
Decoding the Gene Variants of Two Native Probiotic Lactiplantibacillus plantarum Strains through Whole-Genome Resequencing: Insights into Bacterial Adaptability to Stressors and Antimicrobial Strength


Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains
2.2. Whole-Genome Sequencing, Gene Prediction, and Functional Annotation
2.3. Genome and Pan-Genome Comparison Analysis
2.4. Identification of Genomic Islands (GIs) and Insertion Sequences (ISs) within the Genomes of Native Strains
2.5. SNPs and Indel Discovery, Transition and Transversion Information, and Variant Annotation
2.6. Detection of Biosynthetic Gene Clusters (BGCs) and Genes Involved in the Adaptability to Several Stressors
3. Results and Discussion
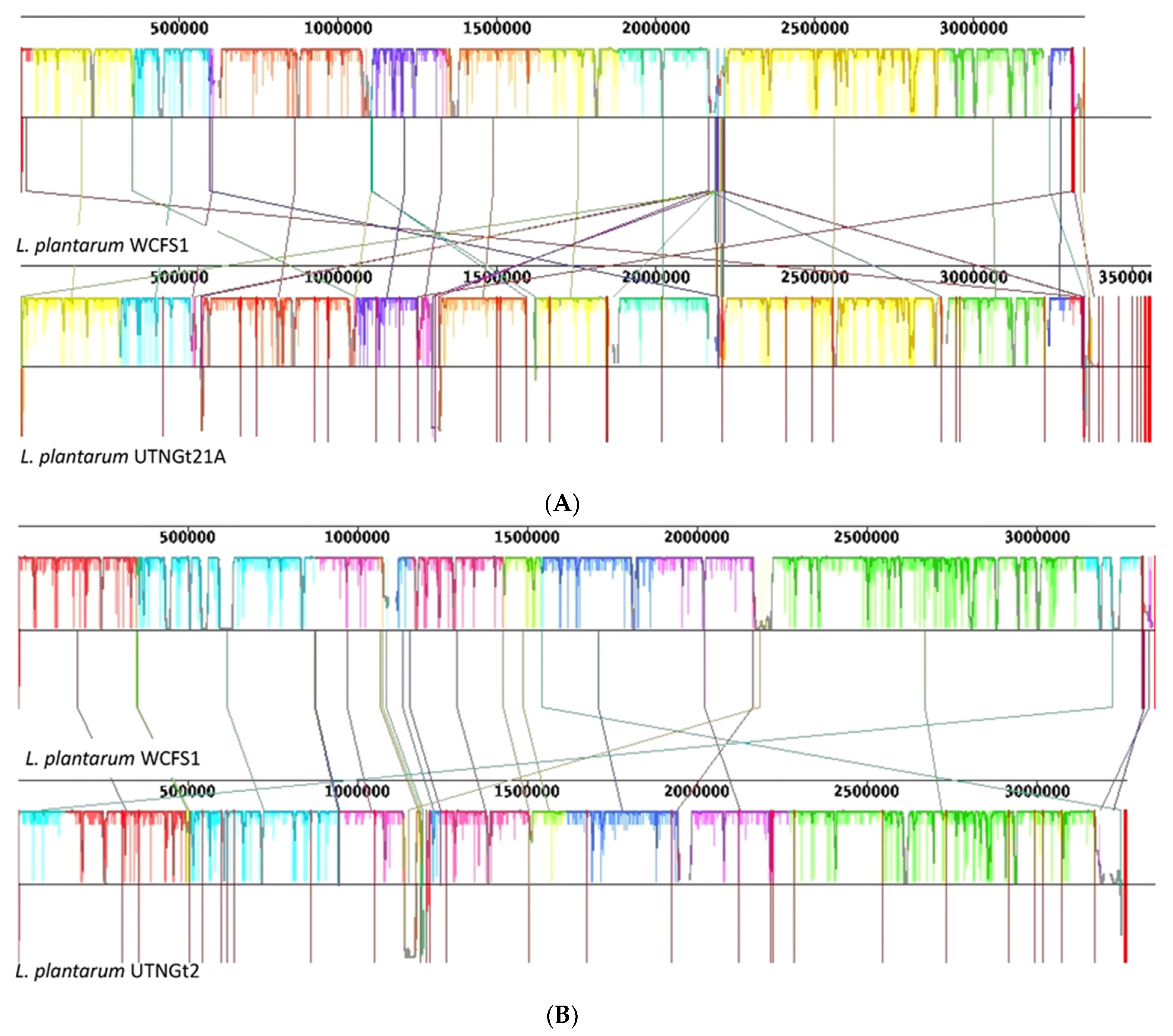
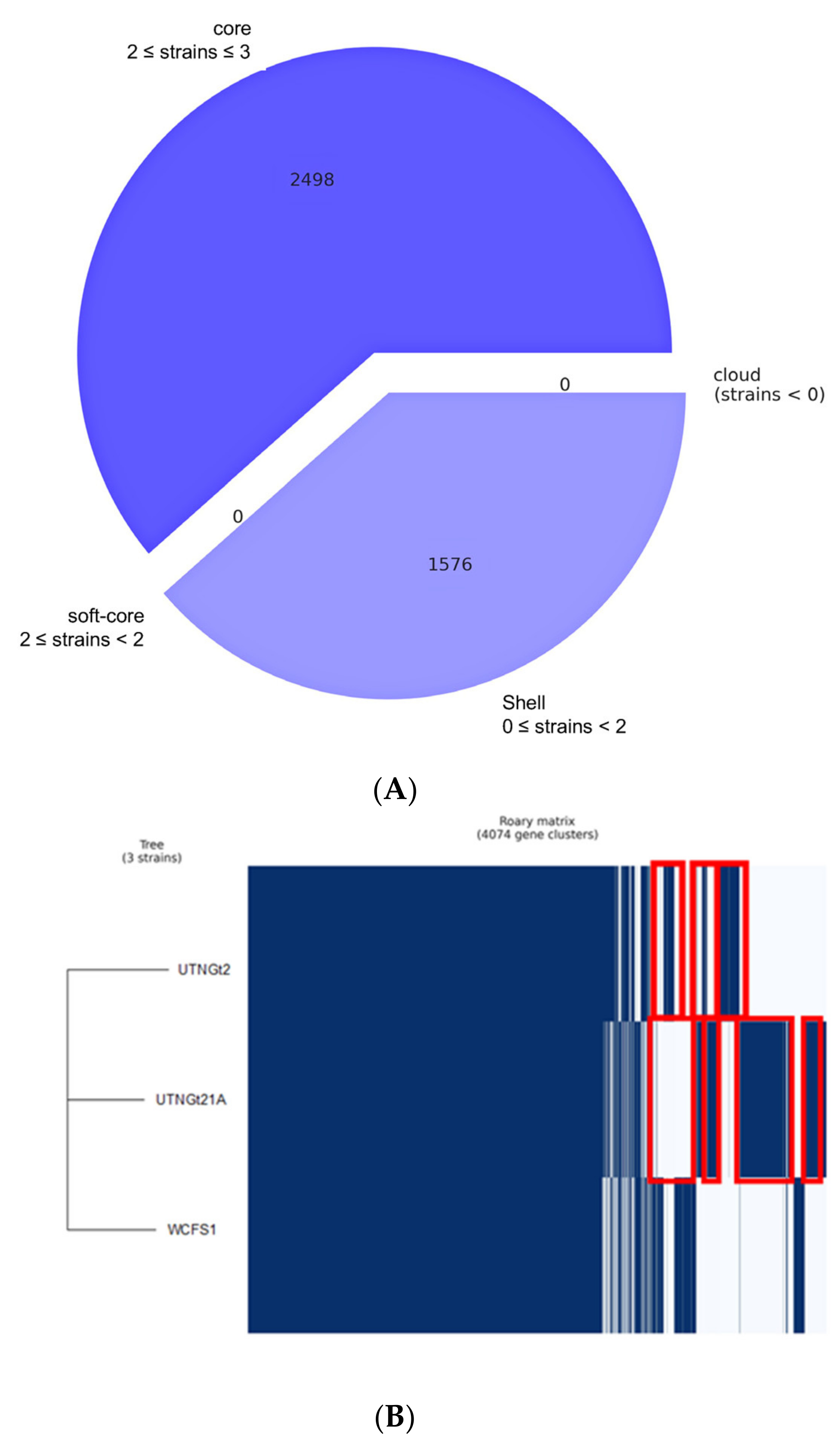
3.1. Comparative Genome and Pan-Genome Analysis Reveals the High Genetic and Niche-Specific Variation of Native Strains
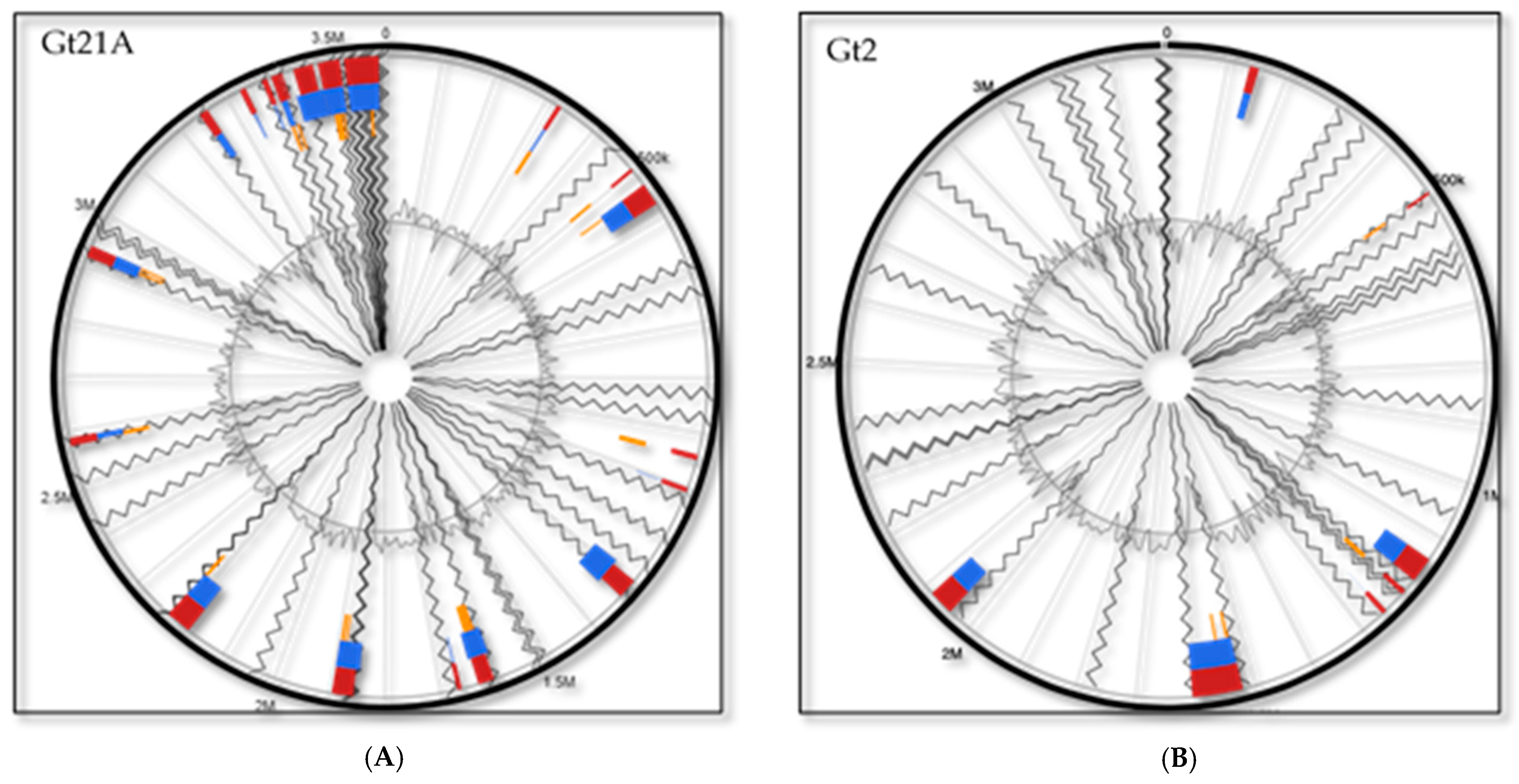
3.2. Differences between the GIs and ISs Might Explain the Strains’ Adaptability to Different Niches
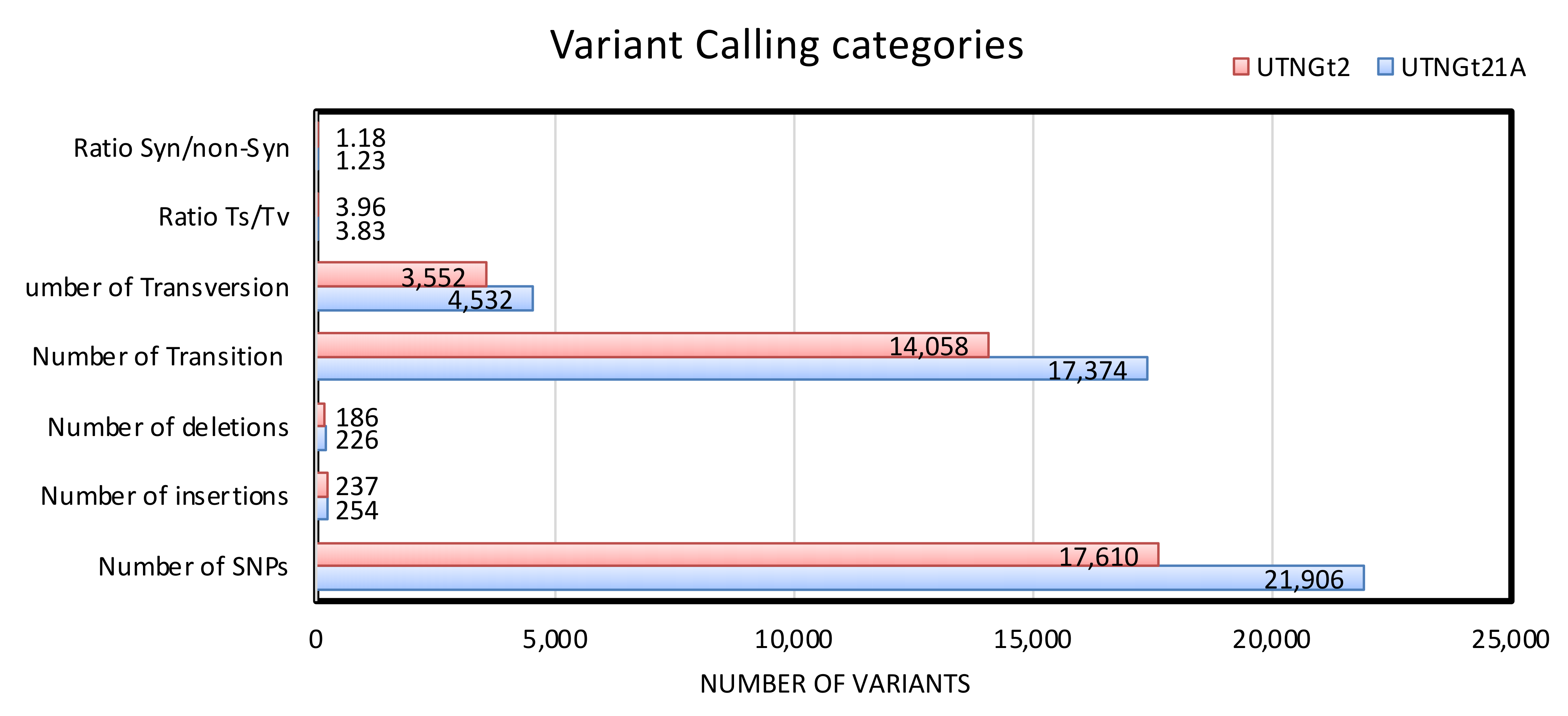
3.3. Detection of Variants (Insertions, Deletions, SNPs) and Their Impact on the Genomic Architecture
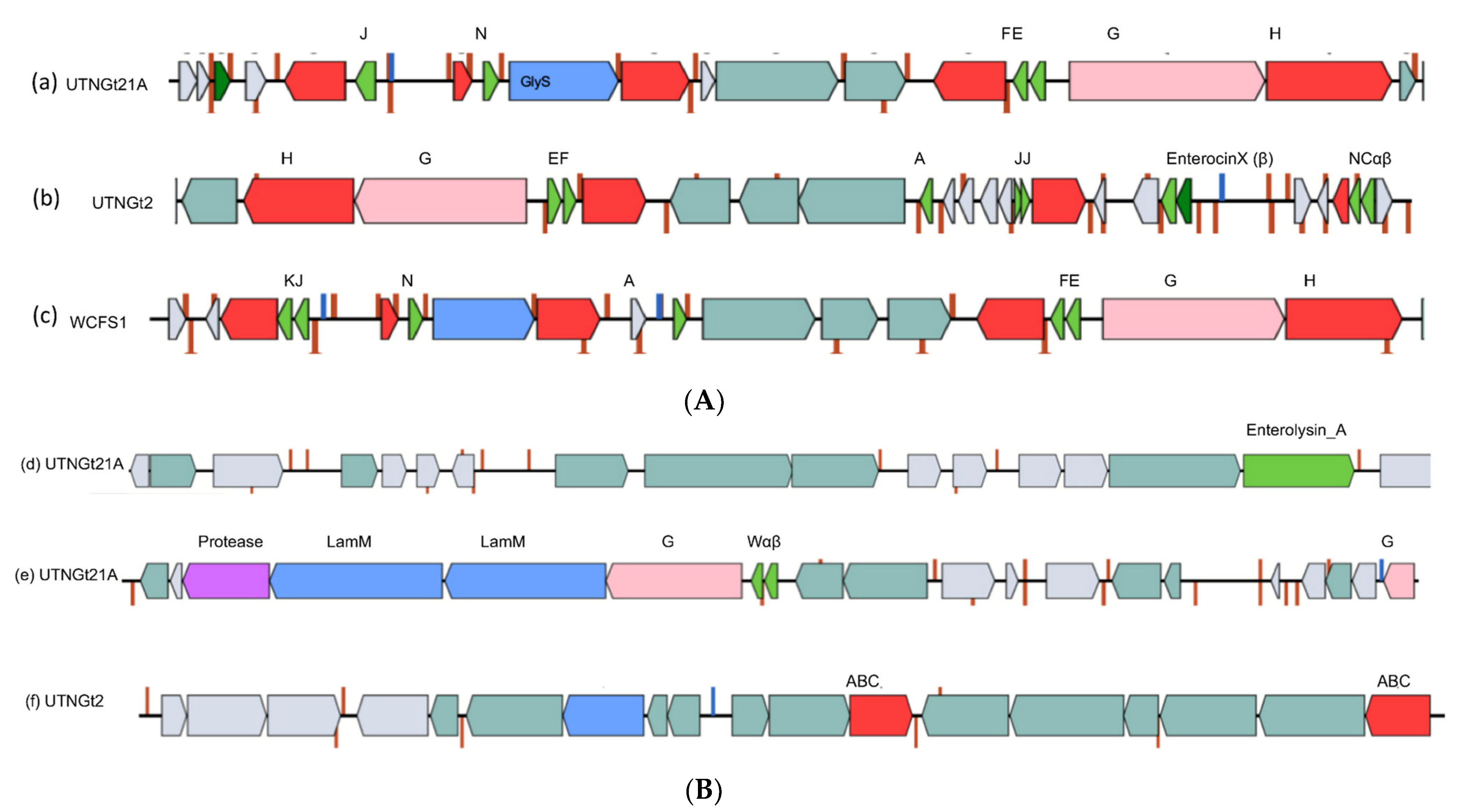
3.4. BGC Orgnization and Detection of Gene Variants Might Explain the Inhibitory Strength of the Native Strains
3.5. Gene Variants Might Play Important Roles in the Strains’ Adaptability to Different Stressors and Overall Probiotic Performance
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Sebastianes, F.L.S.; de Azevedo, J.L.; Souza-Motta, C.M. Diversity and biotechnological potential of endophytic microorganisms associated with tropical mangrove forests. In Diversity and Benefits of Microorganisms from the Tropics; de Azevedo, J.L., Quecine, M.C., Eds.; Springer: Berlin/Heidelberg, Germany, 2017; pp. 37–57. [Google Scholar] [CrossRef]
- Abe Sato, S.T.; Marques, J.M.; da Luz de Freitas, A.; Sanches Progênio, R.C.; Nunes, M.R.T.; Mota de Vasconcelos Massafra, J.; Gomes Moura, F.; Rogez, H. Isolation and genetic identification of endophytic lactic acid bacteria from the Amazonian açai fruits: Probiotics features of selected strains and their potential to inhibit pathogens. Front. Microbiol. 2021, 11, 610524. [Google Scholar] [CrossRef] [PubMed]
- Jabbar, Z.; Mukhtar, H.; Tayyeb, A.; Manzoor, A. Next-generation sequencing to elucidate adaptive stress response and plantaricin genes among Lactobacillus plantarum strains. Future Microbiol. 2020, 15, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Hernández-González, J.C.; Martínez-Tapia, A.; Lazcano-Hernández, G.; García-Pérez, B.E.; Castrejón-Jiménez, N.S. Bacteriocins from lactic acid bacteria. A powerful alternative as antimicrobials, probiotics, and immunomodulators in veterinary medicine. Animals 2021, 11, 979. [Google Scholar] [CrossRef] [PubMed]
- Douillard, F.P.; Kant, R.; Ritari, J.; Paulin, L.; Palva, A.; de Vos, W.M. Comparative genome analysis of Lactobacillus casei strains isolated from Actimel and Yakult products reveals marked similarities and points to a common origin. Microb. Biotechnol. 2013, 6, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Botta, C.; Acquadro, A.; Greppi, A.; Barchi, L.; Bertolino, M.; Cocolin, L.; Rantsiou, K. Genomic assessment in Lactobacillus plantarum links the butyrogenic pathway with glutamine metabolism. Sci. Rep. 2017, 7, 15975. [Google Scholar] [CrossRef] [Green Version]
- Bachmann, H.; Starrenburg, M.J.; Molenaar, D.; Kleerebezem, M.; van Hylckama Vlieg, J.E.T. Microbial domestication signatures of Lactococcus lactis can be reproduced by experimental evolution. Genome Res. 2012, 22, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Manasian, P.; Bustos, A.S.; Pålsson, B.; Håkansson, A.; Peñarrieta, J.M.; Nilsson, L.; Linares-Pastén, J.A. First evidence of acyl-hydrolase/lipase activity from human probiotic bacteria: Lactobacillus rhamnosusGG and Bifidobacterium longum NCC 2705. Front. Microbiol. 2020, 11, 1534. [Google Scholar] [CrossRef]
- Kleerebezem, M.; Boekhorst, J.; van Kranenburg, R.; Molenaar, D.; Kuipers, O.P.; Leer, R.; Tarchini, R.; Peters, S.A.; Sandbrink, H.M.; Fiers, M.W.; et al. Complete genome sequence of Lactobacillus plantarum WCFS1. Proc. Natl. Acad. Sci. USA 2003, 100, 1990–1995. [Google Scholar] [CrossRef] [Green Version]
- De Vries, M.C.; Vaughan, E.E.; Kleerebezem, M.; de Vos, W.M. Lactobacillus plantarum—Survival, functional and potential probiotic properties in the human intestinal tract. Int. Dairy J. 2006, 16, 1018–1028. [Google Scholar] [CrossRef]
- Martino, M.E.; Bayjanov, J.R.; Caffrey, B.E.; Wels, M.; Joncour, P.; Hughes, S.; Gillet, B.; Kleerebezem, M.; van Hijum, S.A.F.T.; Leulier, F. Nomadic lifestyle of Lactobacillus plantarum revealed by comparative genomics of 54 strains isolated from different habitats. Environ. Microbiol. 2016, 18, 4974–4989. [Google Scholar] [CrossRef]
- Choi, S.; Jin, G.D.; Park, J.; You, I.; Kim, E.B. Pan-Genomics of Lactobacillus plantarum revealed group-specific genomic profiles without habitat association. J. Microbiol. Biotechnol. 2018, 28, 1352–1359. [Google Scholar] [CrossRef]
- Mao, B.; Yin, R.; Li, X.; Cui, S.; Zhang, H.; Zhao, J.; Chen, W. Comparative genomic analysis of Lactiplantibacillus plantarum isolated from different niches. Genes 2021, 12, 241. [Google Scholar] [CrossRef]
- Barraclough, T.; Balbi, K.; Ellis, R. Evolving concepts of bacterial species. Evol. Biol. 2012, 39, 148–157. [Google Scholar] [CrossRef]
- Naeem, M.; Ilyas, M.; Haider, S.; Baig, S.; Saleem, M. Isolation characterization and identification of lactic acid bacteria from fruit juices and their efficacy against antibiotics. Pak. J. Bot. 2012, 44, 323–328. [Google Scholar]
- Di Cagno, R.; Filannino, P.; Gobbetti, M. Vegetable and fruit fermentation by lactic acid bacteria. In Biotechnology of Lactic Acid Bacteria: Novel Applications; Mozzi, F., Raya, R.R., Vignolo, G.M., Eds.; John Wiley & Sons: Chichester, UK, 2015; pp. 216–230. [Google Scholar] [CrossRef]
- Benavidez, A.; Ulcuango, M.; Yépez, L.; Tenea, G.N. Assessment of the in vitro bioactive properties of lactic acid bacteria isolated from native ecological niches of Ecuador. Rev. Argent. Microbiol. 2016, 48, 236–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenea, G.N.; Jarrin-V, P.; Yepez, L. Microbiota of wild fruits from the amazon region of Ecuador: Linking diversity and functional potential of lactic acid bacteria with their origin. In Ecosystem and Biodiversity of Amazonia; Mikkola, H.J., Ed.; IntechOpen: London, UK, 2020. [Google Scholar] [CrossRef]
- Tenea, G.N.; Ortega, C. Genome characterization of Lactiplantibacillus plantarum strain UTNGt2 originated from Theobroma grandiflorum (white cacao) of Ecuadorian Amazon: Antimicrobial peptides from safety to potential applications. Antibiotics 2021, 10, 383. [Google Scholar] [CrossRef]
- Tenea, G.N.; Hurtado, P.; Ortega, C. A novel Weissella cibaria strain UTNGt21O isolated from wild Solanum quitoense Fruit: Genome sequence and characterization of a peptide with highly inhibitory potential toward gram-negative bacteria. Foods 2020, 9, 1242. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [Green Version]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Simon Fraser University Research Computing Group; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef] [PubMed]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 34, D32–D36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast processing of NGS alignment formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Patel, V.M.; Coon, M.; Nguyen, T.; Land, S.J.; Ruden, D.M.; Lu, X. Using Drosophila melanogaster as a model for genotoxic chemical mutational studies with a new program, SnpSift. Front. Genetics 2012, 3, 35. [Google Scholar] [CrossRef] [Green Version]
- De Jong, A.; van Hijum, S.A.; Bijlsma, J.J.; Kok, J.; Kuipers, O.P. BAGEL: A web-based bacteriocin genome mining tool. Nucleic Acids Res. 2006, 34, W273–W279. [Google Scholar] [CrossRef]
- Huang, T.; Xiong, T.; Peng, Z.; Xiao, Y.S.; Liu, Z.G.; Hu, M.; Xie, M.Y. Genomic analysis revealed adaptive mechanism to plant-related fermentation of Lactobacillus plantarum NCU116 and Lactobacillus spp. Genomics 2020, 112, 703–711. [Google Scholar] [CrossRef]
- Turner, M.S.; Hafner, L.M.; Walsh, T.; Giffard, P.M. Identification, characterization and specificity of a cell wall lytic enzyme from Lactobacillus fermentum BR11. FEMS Microbiol. Lett. 2004, 238, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Li, X.; Gu, Q.; Lou, X.-Y.; Zhang, Z.-M.; Song, D.-F.; Zhang, C. Comparative genome analysis of Lactobacillus plantarum ZJ316 reveals its genetic adaptation and potential probiotic profiles. J. Zhejiang Univ. Sci. B 2016, 18, 569–579. [Google Scholar] [CrossRef]
- Evanovich, E.; de Souza Mendonça Mattos, P.J.; Guerreiro, J.F. Comparative Genomic Analysis of Lactobacillus plantarum: An Overview. Int. J. Genom. 2019, 2019, 4973214. [Google Scholar] [CrossRef] [Green Version]
- Burgess, C.; O’connell-Motherway, M.; Sybesma, W.; Hugenholtz, J.; van Sinderen, D. Riboflavin production in Lactococcus lactis: Potential for in situ production of vitamin-enriched foods. Appl. Environ. Microbiol. 2004, 70, 5769–5777. [Google Scholar] [CrossRef] [Green Version]
- Alan, Y. Culture fermentation of Lactobacillus in traditional pickled gherkins: Microbial development, chemical, biogenic amine and metabolite analysis. J. Food. Sci. Technol. 2019, 56, 3930–3939. [Google Scholar] [CrossRef]
- Cai, H.; Thompson, R.; Budinich, M.F.; Broadbent, J.R.; Steele, J.L. Genome sequence and comparative genome analysis of Lactobacillus casei: Insights into their niche-associated evolution. Genome Biol. Evol. 2009, 1, 239–257. [Google Scholar] [CrossRef] [Green Version]
- Molenaar, D.; Bringel, F.; Schuren, F.H.; de Vos, W.M.; Siezen, R.J.; Kleerebezem, M. Exploring Lactobacillus plantarum genome diversity by using microarrays. J. Bacteriol. 2005, 187, 6119–6127. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Raskin, L.; Samuels, D.C.; Shyr, Y.; Guo, Y. Genome measures used for quality control are dependent on gene function and ancestry. Bioinformatics 2015, 31, 318–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deveson, I.W.; Brunck, M.E.; Blackburn, J.; Tseng, E.; Hon, T.; Clark, T.A.; Clark, M.B.; Crawford, J.; Dinger, M.E.; Nielsen, L.K.; et al. Universal alternative splicing of noncoding exons. Cell Syst. 2018, 6, 245–255.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nesmelova, I.V.; Hackett, P.B. DDE transposases: Structural similarity and diversity. Adv. Drug Deliv. Rev. 2010, 62, 1187–1195. [Google Scholar] [CrossRef] [Green Version]
- Smeets, L.C.; Bijlsma, J.J.; Kuipers, E.J.; Vandenbroucke-Grauls, C.M.; Kusters, J.G. The dprA gene is required for natural transformation of Helicobacter pylori. FEMS Immunol. Med. Microbiol. 2000, 27, 99–102. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Zuo, F.; Yu, R.; Zeng, Z.; Ma, H.; Chen, S. Comparative genome-based identification of a cell wall-anchored protein from Lactobacillus plantarum increases adhesion of Lactococcus lactis to human epithelial cells. Sci. Rep. 2015, 5, 14109. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- Tenea, G.N. Peptide extracts from native lactic acid bacteria generate ghost cells and spheroplasts upon interaction with Salmonella enterica, as promising food antimicrobials. BioMed Res. Int. 2020, 2020, 6152356. [Google Scholar] [CrossRef]
- Mbye, M.; Baig, M.A.; AbuQamar, S.F.; El-Tarabily, K.A.; Obaid, R.S.; Osaili, T.M.; Al-Nabulsi, A.A.; Turner, M.S.; Shah, N.P.; Ayyash, M.M. Updates on understanding of probiotic lactic acid bacteria responses to environmental stresses and highlights on proteomic analyses. Compr. Rev. Food Sci. Food Saf. 2020, 19, 1110–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- EFSA FEEDAP Panel. Guidance on the characterization of microorganisms used as feed additives or as production organisms. EFSA J. 2018, 16, 5206. [Google Scholar] [CrossRef]
- De Angelis, M.; Gobbetti, M. Environmental stress responses in Lactobacillus: A review. Proteomics 2004, 4, 106–122. [Google Scholar] [CrossRef] [PubMed]
- Haddaji, N.; Krifi, B.; Lagha, R.; Khouadja, S.; Bakhrouf, A. Effect of high temperature on viability of Lactobacillus casei and analysis of secreted and GroEL proteins profiles. Afr. J. Bacteriol. Res. 2015, 7, 29–35. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhang, J.; Zhao, X.; Zhao, W.; Yu, Z.; Chen, C.; Yang, Z. Complete genome sequencing of exopolysaccharide-producing Lactobacillus plantarum K25 provides genetic evidence for the probiotic functionality and cold endurance capacity of the strain. Biosci. Biotechnol. Biochem. 2018, 82, 1225–1233. [Google Scholar] [CrossRef] [Green Version]
- Altermann, E.; Russell, W.M.; Azcarate-Peril, M.A.; Barrangou, R.; Buck, B.L.; McAuliffe, O.; Souther, N.; Dobson, A.; Duong, T.; Callanan, M.; et al. Complete genome sequence of the probiotic lactic acid bacterium Lactobacillus acidophilus NCFM. Proc. Natl. Acad. Sci. USA 2005, 102, 3906–3912. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.M.; Bongers, R.S.; de Vos, W.M.; Kleerebezem, M. Functional analysis of four bile salt hydrolase and penicillin acylase family members in Lactobacillus plantarum WCFS1. Appl. Environ. Microbiol. 2008, 74, 4719–4726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, F.; Li, Y.; Bumann, M.; Raftis, E.J.; Casey, P.G.; Cooney, J.C.; Walsh, M.A.; O’Toole, P.W. Allelic variation of bile salt hydrolase genes in Lactobacillus salivarius does not determine bile resistance levels. J. Bacteriol. 2009, 191, 5743–5757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamon, E.; Horvatovich, P.; Izquierdo, E.; Bringel, F.; Marchioni, E.; Aoudé-Werner, D.; Ennahar, S. Comparative proteomic analysis of Lactobacillus plantarum for the identification of key proteins in bile tolerance. BMC Microbiol. 2011, 11, 63. [Google Scholar] [CrossRef] [Green Version]
- Castaldo, C.; Siciliano, R.A.; Muscariello, L.; Marasco, R.; Sacco, M. CcpA affects expression of the groESL and dnaK operons in Lactobacillus plantarum. Micro. Cell Fact. 2006, 5, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorv, J.S.; Rose, D.R.; Glick, B.R. Bacterial ice crystal controlling proteins. Scientifica 2014, 2014, 976895. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Bae, D.-W.; Lim, K.; Griffiths, M.W.; Oh, S. Cold stress improves the ability of Lactobacillus plantarum l67 to survive freezing. Int. J. Food Microbiol. 2014, 191, 135–143. [Google Scholar] [CrossRef]
- Polo, L.; Manes-Lazaro, R.; Olmeda, I.; Cruz-Pio, L.E.; Medina, A.; Ferrer, S.; Pardo, I. Influence of freezing temperatures prior to freeze-drying on viability of yeasts and lactic acid bacteria isolated from wine. J. Appl. Microbiol. 2017, 122, 1603–1614. [Google Scholar] [CrossRef] [Green Version]
- Wood, J.M. Bacterial responses to osmotic challenges. J. Gen. Physiol. 2015, 145, 381–388. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Library Name | Ref. Length | Mapped Sites (≥1x) | Total Reads | Mapped Reads | Mapped Bases | Mean Depth |
|---|---|---|---|---|---|---|
| UTNGt21A | 3,348,624 | 3,028,007 (90.43%) | 13,121,820 | 10,291,789 (78.43%) | 941,582,288 | 281.18 |
| UTNGt2 | 3,348,624 | 2,979,050 (88.96%) | 11,733,026 | 10,428,975 (88.89%) | 957,588,188 | 285.96 |
| Type of Variant Annotation | Description | Impact * | Library Name | |||
|---|---|---|---|---|---|---|
| UTNGt21A | UTNGt2 | |||||
| Count | Ratio (%) | Count | Ratio (%) | |||
| synonymous_variant | Variant causes a codon that produces the same amino acid (e.g., Ttg/Ctg, L/L) | Low | 12,350 | 71.51 | 9772 | 72.01 |
| missense_variant | Variant causes a codon that produces a different amino acid (e.g., Tgg/Cgg, W/R) | Moderate | 4740 | 27.45 | 3664 | 27 |
| frameshift_variant | Insertion or deletion causes a frame shift (e.g., an indel’s size is not a multiple of 3). | High | 61 | 0.35 | 33 | 0.24 |
| stop_gained | Variant causes a STOP codon (e.g., Cag/Tag, Q/*) | High | 32 | 0.19 | 21 | 0.16 |
| splice_region and stop_retained_variant | A sequence variant in which a change has occurred within the region of the splice site, either within 1–3 bases of the exon or 3–8 bases of the intron/Variant causes stop codon to be mutated into another stop codon (the new codon produces a different AA). (e.g., Atg/Ctg, M/L (ATG and CTG can be START codons)) | Low | 18 | 0.1 | 22 | 0.15 |
| conservative_inframe_deletion | One or many codons are deleted (e.g., a deletion multiple of three at a codon boundary). | Moderate | 15 | 0.09 | 10 | 0.15 |
| disruptive_inframe_insertion | One or many codons are inserted (e.g., an insertion multiple of three at a codon boundary). | Moderate | 12 | 0.07 | 9 | 0.08 |
| stop_lost and splice_region_variant | Variant causes stop codon to be mutated into a non-stop codon (e.g., Tga/Cga, */R)/A sequence variant in which a change has occurred within the region of the splice site, either within 1–3 bases of the exon or 3–8 bases of the intron. | High | 9 | 0.05 | 5 | 0.07 |
| disruptive_inframe_deletion | One codon is changed and one or many codons are inserted (e.g., an insert of a multiple of three in size, not at a codon boundary). | Modifier | 6 | 0.03 | 6 | 0.04 |
| conservative_inframe_insertion | Inversion of a large chromosome segment (over 1%, or 1,000,000 bases). | Moderate | 6 | 0.03 | 6 | 0.04 |
| non_coding_transcript_exon_variant | Region that does not code for any protein or does not carry genetic code. | Low | 0 | 0 | 6 | 0.04 |
| Stress Factor | Gene (locus WCFS1) | % Identity (EggNOG Annotation)/No. of Variants Relative to the REFERENCE WCFS1 | ||||
|---|---|---|---|---|---|---|
| Protein Product | UTNGt21A | UTNGt2 | ||||
| pH | atpC (lp_2363) | ATP synthase epsilon chain | 67.60 | (-) | 67.60 | 1 |
| atpD (lp_2364) | ATP synthase subunit beta | 84.79 | 2 | 84.79 | 1 | |
| atpG (lp_2365) | ATP synthase gamma chain | 64.19 | (-) | 64.19 | 2 | |
| atpA (lp_2366) | ATP synthase subunit alpha | 81.34 | 3 | 81.34 | 2 | |
| atpH (lp_2367) | ATP synthase subunit delta | 45.55 | (-) | 45.55 | 1 | |
| atpF (lp_2368) | ATP synthase subunit b | 57.64 | (-) | 57.64 | 1 | |
| atpE (lp_2369) | ATP synthase subunit c | 82.69 | (-) | 82.69 | (-) | |
| atpB (lp_2370) | ATP synthase subunit a | 54.85 | (-) | 54.85 | (-) | |
| lepA_1 (lp_2015) | Elongation factor 4 | 56.47 | 3 | 82.75 | 5 | |
| lepA_2 (lp_3120) | Elongation factor 4 | 82.75 | (-) | 56.63 | 12 | |
| Bile salt hydrolase | yxeI_1 | Putative protein YxeI (Choloylglycine hydrolase) | 42.98 | (-) | 42.98 | (-) |
| yxeI_2 | Putative protein YxeI (Choloylglycine hydrolase) | 40.54 | (-) | 34.85 | (-) | |
| yxeI_3 | Putative protein YxeI (Choloylglycine hydrolase) | 34.85 | (-) | 40.55 | (-) | |
| cbh (lp_3536) | Conjugated bile acid hydrolase | 67.28 | (-) | 67.28 | (-) | |
| Temperature | hsp2 (lp_2668) | 18 kDa heat shock protein | 44.96 | 3 | 42.05 | 3 |
| hrcA (lp_2029) | Heat-inducible transcription repressor HrcA | 58.90 | (-) | 58.90 | (-) | |
| grpE (lp_2028) | Protein GrpE | 58.89 | (-) | 58.89 | 1 | |
| dnaK (lp_2027) | Chaperone protein DnaK | 84.33 | (-) | 84.33 | 3 | |
| dnaJ (lp_2026) | Chaperone protein DnaJ | 71.12 | 3 | 71.12 | 3 | |
| Gt21A_00947Gt21A_01250Gt2_02817 | 18 kDa heat shock protein | 44.9633.82 | (-) | 44.96 | (-) | |
| hslR | Heat shock protein 15 | 70.79 | (-) | 71.91 | (-) | |
| groL (lp_0728) | 60 kDa chaperonin | 84.89 | 2 | 84.89 | 2 | |
| groS (lp_0727) | 10 kDa chaperonin | 69.14 | (-) | 69.14 | 1 | |
| hslO (lp_0548) | 33 kDa chaperonin | 69.61 | 2 | 69.61 | (-) | |
| hsp 1 (lp_0129) | Hypothetical small heat shock protein | 45.28 | 1 | 45.28 | 3 | |
| ccpA_1 | Catabolite control protein A | 49.33 | 3 | 49.33 | 4 | |
| ccpA_2 | Catabolite control protein A | 44.09 | 44.09 | |||
| ccpA_3 | Catabolite control protein A | 65.76 | 65.76 | |||
| ccpA_4 | Catabolite control protein A | (-) | 44.14 | |||
| ccpB | Catabolite control protein B | 44.92 | 1 | 47.60 | 2 | |
| cspP (lp_1160) | Cold shock protein 1 | 78.78 | 1 | 78.78 | 1 | |
| cspL (lp_0031) | Cold shock protein 2 | 81.81 | (-) | 81.81 | (-) | |
| cspLA | Cold shock-like protein CspLA | 86.36 | (-) | 86.36 | (-) | |
| Osmosis | opuCD (lp_1610) | Carnitine transport permease protein OpuCD | 73.15 | 2 | 73.17 | 2 |
| opuCC (lp_1609) | Glycine betaine/carnitine/choline-binding protein OpuCC | 63.10 | 5 | 63.49 | 2 | |
| opuCB_1 (lp_1608) | Carnitine transport permease protein OpuCB | 98.21 | 5 | 75.00 | 3 | |
| opuCA (lp_1607) | Carnitine transport ATP-binding protein OpuCA | 68.62 | 3 | 68.62 | 3 | |
| opuCB_2 | Carnitine transport permease protein OpuCB | 75.00 | (-) | (-) | (-) | |
| choS (lp_0367) | Glycine betaine/carnitine/choline-binding protein | 73.14 | 6 | 73.14 | 24 | |
| choQ (lp_0368) | Glycine betaine/carnitine/choline-binding protein | 68.62 | 3 | 68.62 | (-) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tenea, G.N. Decoding the Gene Variants of Two Native Probiotic Lactiplantibacillus plantarum Strains through Whole-Genome Resequencing: Insights into Bacterial Adaptability to Stressors and Antimicrobial Strength. Genes 2022, 13, 443. https://doi.org/10.3390/genes13030443
Tenea GN. Decoding the Gene Variants of Two Native Probiotic Lactiplantibacillus plantarum Strains through Whole-Genome Resequencing: Insights into Bacterial Adaptability to Stressors and Antimicrobial Strength. Genes. 2022; 13(3):443. https://doi.org/10.3390/genes13030443
Chicago/Turabian StyleTenea, Gabriela N. 2022. "Decoding the Gene Variants of Two Native Probiotic Lactiplantibacillus plantarum Strains through Whole-Genome Resequencing: Insights into Bacterial Adaptability to Stressors and Antimicrobial Strength" Genes 13, no. 3: 443. https://doi.org/10.3390/genes13030443