
A Novel Missense Mutation in TNNI3K Causes Recessively Inherited Cardiac Conduction Disease in a Consanguineous Pakistani Family


 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Subjects, DNA Extraction, and Clinical Evaluation
2.2. Whole Exome Capture and Sequencing
2.3. Sanger Sequencing Analysis
2.4. Molecular Dynamic Simulations
3. Results
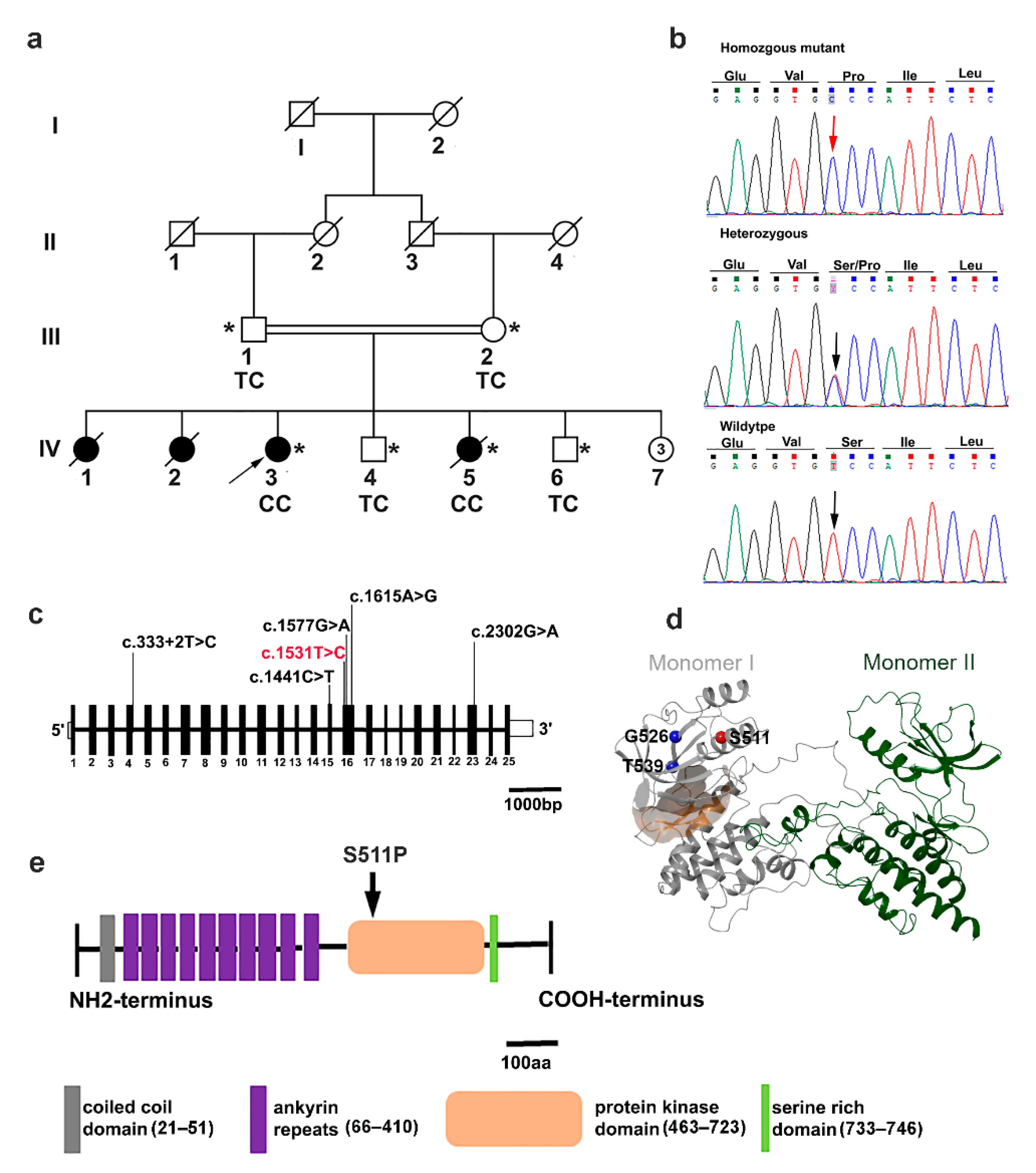
3.1. Clinical Features
3.2. Exome Sequencing Reveals a Pathogenic TNNI3K Variant
3.3. In Silico Molecular Dynamic Simulation
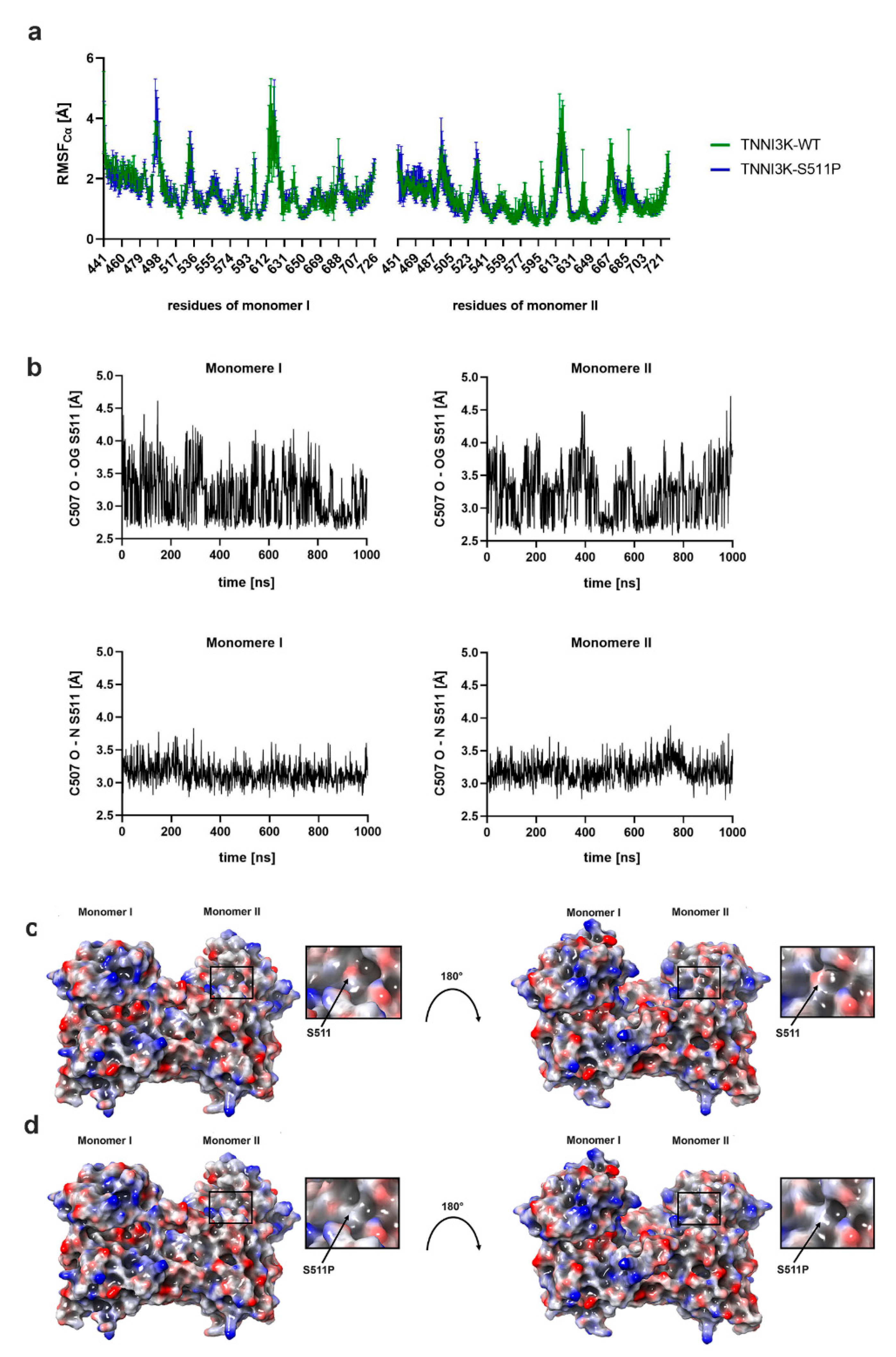
3.3.1. p.Ser511Pro Influences TNNI3K Structural Fluctuation
3.3.2. p.Ser511Pro Affects the Local Hydrogen Bonding Network and Protein Surface Hydrophobicity of TNNI3K
3.3.3. p.Ser511Pro Affects the Structure of Helix 2 and the ATP-Binding Pocket
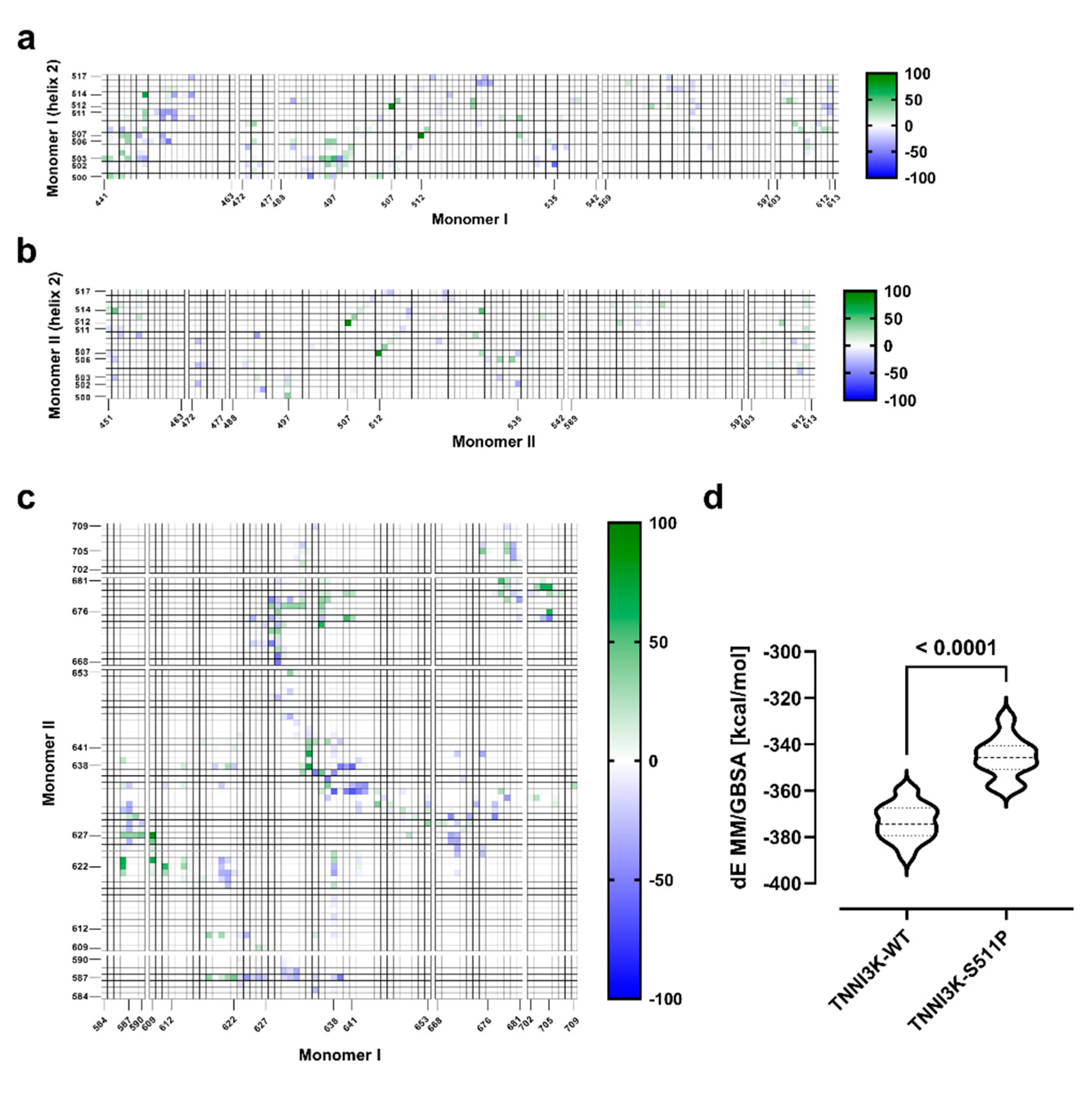
3.3.4. p.Ser511Pro Creates a Less Favorable Binding Site at the Dimer Interface
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Michaëlsson, M.; Jonzon, A.; Riesenfeld, T. Isolated congenital complete atrioventricular block in adult life. A prospective study. Circulation 1995, 92, 442–449. [Google Scholar] [CrossRef]
- Smits, J.P.; Veldkamp, M.W.; Wilde, A.A. Mechanisms of inherited cardiac conduction disease. Europace 2005, 7, 122–137. [Google Scholar] [CrossRef] [PubMed]
- Christoffels, V.M.; Moorman, A.F. Development of the cardiac conduction system: Why are some regions of the heart more arrhythmogenic than others? Circ. Arrhythm. Electrophysiol. 2009, 2, 195–207. [Google Scholar] [CrossRef] [Green Version]
- Munshi, N.V. Gene regulatory networks in cardiac conduction system development. Circ. Res. 2012, 110, 1525–1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedley, P.L.; Jørgensen, P.; Schlamowitz, S.; Wangari, R.; Moolman-Smook, J.; Brink, P.A.; Kanters, J.K.; Corfield, V.A.; Christiansen, M. The genetic basis of long QT and short QT syndromes: A mutation update. Hum. Mutat. 2009, 30, 1486–1511. [Google Scholar] [CrossRef] [PubMed]
- Arnolds, D.E.; Chu, A.; McNally, E.M.; Nobrega, M.A.; Moskowitz, I.P. The emerging genetic landscape underlying cardiac conduction system function. Birth Defects Res. A Clin. Mol. Teratol. 2011, 91, 578–585. [Google Scholar] [CrossRef] [Green Version]
- Neu, A.; Eiselt, M.; Paul, M.; Sauter, K.; Stallmeyer, B.; Isbrandt, D.; Schulze-Bahr, E. A homozygous SCN5A mutation in a severe, recessive type of cardiac conduction disease. Hum. Mutat. 2010, 31, E1609–E1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schott, J.J.; Alshinawi, C.; Kyndt, F.; Probst, V.; Hoorntje, T.M.; Hulsbeek, M.; Wilde, A.A.; Escande, D.; Mannens, M.M.; Le Marec, H. Cardiac conduction defects associate with mutations in SCN5A. Nat. Genet. 1999, 23, 20–21. [Google Scholar] [CrossRef]
- Schott, J.J.; Benson, D.W.; Basson, C.T.; Pease, W.; Silberbach, G.M.; Moak, J.P.; Maron, B.J.; Seidman, C.E.; Seidman, J.G. Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science 1998, 281, 108–111. [Google Scholar] [CrossRef]
- Fatkin, D.; MacRae, C.; Sasaki, T.; Wolff, M.R.; Porcu, M.; Frenneaux, M.; Atherton, J.; Vidaillet, H.J., Jr.; Spudich, S.; De Girolami, U.; et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med. 1999, 341, 1715–1724. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.H.; Xu, S.J.; Bendahhou, S.; Wang, X.L.; Wang, Y.; Xu, W.Y.; Jin, H.W.; Sun, H.; Su, X.Y.; Zhuang, Q.N.; et al. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science 2003, 299, 251–254. [Google Scholar] [CrossRef]
- Gollob, M.H.; Jones, D.L.; Krahn, A.D.; Danis, L.; Gong, X.Q.; Shao, Q.; Liu, X.; Veinot, J.P.; Tang, A.S.; Stewart, A.F.; et al. Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N. Engl. J. Med. 2006, 354, 2677–2688. [Google Scholar] [CrossRef] [PubMed]
- Hodgson-Zingman, D.M.; Karst, M.L.; Zingman, L.V.; Heublein, D.M.; Darbar, D.; Herron, K.J.; Ballew, J.D.; de Andrade, M.; Burnett, J.C., Jr.; Olson, T.M. Atrial natriuretic peptide frameshift mutation in familial atrial fibrillation. N. Engl. J. Med. 2008, 359, 158–165. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, H.; Koopmann, T.T.; Le Scouarnec, S.; Yang, T.; Ingram, C.R.; Schott, J.J.; Demolombe, S.; Probst, V.; Anselme, F.; Escande, D.; et al. Sodium channel β1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J. Clin. Investig. 2008, 118, 2260–2268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruse, M.; Schulze-Bahr, E.; Corfield, V.; Beckmann, A.; Stallmeyer, B.; Kurtbay, G.; Ohmert, I.; Schulze-Bahr, E.; Brink, P.; Pongs, O. Impaired endocytosis of the ion channel TRPM4 is associated with human progressive familial heart block type I. J. Clin. Investig. 2009, 119, 2737–2744. [Google Scholar] [CrossRef]
- Theis, J.L.; Zimmermann, M.T.; Larsen, B.T.; Rybakova, I.N.; Long, P.A.; Evans, J.M.; Middha, S.; de Andrade, M.; Moss, R.L.; Wieben, E.D.; et al. TNNI3K mutation in familial syndrome of conduction system disease, atrial tachyarrhythmia and dilated cardiomyopathy. Hum. Mol. Genet. 2014, 23, 5793–5804. [Google Scholar] [CrossRef]
- Baruteau, A.E.; Probst, V.; Abriel, H. Inherited progressive cardiac conduction disorders. Curr. Opin. Cardiol. 2015, 30, 33–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Meng, X.M.; Wei, Y.J.; Zhao, X.W.; Liu, D.Q.; Cao, H.Q.; Liew, C.C.; Ding, J.F. Cloning and characterization of a novel cardiac-specific kinase that interacts specifically with cardiac troponin I. J. Mol. Med. 2003, 81, 297–304. [Google Scholar] [CrossRef]
- Wang, X.; Wang, J.; Su, M.; Wang, C.; Chen, J.; Wang, H.; Song, L.; Zou, Y.; Zhang, L.; Zhang, Y.; et al. TNNI3K, a cardiac-specific kinase, promotes physiological cardiac hypertrophy in transgenic mice. PLoS ONE 2013, 8, e58570. [Google Scholar] [CrossRef] [Green Version]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Cao, H.; Liu, Z.; Ding, J.; Meng, X. Identification of the dual specificity and the functional domains of the cardiac-specific protein kinase TNNI3K. Gen. Physiol. Biophys. 2007, 26, 104–109. [Google Scholar]
- Cheng, J.; Yu, L.; Zhang, D.; Huang, Q.; Spencer, D.; Su, B. Dimerization through the catalytic domain is essential for MEKK2 activation. J. Biol. Chem. 2005, 280, 13477–13482. [Google Scholar] [CrossRef] [Green Version]
- Lai, Z.-F.; Chen, Y.-Z. Evidence, hypotheses and significance of MAP kinase TNNI3K interacting with its partners. World J. Hypertens. 2012, 2, 22–28. [Google Scholar] [CrossRef]
- Lal, H.; Ahmad, F.; Parikh, S.; Force, T. Troponin I-interacting protein kinase: A novel cardiac-specific kinase, emerging as a molecular target for the treatment of cardiac disease. Circ. J. 2014, 78, 1514–1519. [Google Scholar] [CrossRef] [Green Version]
- Fan, L.-L.; Huang, H.; Jin, J.-Y.; Li, J.-J.; Chen, Y.-Q.; Zhao, S.-P.; Xiang, R. Whole exome sequencing identifies a novel mutation (c. 333+ 2T> C) of TNNI3K in a Chinese family with dilated cardiomyopathy and cardiac conduction disease. Gene 2018, 648, 63–67. [Google Scholar] [CrossRef]
- Podliesna, S.; Delanne, J.; Miller, L.; Tester, D.J.; Uzunyan, M.; Yano, S.; Klerk, M.; Cannon, B.C.; Khongphatthanayothin, A.; Laurent, G.; et al. Supraventricular tachycardias, conduction disease, and cardiomyopathy in 3 families with the same rare variant in TNNI3K (p.Glu768Lys). Heart Rhythm 2019, 16, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, D.; Li, M.; Wu, K.; Liu, N.; Zhao, C.; Shi, X.; Liu, Q. Identification of a nonsense mutation in TNNI3K associated with cardiac conduction disease. J. Clin. Lab. Anal. 2020, 34, e23418. [Google Scholar] [CrossRef] [PubMed]
- Patterson, M.; Barske, L.; Van Handel, B.; Rau, C.D.; Gan, P.; Sharma, A.; Parikh, S.; Denholtz, M.; Huang, Y.; Yamaguchi, Y. Frequency of mononuclear diploid cardiomyocytes underlies natural variation in heart regeneration. Nat. Genet. 2017, 49, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Lawhorn, B.G.; Philp, J.; Zhao, Y.; Louer, C.; Hammond, M.; Cheung, M.; Fries, H.; Graves, A.P.; Shewchuk, L.; Wang, L. Identification of purines and 7-deazapurines as potent and selective type I inhibitors of troponin I-interacting kinase (TNNI3K). J. Med. Chem. 2015, 58, 7431–7448. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Fromer, M.; Purcell, S.M. Using XHMM software to detect copy number variation in whole-exome sequencing data. Curr. Protoc. Hum. Genet. 2014, 81, 7–23. [Google Scholar] [CrossRef] [Green Version]
- Rimmer, A.; Phan, H.; Mathieson, I.; Iqbal, Z.; Twigg, S.R.; Wilkie, A.O.; McVean, G.; Lunter, G. Integrating mapping-, assembly-and haplotype-based approaches for calling variants in clinical sequencing applications. Nat. Genet. 2014, 46, 912–918. [Google Scholar] [CrossRef] [Green Version]
- Gudbjartsson, D.F.; Jonasson, K.; Frigge, M.L.; Kong, A. Allegro, a new computer program for multipoint linkage analysis. Nat. Genet. 2000, 25, 12–13. [Google Scholar] [CrossRef] [PubMed]
- Krumm, N.; Sudmant, P.H.; Ko, A.; O’Roak, B.J.; Malig, M.; Coe, B.P.; Quinlan, A.R.; Nickerson, D.A.; Eichler, E.E.; Project, N.E.S. Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012, 22, 1525–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plagnol, V.; Curtis, J.; Epstein, M.; Mok, K.Y.; Stebbings, E.; Grigoriadou, S.; Wood, N.W.; Hambleton, S.; Burns, S.O.; Thrasher, A.J. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics 2012, 28, 2747–2754. [Google Scholar] [CrossRef] [Green Version]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef] [PubMed]
- Noderer, W.L.; Flockhart, R.J.; Bhaduri, A.; Diaz de Arce, A.J.; Zhang, J.; Khavari, P.A.; Wang, C.L. Quantitative analysis of mammalian translation initiation sites by FACS-seq. Mol. Syst. Biol. 2014, 10, 748. [Google Scholar] [CrossRef]
- Liu, X.; Jian, X.; Boerwinkle, E. dbNSFP: A lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 2011, 32, 894–899. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger Release 2020-3: Protein Preparation Wizard; Epik, Schrödinger, LLC, New York, NY, 2020; Impact, Schrödinger, LLC, New York, NY; Prime, Schrödinger, LLC, New York, NY, 2020.
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-3: Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2020. Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, 2020.
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the SC’06—2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar]
- Schrödinger Release 2020-3: Maestro, Schrödinger, LLC, New York, NY, 2020.
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. II. The accuracy of ranking poses generated from docking. J. Comput. Chem. 2011, 32, 866–877. [Google Scholar] [CrossRef] [Green Version]
- Beard, H.; Cholleti, A.; Pearlman, D.; Sherman, W.; Loving, K.A. Applying physics-based scoring to calculate free energies of binding for single amino acid mutations in protein-protein complexes. PLoS ONE 2013, 8, e82849. [Google Scholar] [CrossRef] [Green Version]
- Zou, X.Q.; Sun, Y.X.; Kuntz, I.D. Inclusion of solvation in ligand binding free energy calculations using the generalized-born model. J. Am. Chem. Soc. 1999, 121, 8033–8043. [Google Scholar] [CrossRef]
- Wendler, P.; Ciniawsky, S.; Kock, M.; Kube, S. Structure and function of the AAA+ nucleotide binding pocket. Biochim. Biophys. Acta 2012, 1823, 2–14. [Google Scholar] [CrossRef] [Green Version]
- Trevino, S.R.; Scholtz, J.M.; Pace, C.N. Amino acid contribution to protein solubility: Asp, Glu, and Ser contribute more favorably than the other hydrophilic amino acids in RNase Sa. J. Mol. Biol. 2007, 366, 449–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, R.J.; Minton, A.P. Protein aggregation in crowded environments. Biol. Chem. 2006, 387, 485–497. [Google Scholar] [CrossRef]
- Gallati, S. Disease-modifying genes and monogenic disorders: Experience in cystic fibrosis. Appl. Clin. Genet. 2014, 7, 133. [Google Scholar] [CrossRef] [Green Version]
- Quintana-Murci, L. Understanding rare and common diseases in the context of human evolution. Genome Biol. 2016, 17, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monies, D.; Maddirevula, S.; Kurdi, W.; Alanazy, M.H.; Alkhalidi, H.; Al-Owain, M.; Sulaiman, R.A.; Faqeih, E.; Goljan, E.; Ibrahim, N. Autozygosity reveals recessive mutations and novel mechanisms in dominant genes: Implications in variant interpretation. Genet. Med. 2017, 19, 1144–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baig, S.M.; Koschak, A.; Lieb, A.; Gebhart, M.; Dafinger, C.; Nürnberg, G.; Ali, A.; Ahmad, I.; Sinnegger-Brauns, M.J.; Brandt, N. Loss of Ca v 1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nat. Neurosci. 2011, 14, 77–84. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Subject ID | Sex | Age (y) | Age at Diagnosis (y) | HR b.p.m. | Arrhy-thmia | Pacemaker Insertion | 24 h ECG Holter | Echocardio-graphy | PR Interval | QRs Interval | HF | Other Associated Phenotypes | Health Status | TNNI3K Genotype |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| III-1 | M | 60 | - | 67 | N | N | NAD | NAD | No AV block, 130 ms | Normal duration | N | - | U | TC |
| III-2 | F | 50 | - | 71 | N | N | NAD | NAD | No AV block, 140 ms | PRWP | N | - | U | TC |
| IV-1 | F | 26 | 16 | - | - | N | - | - | - | - | SCA | - | A | - |
| IV-2 | F | 23 | 14 | - | - | N | - | - | - | - | SCA | - | A | - |
| IV-3 | F | 22 | 13 | 80 | N | N | SB (55 beats/min) Tach (160 beats/min) | NAD | No AV block, 130 ms | RBBB, LAFB, PRWP, and BFB | N | Body posture defect | A | CC |
| IV-4 | M | 17 | - | - | N | N | NAD | - | No AV block, 160 ms | LVH | N | - | U | TC |
| IV-5 | F | 15 | 12 | 80 | N | N | SB (56 beats/min) Tach (160 beats/min) | VSD | - | - | SCA | - | A | CC |
| IV-6 | M | 10 | - | 77 | N | N | NAD | NAD | No AV block, 140 ms | LAD, LAFB | N | - | U | TC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramzan, S.; Tennstedt, S.; Tariq, M.; Khan, S.; Noor Ul Ayan, H.; Ali, A.; Munz, M.; Thiele, H.; Korejo, A.A.; Mughal, A.R.; et al. A Novel Missense Mutation in TNNI3K Causes Recessively Inherited Cardiac Conduction Disease in a Consanguineous Pakistani Family. Genes 2021, 12, 1282. https://doi.org/10.3390/genes12081282
Ramzan S, Tennstedt S, Tariq M, Khan S, Noor Ul Ayan H, Ali A, Munz M, Thiele H, Korejo AA, Mughal AR, et al. A Novel Missense Mutation in TNNI3K Causes Recessively Inherited Cardiac Conduction Disease in a Consanguineous Pakistani Family. Genes. 2021; 12(8):1282. https://doi.org/10.3390/genes12081282
Chicago/Turabian StyleRamzan, Shafaq, Stephanie Tennstedt, Muhammad Tariq, Sheraz Khan, Hafiza Noor Ul Ayan, Aamir Ali, Matthias Munz, Holger Thiele, Asad Aslam Korejo, Abdul Razzaq Mughal, and et al. 2021. "A Novel Missense Mutation in TNNI3K Causes Recessively Inherited Cardiac Conduction Disease in a Consanguineous Pakistani Family" Genes 12, no. 8: 1282. https://doi.org/10.3390/genes12081282