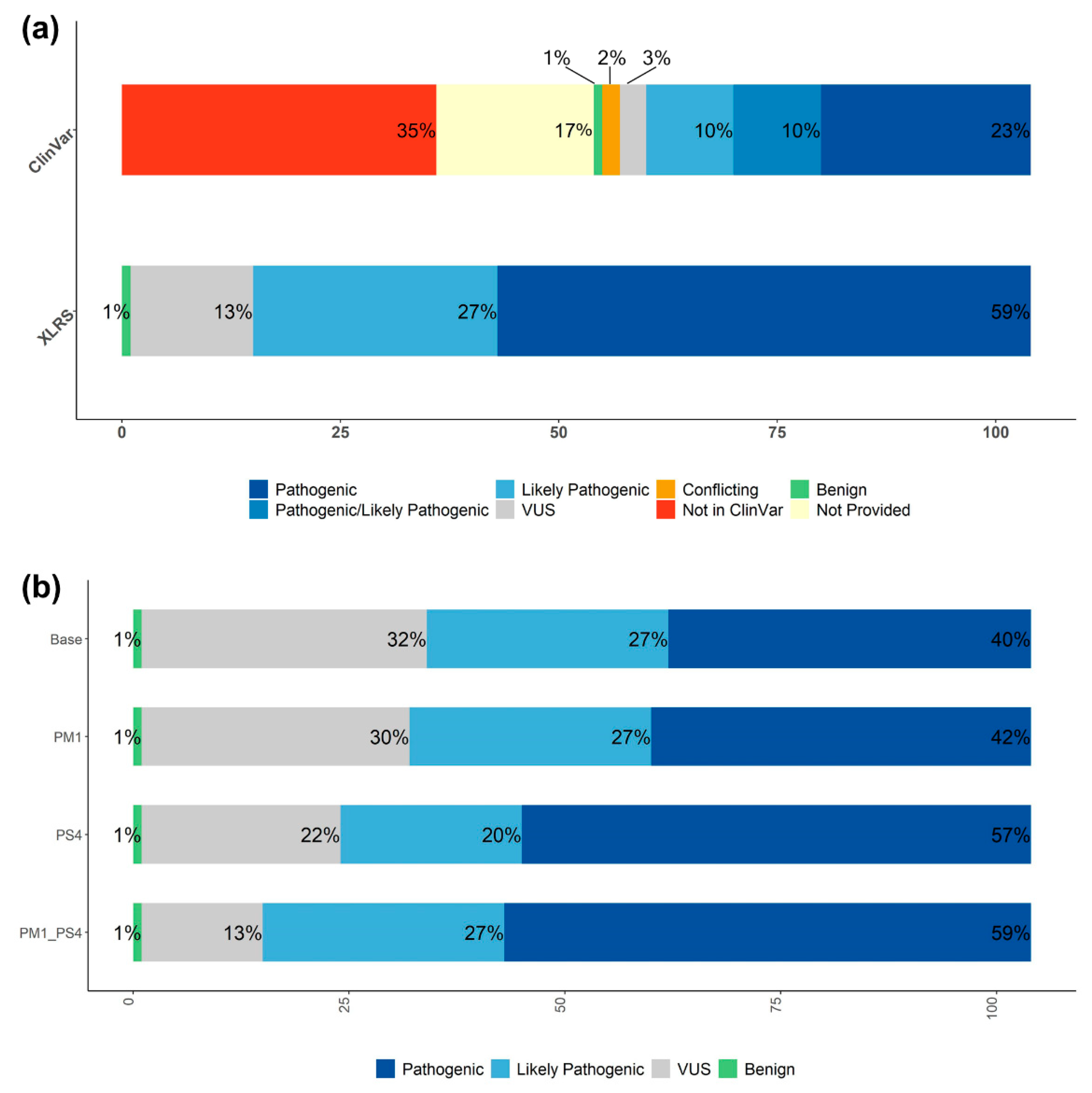
3.4. Systematic RS1 Variant Classification and Interpretation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Migeon, B.R. X-linked diseases: Susceptible females. Genet. Med. 2020, 22, 1156–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migeon, B.R. The role of X inactivation and cellular mosaicism in women’s health and sex-specific diseases. JAMA 2006, 295, 1428–1433. [Google Scholar] [CrossRef] [PubMed]
- Shinagawa, J.; Moteki, H.; Nishio, S.Y.; Noguchi, Y.; Usami, S.I. Haplotype Analysis of GJB2 Mutations: Founder Effect or Mutational Hot Spot? Genes 2020, 11, 250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeves, M.J.; Goetz, K.E.; Guan, B.; Ullah, E.; Blain, D.; Zein, W.M.; Tumminia, S.J.; Hufnagel, R.B. Genotype-phenotype associations in a large PRPH2-related retinopathy cohort. Hum. Mutat. 2020, 41, 1528–1539. [Google Scholar] [CrossRef]
- Tsang, S.H.; Sharma, T. X-linked Juvenile Retinoschisis. Adv. Exp. Med. Biol. 2018, 1085, 43–48. [Google Scholar] [CrossRef]
- Sergeev, Y.V.; Vitale, S.; Sieving, P.A.; Vincent, A.; Robson, A.G.; Moore, A.T.; Webster, A.R.; Holder, G.E. Molecular modeling indicates distinct classes of missense variants with mild and severe XLRS phenotypes. Hum. Mol. Genet. 2013, 22, 4756–4767. [Google Scholar] [CrossRef] [Green Version]
- Bush, R.A.; Wei, L.L.; Sieving, P.A. Convergence of Human Genetics and Animal Studies: Gene Therapy for X-Linked Retinoschisis. Cold Spring Harb. Perspect. Med. 2015, 5, a017368. [Google Scholar] [CrossRef] [Green Version]
- The Retinoschisis Consortium. Functional implications of the spectrum of mutations found in 234 cases with X-linked juvenile retinoschisis. Hum. Mol. Genet. 1998, 7, 1185–1192. [Google Scholar] [CrossRef] [Green Version]
- Cukras, C.; Wiley, H.E.; Jeffrey, B.G.; Sen, H.N.; Turriff, A.; Zeng, Y.; Vijayasarathy, C.; Marangoni, D.; Ziccardi, L.; Kjellstrom, S.; et al. Retinal AAV8-RS1 Gene Therapy for X-Linked Retinoschisis: Initial Findings from a Phase I/IIa Trial by Intravitreal Delivery. Mol. Ther. 2018, 26, 2282–2294. [Google Scholar] [CrossRef] [Green Version]
- Sieving, P.A.; Collins, F.S. Genetic ophthalmology and the era of clinical care. JAMA 2007, 297, 733–736. [Google Scholar] [CrossRef]
- Brooks, B.P.; Macdonald, I.M.; Tumminia, S.J.; Smaoui, N.; Blain, D.; Nezhuvingal, A.A.; Sieving, P.A. Genomics in the era of molecular ophthalmology: Reflections on the National Ophthalmic Disease Genotyping Network (eyeGENE). Arch. Ophthalmol. 2008, 126, 424–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blain, D.; Goetz, K.E.; Ayyagari, R.; Tumminia, S.J. eyeGENE®: A vision community resource facilitating patient care and paving the path for research through molecular diagnostic testing. Clin. Genet. 2013, 84, 190–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetz, K.E.; Reeves, M.J.; Tumminia, S.J.; Brooks, B.P. eyeGENE®: A novel approach to combine clinical testing and researching genetic ocular disease. Curr. Opin. Ophthalmol. 2012, 23, 355–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Rodelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shiel, J.A.; Thomas, N.S.; Abeysinghe, S.; Krawczak, M.; Cooper, D.N. Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat. 2003, 21, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarvik, G.P.; Browning, B.L. Consideration of Cosegregation in the Pathogenicity Classification of Genomic Variants. Am. J. Hum. Genet. 2016, 98, 1077–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, G.; Cooper, T.A. Minigene reporter for identification and analysis of cis elements and trans factors affecting pre-mRNA splicing. Biotechniques 2006, 41, 177–181. [Google Scholar] [CrossRef] [Green Version]
- Guan, B.; Welch, J.M.; Sapp, J.C.; Ling, H.; Li, Y.; Johnston, J.J.; Kebebew, E.; Biesecker, L.G.; Simonds, W.F.; Marx, S.J.; et al. GCM2-Activating Mutations in Familial Isolated Hyperparathyroidism. Am. J. Hum. Genet. 2016, 99, 1034–1044. [Google Scholar] [CrossRef] [Green Version]
- Matise, T.C.; Chen, F.; Chen, W.; De La Vega, F.M.; Hansen, M.; He, C.; Hyland, F.C.; Kennedy, G.C.; Kong, X.; Murray, S.S.; et al. A second-generation combined linkage physical map of the human genome. Genome Res. 2007, 17, 1783–1786. [Google Scholar] [CrossRef] [Green Version]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Schuelke, M. An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 2000, 18, 233–234. [Google Scholar] [CrossRef]
- Tolun, G.; Vijayasarathy, C.; Huang, R.; Zeng, Y.; Li, Y.; Steven, A.C.; Sieving, P.A.; Heymann, J.B. Paired octamer rings of retinoschisin suggest a junctional model for cell-cell adhesion in the retina. Proc. Natl. Acad. Sci. USA 2016, 113, 5287–5292. [Google Scholar] [CrossRef] [Green Version]
- Signal, B.; Gloss, B.S.; Dinger, M.E.; Mercer, T.R. Machine learning annotation of human branchpoints. Bioinformatics 2018, 34, 920–927. [Google Scholar] [CrossRef]
- Taggart, A.J.; DeSimone, A.M.; Shih, J.S.; Filloux, M.E.; Fairbrother, W.G. Large-scale mapping of branchpoints in human pre-mRNA transcripts in vivo. Nat. Struct. Mol. Biol. 2012, 19, 719–721. [Google Scholar] [CrossRef] [PubMed]
- Ferla, R.; Calo, V.; Cascio, S.; Rinaldi, G.; Badalamenti, G.; Carreca, I.; Surmacz, E.; Colucci, G.; Bazan, V.; Russo, A. Founder mutations in BRCA1 and BRCA2 genes. Ann. Oncol. 2007, 18 (Suppl. S6), vi93–vi98. [Google Scholar] [CrossRef] [PubMed]
- Huopaniemi, L.; Rantala, A.; Forsius, H.; Somer, M.; de la Chapelle, A.; Alitalo, T. Three widespread founder mutations contribute to high incidence of X-linked juvenile retinoschisis in Finland. Eur. J. Hum. Genet. 1999, 7, 368–376. [Google Scholar] [CrossRef] [Green Version]
- Riveiro-Alvarez, R.; Trujillo-Tiebas, M.J.; Gimenez-Pardo, A.; Garcia-Hoyos, M.; Lopez-Martinez, M.A.; Aguirre-Lamban, J.; Garcia-Sandoval, B.; Vazquez-Fernandez del Pozo, S.; Cantalapiedra, D.; Avila-Fernandez, A.; et al. Correlation of genetic and clinical findings in Spanish patients with X-linked juvenile retinoschisis. Investig. Ophthalmol. Vis. Sci. 2009, 50, 4342–4350. [Google Scholar] [CrossRef]
- Wang, T.; Waters, C.T.; Rothman, A.M.; Jakins, T.J.; Romisch, K.; Trump, D. Intracellular retention of mutant retinoschisin is the pathological mechanism underlying X-linked retinoschisis. Hum. Mol. Genet. 2002, 11, 3097–3105. [Google Scholar] [CrossRef] [PubMed]
- Bush, M.; Setiaputra, D.; Yip, C.K.; Molday, R.S. Cog-Wheel Octameric Structure of RS1, the Discoidin Domain Containing Retinal Protein Associated with X-Linked Retinoschisis. PLoS ONE 2016, 11, e0147653. [Google Scholar] [CrossRef] [Green Version]
- Forsius, H.; Krause, U.; Helve, J.; Vuopala, V.; Mustonen, E.; Vainio-Mattila, B.; Fellman, J.; Eriksson, A.W. Visual acuity in 183 cases of X-chromosomal retinoschisis. Can. J. Ophthalmol. 1973, 8, 385–393. [Google Scholar]
- Miano, M.G.; Testa, F.; Filippini, F.; Trujillo, M.; Conte, I.; Lanzara, C.; Millan, J.M.; De Bernardo, C.; Grammatico, B.; Mangino, M.; et al. Identification of novel RP2 mutations in a subset of X-linked retinitis pigmentosa families and prediction of new domains. Hum. Mutat. 2001, 18, 109–119. [Google Scholar] [CrossRef]
- D’Avanzo, F.; Rigon, L.; Zanetti, A.; Tomanin, R. Mucopolysaccharidosis Type II: One Hundred Years of Research, Diagnosis, and Treatment. Int. J. Mol. Sci. 2020, 21, 1258. [Google Scholar] [CrossRef] [Green Version]
- Flanigan, K.M.; Dunn, D.M.; von Niederhausern, A.; Soltanzadeh, P.; Gappmaier, E.; Howard, M.T.; Sampson, J.B.; Mendell, J.R.; Wall, C.; King, W.M.; et al. Mutational spectrum of DMD mutations in dystrophinopathy patients: Application of modern diagnostic techniques to a large cohort. Hum. Mutat. 2009, 30, 1657–1666. [Google Scholar] [CrossRef] [Green Version]
- Kong, X.; Zhong, X.; Liu, L.; Cui, S.; Yang, Y.; Kong, L. Genetic analysis of 1051 Chinese families with Duchenne/Becker Muscular Dystrophy. BMC Med. Genet. 2019, 20, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leroy, B.P.; Budde, B.S.; Wittmer, M.; De Baere, E.; Berger, W.; Zeitz, C. A common NYX mutation in Flemish patients with X linked CSNB. Br. J. Ophthalmol. 2009, 93, 692–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azevedo, O.; Gal, A.; Faria, R.; Gaspar, P.; Miltenberger-Miltenyi, G.; Gago, M.F.; Dias, F.; Martins, A.; Rodrigues, J.; Reimao, P.; et al. Founder effect of Fabry disease due to p.F113L mutation: Clinical profile of a late-onset phenotype. Mol. Genet. Metab. 2020, 129, 150–160. [Google Scholar] [CrossRef]
- Huang, L.; Sun, L.; Wang, Z.; Chen, C.; Wang, P.; Sun, W.; Luo, X.; Ding, X. Clinical manifestation and genetic analysis in Chinese early onset X-linked retinoschisis. Mol. Genet. Genom. Med. 2020, 8, e1421. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bender, C.; Woo, E.G.; Guan, B.; Ullah, E.; Feng, E.; Turriff, A.; Tumminia, S.J.; Sieving, P.A.; Cukras, C.A.; Hufnagel, R.B. Predominant Founder Effect among Recurrent Pathogenic Variants for an X-Linked Disorder. Genes 2022, 13, 675. https://doi.org/10.3390/genes13040675
Bender C, Woo EG, Guan B, Ullah E, Feng E, Turriff A, Tumminia SJ, Sieving PA, Cukras CA, Hufnagel RB. Predominant Founder Effect among Recurrent Pathogenic Variants for an X-Linked Disorder. Genes. 2022; 13(4):675. https://doi.org/10.3390/genes13040675
Chicago/Turabian StyleBender, Chelsea, Elizabeth Geena Woo, Bin Guan, Ehsan Ullah, Eric Feng, Amy Turriff, Santa J. Tumminia, Paul A. Sieving, Catherine A. Cukras, and Robert B. Hufnagel. 2022. "Predominant Founder Effect among Recurrent Pathogenic Variants for an X-Linked Disorder" Genes 13, no. 4: 675. https://doi.org/10.3390/genes13040675