Whole Exome Sequencing in Coloboma/Microphthalmia: Identification of Novel and Recurrent Variants in Seven Genes

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Genes of Interest
2.3. Whole-Exome Sequencing and Data Analysis
2.4. Segregation Analysis
2.5. CNV Analysis and Breakpoint Assessment
2.6. Functional Analyses by RT-PCR of Potential Splice Site Variants in CHD7, ACTG1, and EFTUD2
2.7. Functional Analysis by Minigene Assay of a Potential Splice Site Variant in EFTUD2
3. Results
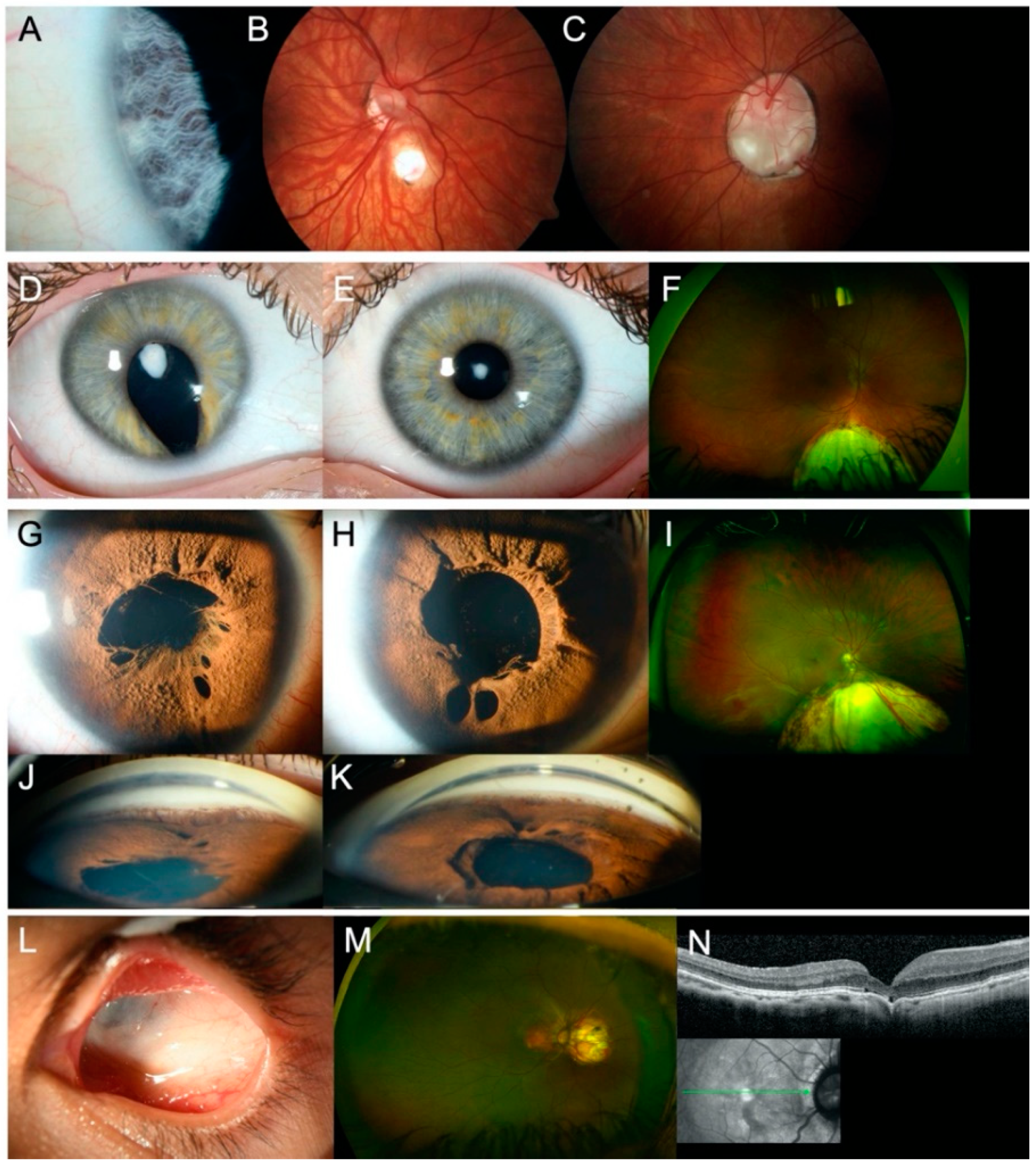
3.1. Sequence Variants and Clinical Findings in Coloboma
3.2. Sequence Variants and Clinical Findings in Colobomatous Microphthalmia
3.3. Additional Sequence Variants
4. Discussion
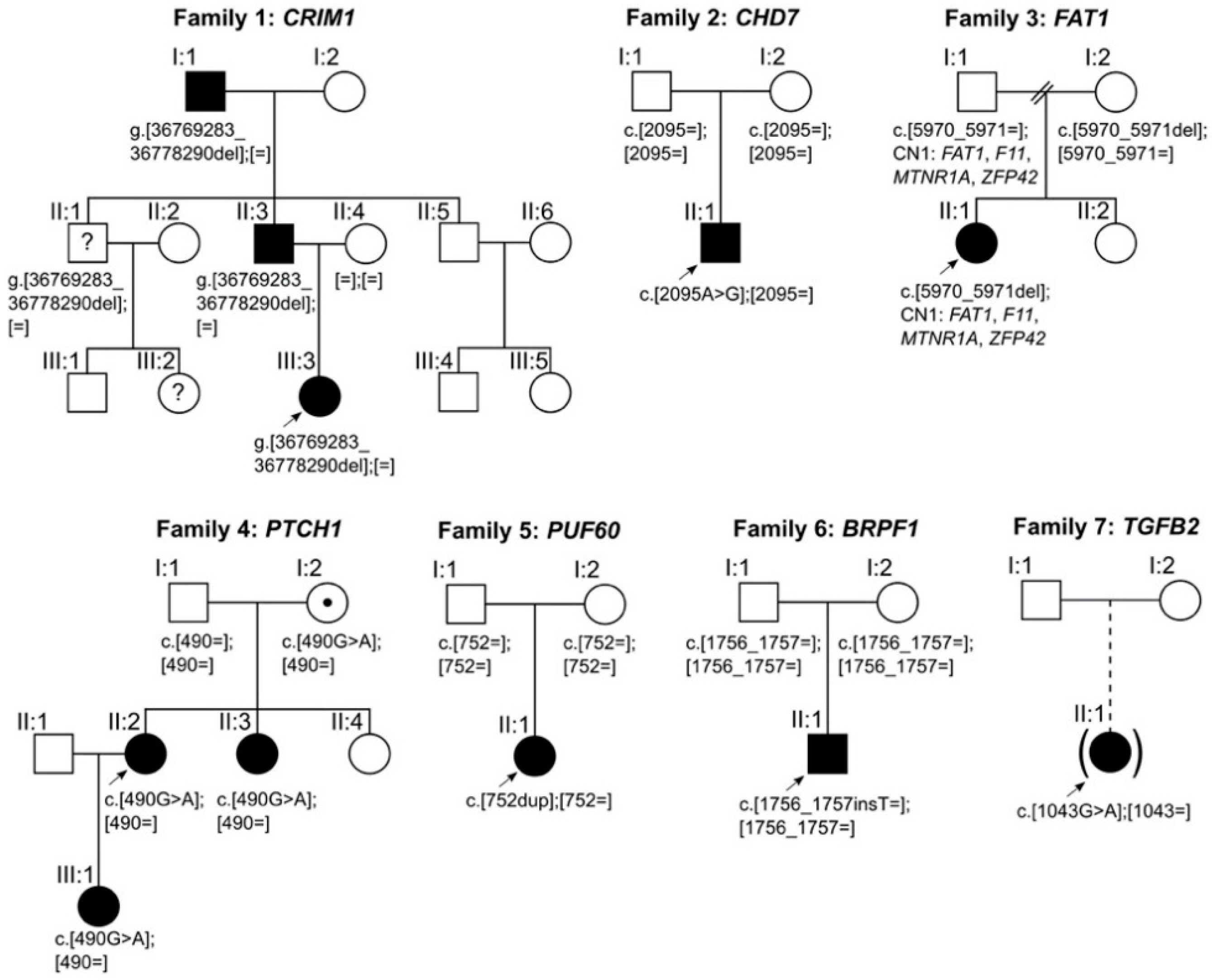
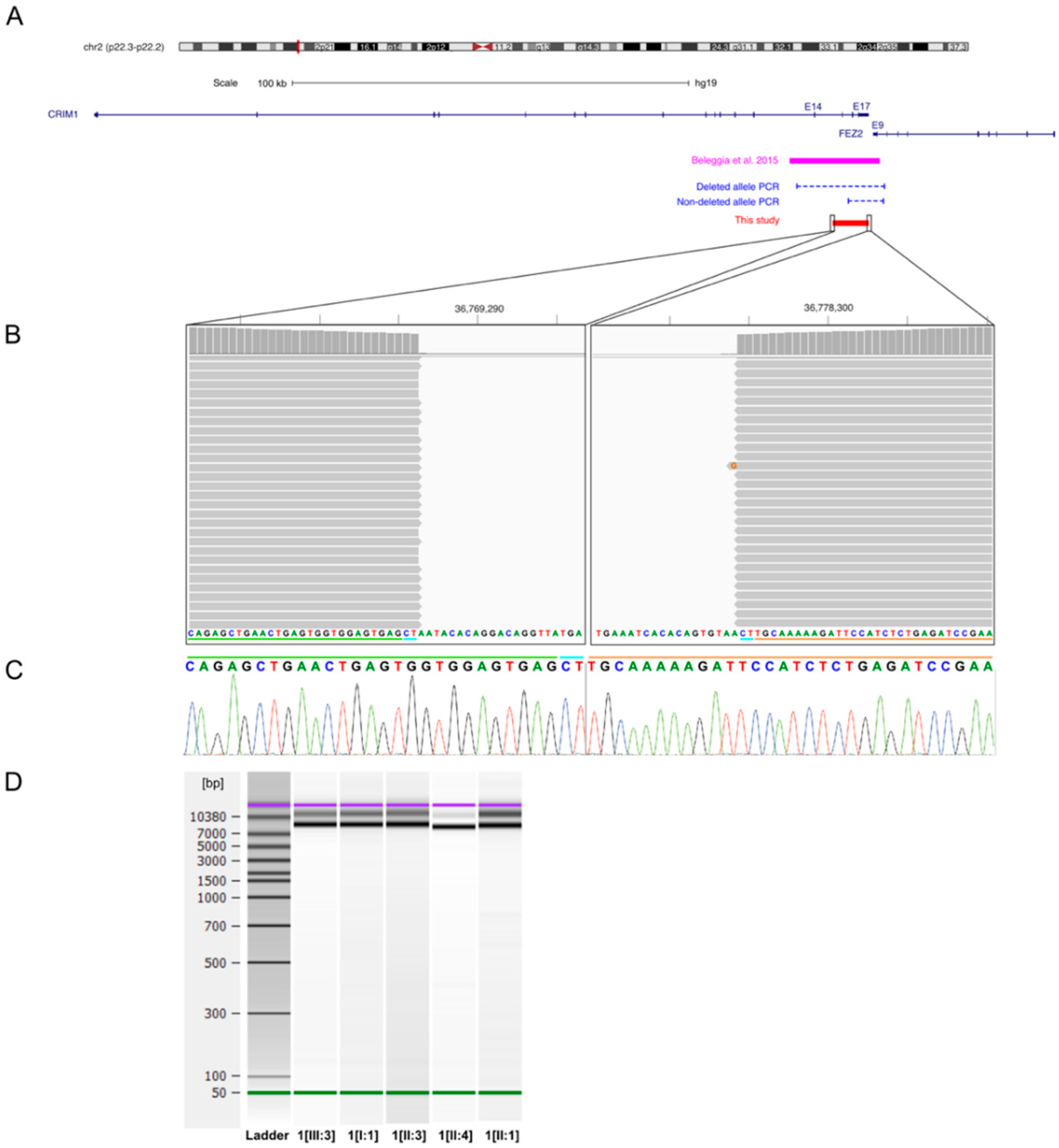
4.1. CRIM1
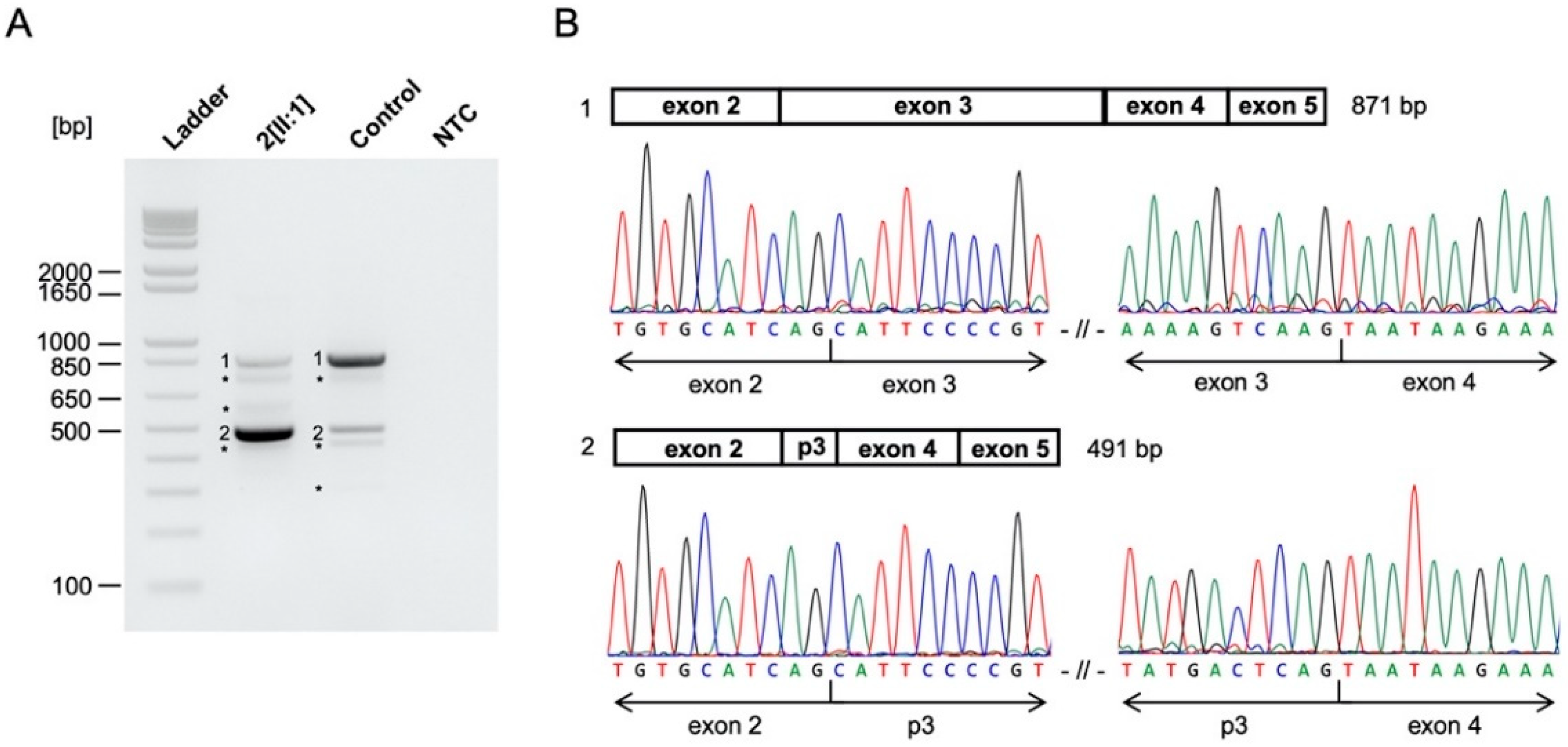
4.2. CHD7
4.3. FAT1
4.4. PTCH1
4.5. PUF60
4.6. BRPF1
4.7. TGFB2
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Williamson, K.A.; Fitzpatrick, D.R. The genetic architecture of microphthalmia, anophthalmia and coloboma. Eur. J. Med. Genet. 2014, 57, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Alsomiry, A.S.; Gregory-Evans, C.Y.; Gregory-Evans, K. An update on the genetics of ocular coloboma. Qual. Life Res. 2019, 138, 865–880. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Blain, D.; Bertuzzi, S.; Brooks, B.P. Uveal coloboma: Clinical and basic science update. Curr. Opin. Ophthalmol. 2006, 17, 447–470. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.H.; Kousseff, B.G.; Ross, E.A.; Longbottom, J. Complex Microphthalmos. Arch. Ophthalmol. 1989, 107, 1619–1624. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.H.; Kousseff, B.G.; Ross, E.A.; Longbottom, J. Simple Microphthalmos. Arch. Ophthalmol. 1989, 107, 1625–1630. [Google Scholar] [CrossRef]
- Davenport, M.P.; Fitzpatrick, D.; Hanson, I.; Williamson, K.; Van Heyningen, V.; Fleck, B.; Jones, I.; Chalmers, J.; Campbell, H. National study of microphthalmia, anophthalmia, and coloboma (MAC) in Scotland: Investigation of genetic aetiology. J. Med. Genet. 2002, 39, 16–22. [Google Scholar] [CrossRef] [Green Version]
- Källén, B.; Tornqvist, K. The epidemiology of anophthalmia and microphthalmia in Sweden. Eur. J. Epidemiol. 2005, 20, 345–350. [Google Scholar] [CrossRef] [Green Version]
- Plaisancié, J.; Ceroni, F.; Holt, R.; Seco, C.Z.; Calvas, P.; Chassaing, N.; Ragge, N.K. Genetics of anophthalmia and microphthalmia. Part 1: Non-syndromic anophthalmia/microphthalmia. Qual. Life Res. 2019, 138, 799–830. [Google Scholar] [CrossRef]
- Hornby, S.J.; Gilbert, C.E.; Rahi, J.; Sil, A.K.; Xiao, Y.; Dandona, L.; Foster, A. Regional variation in blindness in children due to microphthalmos, anophthalmos and coloboma. Ophthalmic Epidemiol. 2000, 7, 127–138. [Google Scholar] [CrossRef]
- Bermejo, E.; Martínez-Frías, M. Congenital eye malformations: Clinical-epidemiological analysis of 1,124,654 consecutive births in Spain. Am. J. Med. Genet. 1998, 75, 497–504. [Google Scholar] [CrossRef]
- Shaw, G.; Carmichael, S.L.; Yang, W.; Harris, J.A.; Finnell, R.H.; Lammer, E.J. Epidemiologic characteristics of anophthalmia and bilateral microphthalmia among 2.5 million births in California, 1989–1997. Am. J. Med. Genet. Part A 2005, 137, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, A.; Bianchi, F.; Calabro, A.; Calzolari, E.; Clementi, M.; Mastroiacovo, P.; Meli, P.; Petrelli, G.; Tenconi, R. Anophthalmia and benomyl in Italy: A multicenter study based on 940,615 newborns. Reprod. Toxicol. 1994, 8, 397–403. [Google Scholar] [CrossRef]
- Hu, D.N. Prevalence and mode of inheritance of major genetic eye diseases in China. Acta Genet. Sin. 1988, 15, 231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.P.; Taylor, A.E.; Sowden, J.C.; Ragge, N.K.; Russell-Eggitt, I.; Rahi, J.S.; Gilbert, C.E. Anophthalmos, Microphthalmos, and Typical Coloboma in the United Kingdom: A Prospective Study of Incidence and Risk. Investig. Opthalmol. Vis. Sci. 2011, 52, 558–564. [Google Scholar] [CrossRef]
- Reis, L.M.; Semina, E.V. Conserved genetic pathways associated with microphthalmia, anophthalmia, and coloboma. Birth Defects Res. Part C Embryo Today Rev. 2015, 105, 96–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaisancie, J.; Calvas, P.; Chassaing, N. Genetic Advances in Microphthalmia. J. Pediatr. Genet. 2016, 5, 184–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerth-Kahlert, C.; Williamson, K.; Ansari, M.; Rainger, J.K.; Hingst, V.; Zimmermann, T.; Tech, S.; Guthoff, R.F.; Van Heyningen, V.; Fitzpatrick, D.R. Clinical and mutation analysis of 51 probands with anophthalmia and/or severe microphthalmia from a single center. Mol. Genet. Genom. Med. 2013, 1, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Widen, S.A.; Williamson, K.A.; Ratnapriya, R.; Gerth-Kahlert, C.; Rainger, J.; Alur, R.P.; Strachan, E.; Manjunath, S.H.; Balakrishnan, A.; et al. A secreted WNT-ligand-binding domain of FZD5 generated by a frameshift mutation causes autosomal dominant coloboma. Hum. Mol. Genet. 2016, 25, 1382–1391. [Google Scholar] [CrossRef]
- Raca, G.; Jackson, C.A.; Warman, B.; Bair, T.; Schimmenti, L.A. Next generation sequencing in research and diagnostics of ocular birth defects. Mol. Genet. Metab. 2010, 100, 184–192. [Google Scholar] [CrossRef] [Green Version]
- Lang, E.; Koller, S.; Bähr, L.; Töteberg-Harms, M.; Atac, D.; Roulez, F.; Bahr, A.; Steindl, K.; Feil, S.; Berger, W.; et al. Exome Sequencing in a Swiss Childhood Glaucoma Cohort Reveals CYP1B1 and FOXC1 Variants as Most Frequent Causes. Transl. Vis. Sci. Technol. 2020, 9, 47. [Google Scholar] [CrossRef]
- Mathe, E.; Olivier, M.; Kato, S.; Ishioka, C.; Hainaut, P.; Tavtigian, S.V. Computational approaches for predicting the biological effect of p53 missense mutations: A comparison of three sequence analysis based methods. Nucleic Acids Res. 2006, 34, 1317–1325. [Google Scholar] [CrossRef]
- Shihab, H.A.; Rogers, M.F.; Gough, J.; Mort, M.E.; Cooper, D.N.; Day, I.N.M.; Gaunt, T.R.; Campbell, C. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 2015, 31, 1536–1543. [Google Scholar] [CrossRef] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Bergman, J.E.H.; Janssen, N.; Van Der Sloot, A.M.; De Walle, H.E.K.; Schoots, J.; Rendtorff, N.D.; Tranebjaerg, L.; Hoefsloot, L.H.; Van Ravenswaaij-Arts, C.M.A.; Hofstra, R. A novel classification system to predict the pathogenic effects of CHD7 missense variants in CHARGE syndrome. Hum. Mutat. 2012, 33, 1251–1260. [Google Scholar] [CrossRef]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef] [Green Version]
- Maggi, J.; Koller, S.; Bähr, L.; Feil, S.; Pfiffner, F.K.; Hanson, J.V.M.; Maspoli, A.; Gerth-Kahlert, C.; Berger, W. Long-Range PCR-Based NGS Applications to Diagnose Mendelian Retinal Diseases. MedRxiv 2020. [Google Scholar] [CrossRef]
- Gamundi, M.J.; Hernan, I.; Muntanyola, M.; Maseras, M.; Álvarez, R.; Dopazo, A.; Borrego, S.; Carballo, M.; López-Romero, P. Transcriptional expression ofcis-acting andtrans-acting splicing mutations cause autosomal dominant retinitis pigmentosa. Hum. Mutat. 2008, 29, 869–878. [Google Scholar] [CrossRef]
- De Heer, A.-M.R.; Collin, R.W.; Huygen, P.L.M.; Schraders, M.; Oostrik, J.; Rouwette, M.; Kunst, H.P.; Kremer, H.; Cremers, C.W. Progressive Sensorineural Hearing Loss and Normal Vestibular Function in a Dutch DFNB7/11 Family with a Novel Mutation in TMC1. Audiol. Neurotol. 2010, 16, 93–105. [Google Scholar] [CrossRef]
- Jongmans, M.C.J.; Admiraal, R.J.; Van Der Donk, K.P.; Vissers, L.E.L.M.; Baas, A.F.; Kapusta, L.; Van Hagen, J.M.; Donnai, D.; De Ravel, T.J.; Veltman, J.A.; et al. CHARGE syndrome: The phenotypic spectrum of mutations in the CHD7 gene. J. Med. Genet. 2005, 43, 306–314. [Google Scholar] [CrossRef] [Green Version]
- Amato, L.G.L.; Montenegro, L.R.; Lerario, A.M.; Jorge, A.A.L.; Junior, G.G.; Schnoll, C.; Renck, A.C.; Trarbach, E.B.; Costa, E.M.F.; Mendonca, B.B.; et al. New genetic findings in a large cohort of congenital hypogonadotropic hypogonadism. Eur. J. Endocrinol. 2019, 181, 103–119. [Google Scholar] [CrossRef]
- Janssen, N.; Bergman, J.E.H.; Swertz, M.A.; Tranebjaerg, L.; Lodahl, M.; Schoots, J.; Hofstra, R.M.W.; Van Ravenswaaij-Arts, C.; Hoefsloot, L.H. Mutation update on the CHD7 gene involved in CHARGE syndrome. Hum. Mutat. 2012, 33, 1149–1160. [Google Scholar] [CrossRef]
- Thomas, M.G.; Kumar, A.; Mohammad, S.; Proudlock, F.A.; Engle, E.C.; Andrews, C.; Chan, W.-M.; Thomas, S.; Gottlob, I. Structural Grading of Foveal Hypoplasia Using Spectral-Domain Optical Coherence Tomography. Ophthalmology 2011, 118, 1653–1660. [Google Scholar] [CrossRef] [Green Version]
- Beleggia, F.; Filippo, B.; Fan, J.; Elcioglu, N.H.; Toker, E.; Wieland, T.; Maumenee, I.H.; Akarsu, N.A.; Meitinger, T.; Strom, T.M.; et al. CRIM1 haploinsufficiency causes defects in eye development in human and mouse. Hum. Mol. Genet. 2015, 24, 2267–2273. [Google Scholar] [CrossRef] [Green Version]
- Bateman, J.B.; Maumenee, I.H. Colobomatous macrophthalmia with microcornea. Ophthalmic Paediatr. Genet. 1984, 4, 59–66. [Google Scholar] [CrossRef]
- Ponferrada, V.G.; Fan, J.; Vallance, J.E.; Hu, S.; Mamedova, A.; Rankin, S.A.; Kofron, M.; Zorn, A.M.; Hegde, R.S.; Lang, R.A. CRIM1 Complexes with ß-catenin and Cadherins, Stabilizes Cell-Cell Junctions and Is Critical for Neural Morphogenesis. PLoS ONE 2012, 7, e32635. [Google Scholar] [CrossRef] [Green Version]
- Toker, A.; Elcioglu, N.; Özcan, E.; Yenice, Ö.; Ogut, M.; Elçioǧlu, N. Colobomatous macrophthalmia with microcornea syndrome: Report of a new pedigree. Am. J. Med. Genet. 2003, 121, 25–30. [Google Scholar] [CrossRef]
- Brajadenta, G.S.; Bilan, F.; Gilbert-Dussardier, B.; Kitzis, A.; Thoreau, V. A functional assay to study the pathogenicity of CHD7 protein variants encountered in CHARGE syndrome patients. Eur. J. Hum. Genet. 2019, 27, 1683–1691. [Google Scholar] [CrossRef]
- Verloes, A. Updated diagnostic criteria for CHARGE syndrome: A proposal. Am. J. Med. Genet. Part A 2005, 306–308. [Google Scholar] [CrossRef]
- Zentner, G.E.; Layman, W.S.; Martin, D.M.; Scacheri, P.C. Molecular and phenotypic aspects ofCHD7mutation in CHARGE syndrome. Am. J. Med. Genet. Part A 2010, 152A, 674–686. [Google Scholar] [CrossRef] [Green Version]
- Gee, H.Y.; Sadowski, C.E.; Aggarwal, P.K.; Porath, J.D.; Yakulov, T.A.; Schueler, M.; Lovric, S.; Ashraf, S.; Braun, D.A.; Halbritter, J.; et al. FAT1 mutations cause a glomerulotubular nephropathy. Nat. Commun. 2016, 7, 10822. [Google Scholar] [CrossRef] [Green Version]
- Lahrouchi, N.; George, A.; Ratbi, I.; Schneider, R.; Elalaoui, S.C.; Moosa, S.; Bharti, S.; Sharma, R.; Abu-Asab, M.; Onojafe, F.; et al. Homozygous frameshift mutations in FAT1 cause a syndrome characterized by colobomatous-microphthalmia, ptosis, nephropathy and syndactyly. Nat. Commun. 2019, 10, 1180. [Google Scholar] [CrossRef]
- Liegel, R.P.; Handley, M.T.; Ronchetti, A.; Brown, S.; Langemeyer, L.; Linford, A.; Chang, B.; Morris-Rosendahl, D.J.; Carpanini, S.; Posmyk, R.; et al. Loss-of-Function Mutations in TBC1D20 Cause Cataracts and Male Infertility in blind sterile Mice and Warburg Micro Syndrome in Humans. Am. J. Hum. Genet. 2013, 93, 1001–1014. [Google Scholar] [CrossRef] [Green Version]
- Puppo, F.; Dionnet, E.; Gaillard, M.-C.; Gaildrat, P.; Castro, C.; Vovan, C.; Bertaux, K.; Bernard, R.; Attarian, S.; Goto, K.; et al. Identification of Variants in the 4q35 GeneFAT1in Patients with a Facioscapulohumeral Dystrophy-Like Phenotype. Hum. Mutat. 2015, 36, 443–453. [Google Scholar] [CrossRef]
- Pai, Y.-J.; Abdullah, N.; Mohd-Zin, S.; Mohammed, R.S.; Rolo, A.; Greene, N.D.; Abdul-Aziz, N.M.; Copp, A.J. Epithelial fusion during neural tube morphogenesis. Birth Defects Res. Part A Clin. Mol. Teratol. 2012, 94, 817–823. [Google Scholar] [CrossRef] [Green Version]
- Tanoue, T.; Takeichi, M. Mammalian Fat1 cadherin regulates actin dynamics and cell–cell contact. J. Cell Biol. 2004, 165, 517–528. [Google Scholar] [CrossRef]
- Sugiyama, Y.; Shelley, E.J.; Badouel, C.; McNeill, H.; McAvoy, J.W. Atypical Cadherin Fat1 Is Required for Lens Epithelial Cell Polarity and Proliferation but Not for Fiber Differentiation. Investig. Opthalmol. Vis. Sci. 2015, 56, 4099–4107. [Google Scholar] [CrossRef] [Green Version]
- Vona, B.; Nanda, I.; Neuner, C.; Schröder, J.; Kalscheuer, V.M.; Shehata-Dieler, W.; Haaf, T. Terminal chromosome 4q deletion syndrome in an infant with hearing impairment and moderate syndromic features: Review of literature. BMC Med. Genet. 2014, 15, 72. [Google Scholar] [CrossRef] [Green Version]
- Caruso, N.; Herberth, B.; Bartoli, M.; Puppo, F.; Dumonceaux, J.; Zimmermann, A.; Denadai, S.; Lebossé, M.; Roche, S.; Geng, L.; et al. Deregulation of the Protocadherin Gene FAT1 Alters Muscle Shapes: Implications for the Pathogenesis of Facioscapulohumeral Dystrophy. PLoS Genet. 2013, 9, e1003550. [Google Scholar] [CrossRef] [Green Version]
- Saburi, S.; Hester, I.; Goodrich, L.; McNeill, H. Functional interactions between Fat family cadherins in tissue morphogenesis and planar polarity. Development 2012, 139, 1806–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, H.N.; Williamson, K.; Fitzpatrick, D.R. The genetic architecture of aniridia and Gillespie syndrome. Qual. Life Res. 2019, 138, 881–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragge, N.K.; Salt, A.; Collin, J.R.O.; Michalski, A.; Farndon, P.A. Gorlin syndrome: The PTCH gene links ocular developmental defects and tumour formation. Br. J. Ophthalmol. 2005, 89, 988–991. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, N.; Davis, E.E.; McKnight, K.L.; Niederriter, A.R.; Causse, A.; David, V.; Desmaison, A.; Lamarre, S.; Vincent-Delorme, C.; Pasquier, L.; et al. Targeted resequencing identifiesPTCH1as a major contributor to ocular developmental anomalies and extends the SOX2 regulatory network. Genome Res. 2016, 26, 474–485. [Google Scholar] [CrossRef] [Green Version]
- Bree, A.F.; Shah, M.R.; the BCNS Colloquium Group. Consensus statement from the first international colloquium on basal cell nevus syndrome (BCNS). Am. J. Med. Genet. Part A 2011, 155, 2091–2097. [Google Scholar] [CrossRef]
- Black, G.C.; Mazerolle, C.J.; Wang, Y.; Campsall, K.D.; Petrin, D.; Leonard, B.C.; Damji, K.F.; Evans, D.G.R.; McLeod, D.; Wallace, V.A. Abnormalities of the vitreoretinal interface caused by dysregulated Hedgehog signaling during retinal development. Hum. Mol. Genet. 2003, 12, 3269–3276. [Google Scholar] [CrossRef] [Green Version]
- Ågren, M.; Kogerman, P.; Kleman, M.I.; Wessling, M.; Toftgård, R. Expression of the PTCH1 tumor suppressor gene is regulated by alternative promoters and a single functional Gli-binding site. Gene 2004, 330, 101–114. [Google Scholar] [CrossRef]
- Hernando, C.; Plaja, A.; Rigola, M.A.; Pérez, M.M.; Vendrell, T.; Egocue, J.; Fuster, C. Comparative genomic hybridisation shows a partial de novo deletion 16p11.2 in a neonate with multiple congenital malformations. J. Med. Genet. 2002, 39, 24e. [Google Scholar] [CrossRef] [Green Version]
- Shinawi, M.; Liu, P.; Kang, S.-H.L.; Shen, J.; Belmont, J.W.; Scott, D.A.; Probst, F.J.; Craigen, W.J.; Graham, B.H.; Pursley, A.; et al. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J. Med. Genet. 2009, 47, 332–341. [Google Scholar] [CrossRef]
- Schaaf, C.P.; Goin-Kochel, R.P.; Nowell, K.P.; Hunter, J.V.; Aleck, K.A.; Cox, S.; Patel, A.; Bacino, C.A.; Shinawi, M. Expanding the clinical spectrum of the 16p11.2 chromosomal rearrangements: Three patients with syringomyelia. Eur. J. Hum. Genet. 2010, 19, 152–156. [Google Scholar] [CrossRef]
- Lin, S.; Shi, S.; Zhou, Y.; Ji, Y.; Huang, P.; Wu, J.; Chen, B.; Luo, Y. Intrauterine phenotypic features associated with 16p11.2 recurrent microdeletions. Prenat. Diagn. 2018, 38, 381–389. [Google Scholar] [CrossRef]
- Bardakjian, T.M.; Kwok, S.; Slavotinek, A.M.; Schneider, A.S. Clinical report of microphthalmia and optic nerve coloboma associated with a de novo microdeletion of chromosome 16p11.2. Am. J. Med. Genet. Part A 2010, 152, 3120–3123. [Google Scholar] [CrossRef]
- Graziano, C.; Gusson, E.; Severi, G.; Isidori, F.; Wischmeijer, A.; Brugnara, M.; Seri, M.; Rossi, C. A de novo PUF60 mutation in a child with a syndromic form of coloboma and persistent fetal vasculature. Ophthalmic Genet. 2017, 38, 590–592. [Google Scholar] [CrossRef]
- Low, K.J.; Ansari, M.; Jamra, R.A.; Clarke, A.; El Chehadeh, S.; Fitzpatrick, D.R.; Greenslade, M.; Henderson, A.; Hurst, J.; Keller, K.; et al. PUF60 variants cause a syndrome of ID, short stature, microcephaly, coloboma, craniofacial, cardiac, renal and spinal features. Eur. J. Hum. Genet. 2017, 25, 552–559. [Google Scholar] [CrossRef] [Green Version]
- Dauber, A.; Golzio, C.; Guenot, C.; Jodelka, F.M.; Kibaek, M.; Kjaergaard, S.; Leheup, B.; Martinet, D.; Nowaczyk, M.J.; Rosenfeld, J.A.; et al. SCRIB and PUF60 Are Primary Drivers of the Multisystemic Phenotypes of the 8q24.3 Copy-Number Variant. Am. J. Hum. Genet. 2013, 93, 798–811. [Google Scholar] [CrossRef] [Green Version]
- Demeulenaere, S.; Beysen, D.; De Veuster, I.; Reyniers, E.; Kooy, R.F.; Meuwissen, M.E. Novel BRPF1 mutation in a boy with intellectual disability, coloboma, facial nerve palsy and hypoplasia of the corpus callosum. Eur. J. Med. Genet. 2019, 62, 103691. [Google Scholar] [CrossRef]
- Mattioli, F.; Schaefer, E.; Magee, A.; Mark, P.R.; Mancini, G.M.; Dieterich, K.; Von Allmen, G.; Alders, M.; Coutton, C.; Van Slegtenhorst, M.; et al. Mutations in Histone Acetylase Modifier BRPF1 Cause an Autosomal-Dominant Form of Intellectual Disability with Associated Ptosis. Am. J. Hum. Genet. 2016, 100, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Yan, K.; Rousseau, J.; Littlejohn, R.O.; Kiss, C.; Lehman, A.; Rosenfeld, J.A.; Stumpel, C.T.; Stegmann, A.P.; Robak, L.; Scaglia, F.; et al. Mutations in the Chromatin Regulator Gene BRPF1 Cause Syndromic Intellectual Disability and Deficient Histone Acetylation. Am. J. Hum. Genet. 2017, 100, 91–104. [Google Scholar] [CrossRef] [Green Version]
- You, L.; Zou, J.; Zhao, H.; Bertos, N.R.; Park, M.; Wang, E.; Yang, X.-J. Deficiency of the Chromatin Regulator Brpf1 Causes Abnormal Brain Development. J. Biol. Chem. 2015, 290, 7114–7129. [Google Scholar] [CrossRef] [Green Version]
- Gago-Díaz, M.; Blanco-Verea, A.; Teixidó-Turà, G.; Valenzuela, I.; Del Campo, M.; Borregan, M.; Sobrino, B.; Amigo, J.; García-Dorado, D.; Evangelista, A.; et al. Whole exome sequencing for the identification of a new mutation in TGFB2 involved in a familial case of non-syndromic aortic disease. Clin. Chim. Acta 2014, 437, 88–92. [Google Scholar] [CrossRef]
- Lindsay, M.E.; Schepers, D.; Bolar, N.A.; Doyle, J.J.; Gallo, E.; Fert-Bober, J.; Kempers, M.J.; Fishman, E.K.; Chen, Y.; Myers, L.; et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat. Genet. 2012, 44, 922–927. [Google Scholar] [CrossRef]
- MacCarrick, G.; Black, J.H.; Bowdin, S.; El-Hamamsy, I.; Frischmeyer-Guerrerio, P.A.; Guerrerio, A.L.; Sponseller, P.D.; Loeys, B.; Dietz, H.C. Loeys–Dietz syndrome: A primer for diagnosis and management. Genet. Med. 2014, 16, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Knickmeyer, M.D.; Mateo, J.L.; Eckert, P.; Roussa, E.; Rahhal, B.; Zuniga, A.; Krieglstein, K.; Wittbrodt, J.; Heermann, S. TGFβ-facilitated optic fissure fusion and the role of bone morphogenetic protein antagonism. Open Biol. 2018, 8, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanford, L.P.; Ormsby, I.; De Groot, A.C.G.; Sariola, H.; Friedman, R.; Boivin, G.P.; Cardell, E.L.; Doetschman, T. TGFβ2 Knockout Mice Have Multiple Developmental Defects That Are Non-Overlapping with Other TGFβ Knockout Phenotypes. Development 1997, 124, 2659. [Google Scholar] [PubMed]
- Harding, P.; Brooks, B.P.; Fitzpatrick, D.; Moosajee, M. Anophthalmia including next-generation sequencing-based approaches. Eur. J. Hum. Genet. 2019, 28, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.S.; Fitzpatrick, D.R. Anophthalmia and microphthalmia. Orphanet J. Rare Dis. 2007, 2, 47. [Google Scholar] [CrossRef] [Green Version]
- Chassaing, N.; Causse, A.; Vigouroux, A.; Delahaye-Duriez, A.; Alessandri, J.-L.; Boespflug-Tanguy, O.; Boute-Benejean, O.; Dollfus, H.; Duban-Bedu, B.; Gilbert-Dussardier, B.; et al. Molecular findings and clinical data in a cohort of 150 patients with anophthalmia/microphthalmia. Clin. Genet. 2013, 86, 326–334. [Google Scholar] [CrossRef]
- Deml, B.; Reis, L.M.; Lemyre, E.; Clark, R.D.; Kariminejad, A.; Semina, E.V. Novel mutations in PAX6, OTX2 and NDP in anophthalmia, microphthalmia and coloboma. Eur. J. Hum. Genet. 2015, 24, 535–541. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Rodriguez, J.; Pelcastre, E.L.; Tovilla-Canales, J.L.; Garcia-Ortiz, J.E.; Amato-Almanza, M.; Villanueva-Mendoza, C.; Espinosa-Mattar, Z.; Zenteno, J.C. Mutational screening of CHX10, GDF6, OTX2, RAX and SOX2 genes in 50 unrelated microphthalmia-anophthalmia-coloboma (MAC) spectrum cases. Br. J. Ophthalmol. 2010, 94, 1100–1104. [Google Scholar] [CrossRef]
- Jimenez, N.L.; Flannick, J.; Yahyavi, M.; Li, J.; Bardakjian, T.M.; Tonkin, L.; Schneider, A.; Sherr, E.; Slavotinek, A.M. Targeted ’Next-Generation’ sequencing in anophthalmia and microphthalmia patients confirms SOX2, OTX2 and FOXE3 mutations. BMC Med. Genet. 2011, 12, 172. [Google Scholar] [CrossRef] [Green Version]
- Slavotinek, A.M.; Garcia, S.T.; Chandratillake, G.; Bardakjian, T.; Ullah, E.; Wu, D.; Umeda, K.; Lao, R.; Tang, P.L.-F.; Wan, E.; et al. Exome sequencing in 32 patients with anophthalmia/microphthalmia and developmental eye defects. Clin. Genet. 2015, 88, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Matías-Pérez, D.; García-Montaño, L.A.; Cruz-Aguilar, M.; García-Montalvo, I.A.; Nava-Valdéz, J.; Barragán-Arevalo, T.; Villanueva-Mendoza, C.; Villarroel, C.; Guadarrama-Vallejo, C.; La Cruz, R.V.-D.; et al. Identification of novel pathogenic variants and novel gene-phenotype correlations in Mexican subjects with microphthalmia and/or anophthalmia by next-generation sequencing. J. Hum. Genet. 2018, 63, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Burgess, A.; Shah, K.; Hough, O.; Hynynen, K. Endothelial Cell Metabolism in Normal and Diseased Vasculature. Circ. Res. 2016, 116, 1231. [Google Scholar] [CrossRef] [Green Version]
- Rehm, H.L.; Bale, S.J.; Bayrak-Toydemir, P.; Berg, J.S.; Brown, K.K.; Deignan, J.L.; Friez, M.J.; Funke, B.H.; Hegde, M.R.; Lyon, E. ACMG clinical laboratory standards for next-generation sequencing. Genet. Med. 2013, 15, 733–747. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID † | Sex | Age (yrs) at Examination | Origin ‡ | VA | Coloboma | Microphthalmia | Additional Ocular Anomalies | Extraocular Phenotype | |
|---|---|---|---|---|---|---|---|---|---|
| OD | OS | ||||||||
| 1[III:3] | f | 2 | Portugal, Poland | 0.5/0.25 | NA | I | NA | None | None |
| 1[II:3] | m | 45 | Portugal | 0.1/NLP | IRCh | IRCh | NA | Microcornea (OU) | None |
| 1[I:1] | m | 75 | Portugal | NA | I | NA | NA | Microcornea (OD) | None |
| 2[II:1] | m | 14 | Netherlands | 0.6/0.6 | RCh | RCh | NA | Megalocornea (OD) | SNHL, DORV, VSD, PDA, DD, dysmorphic features |
| 3[II:1] | f | 17 | Switzerland | 0.2/0.6 | IRCh | NA | NA | Anterior polar cataract (OU) | Syndactyly, hearing impairment |
| 4[II:2] | f | 30 | Italy | NLP/0.8 | RCh | RCh | NA | Axenfeld-Rieger anomaly (OU) | None |
| 4[III:1] | f | 0.75 | Italy | 0.25/0.25 | I | IRCh | NA | None | VSD, clinodactyly |
| 4[II:3] | f | 24 | Italy | 1.0/1.0 | NA | NA | NA | Goniodysgenesis (OU) | None |
| 4[I:2] | f | 56 | Italy | 0.6./0.8 | NA | NA | NA | None | Uterine fibroids, keratocystic lesions |
| 5[II:1] | f | 7 | Switzerland, Germany | NLP/0.5 | IRCh | RCh | OD | None | DD, ASD, short stature |
| 6[II:1] | m | 2 | Portugal | 0.16/0.1 | IRCh | IRCh | OS | None | None |
| 7[II:1] | f | 13 | India | 0.5/NLP | RCh a | NA | OS | None | Aortic root enlargement |
| 8[II:1] | f | 12 | Spain | 0.6/1.0 | I | I | NA | Axenfeld-Rieger spectrum (OU) | Tooth displacement |
| 9[II:2] | f | 33 | Switzerland | 0.1/0.1 | RCh | RCh | OU | None | None |
| 9[III:1] | m | 6 | Switzerland | LP/NLP | RCh | RCh | OU | None | None |
| 10[II:1] | f | 3 | Switzerland, Germany | 0.16/0.06 | IRCh | IRCh | OS | None | None |
| 11[II:2] | f | 4 | Switzerland, Belgium | 0.6/0.8 | I | I | NA | Cataracta corticonuclearis (initial partial inferonasal, at age 4 years complete; OD) | None |
| 12[II:2] | f | 2 | Switzerland | 0.05/0.08 | IRCh | IRCh | NA | None | None |
| 13[IV:4] | f | 9 | Switzerland, Austria | 0.05/NLP | IRCh | NA | OS | None | Clinodactyly, mild pigeon toe, mild protruding ears |
| 14[II:1] | m | 19 | Switzerland | 0.4/0.05 | RCh | RCh | NA | None | None |
| 15[II:1] | f | 13 | Italy | 0.4/0.8 | RCh | RCh | NA | None | None |
| ID † | Gene | Gene Function | Reference Sequence | Sequence Variant (hg19) | Predicted Protein Change | Region/Size | gnomAD | Zygosity | ACMG | CADD | First Report |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1[III:3] | CRIM1 | Tether for growth factors, complexes with ß-catenin and cadherins | hg19 | g.36769283_36778290del a | NA | 9,008 bp | NA | het | NA | NA | This study |
| 2[II:1] | CHD7 | Chromatin remodeling | NM_017780.3 | c.2095A>G b | p.Ser699Gly e | exon 3 | 0% | het | vus | 15.4 | [26,31,32,33] |
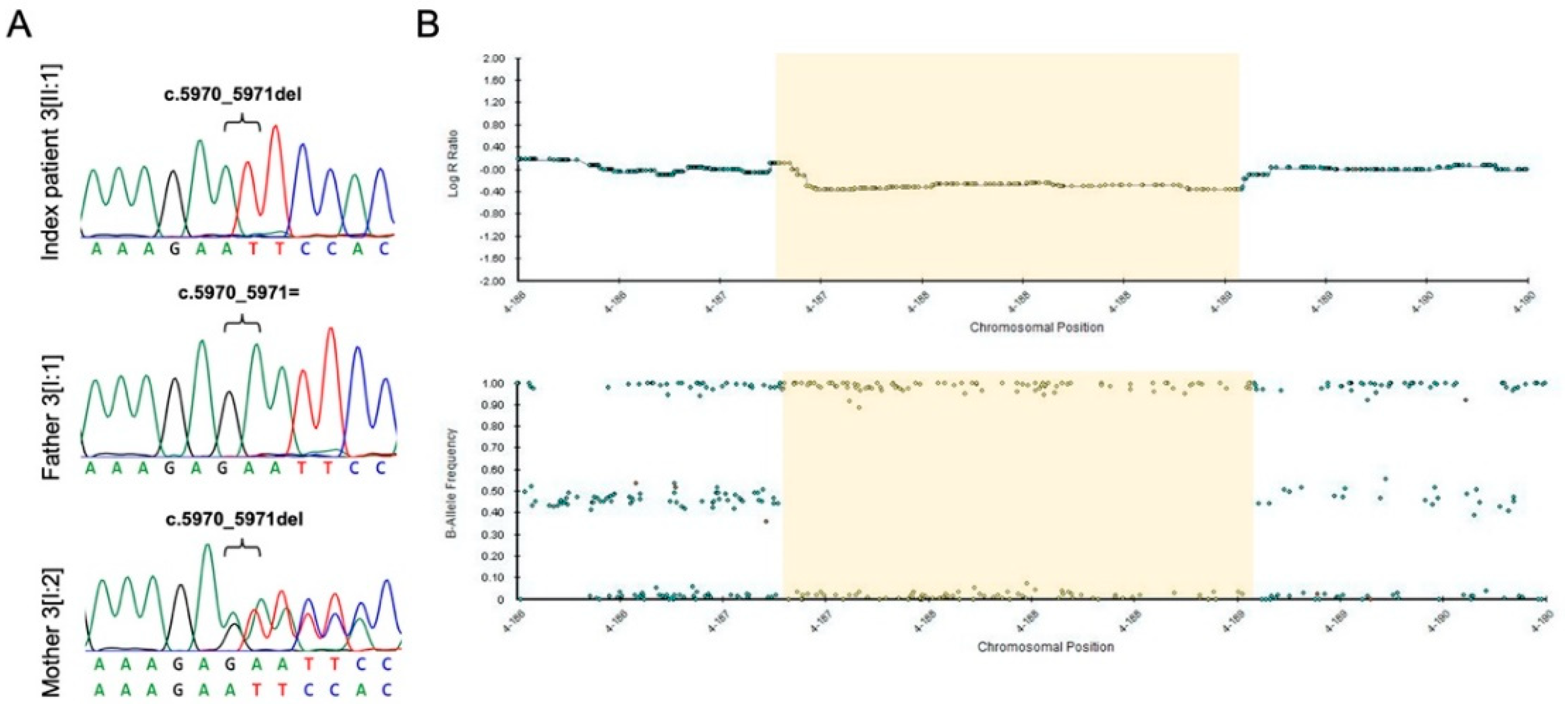
| 3[II:1] | FAT1 | Cell polarity, cell migration, cell–cell adhesion | NM_005245.3 | c.5970_5971delc | p.Asn1991PhefsTer19 | exon 10 | 0% | hemi | vus | NA | This study |
| FAT1, F11, MTNR1A, ZFP42 | hg19 | g.(187179210_187179486_188926200_189012426)del a | NA | 1.8 Mb | NA | het | NA | NA | This study | ||
| 4[II:2] | PTCH1 | Hedgehog receptor | NM_000264.4 | c.490G>A c | p.Glu164Lys | exon 3 | 0% | het | vus | 18.1 | This study |
| 5[II:1] | PUF60 | Transcriptional regulation, pre-mRNA splicing, apoptosis | NM_001136033.2 | c.752dup b | p.Gln252ProfsTer152 | exon 9 | 0% | het | P | NA | This study |
| 6[II:1] | BRPF1 | Chromatin regulator | NM_001003694.1 | c.1756_1757insT b | p.Glu586ValfsTer12 | exon 5 | 0% | het | P | NA | This study |
| 7[II:1] | TGFB2 | Growth factor | NM_001135599.3 | c.1043G>A d | p.Arg348His | exon 7 | 0.00082% | het | LP | 35 | This study |
| ID † | Sex | Gene | Reference Sequence | Sequence Variant | Predicted Protein Change | Region | gnomAD | Zygosity | ACMG | CADD | Segregation |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1[III:3] | f | CRIM1 | NM_016441.2 | c.926C>T | p.Pro309Leu | exon 5 | 0% | het | LB | 25 | paternal |
| 1[II:3] | m | paternal | |||||||||
| 1[I:1] | m | NA | |||||||||
| 1[III:1] | f | BRPF1 | NM_001003694.1 | c.1489G>A | p.Ala497Thr | exon 3 | 0% | het | vus | 25 | maternal |
| 3[II:1] | f | TBX5 | NM_000192.3 | c.349G>T | p.Ala117Ser | exon 4 | 0% | het | vus | 24.7 | maternal |
| FZD7 | NM_003507.1 | c.1154C>T | p.Ala385Val | exon 1 | 0.0004% | het | vus | 32 | maternal | ||
| 4[II:2] | f | PPP1R12A | NM_001143885.1 | c.2014C>G | p.Pro672Ala | exon 15 | 0% | het | vus | 24 | paternal |
| 4[III:1] | f | maternal | |||||||||
| 7[II:1] | f | ACTG1 | NM_001199954.1 | c.803-18dup | p.? | intron 4 | 0% | het | vus | NA | NA |
| 8[II:1] | f | EFTUD2 | NM_001142605.1 | c.765-15C>G | p.? | intron 10 | 0% | het | vus | NA | paternal |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haug, P.; Koller, S.; Maggi, J.; Lang, E.; Feil, S.; Wlodarczyk, A.; Bähr, L.; Steindl, K.; Rohrbach, M.; Gerth-Kahlert, C.; et al. Whole Exome Sequencing in Coloboma/Microphthalmia: Identification of Novel and Recurrent Variants in Seven Genes. Genes 2021, 12, 65. https://doi.org/10.3390/genes12010065
Haug P, Koller S, Maggi J, Lang E, Feil S, Wlodarczyk A, Bähr L, Steindl K, Rohrbach M, Gerth-Kahlert C, et al. Whole Exome Sequencing in Coloboma/Microphthalmia: Identification of Novel and Recurrent Variants in Seven Genes. Genes. 2021; 12(1):65. https://doi.org/10.3390/genes12010065
Chicago/Turabian StyleHaug, Patricia, Samuel Koller, Jordi Maggi, Elena Lang, Silke Feil, Agnès Wlodarczyk, Luzy Bähr, Katharina Steindl, Marianne Rohrbach, Christina Gerth-Kahlert, and et al. 2021. "Whole Exome Sequencing in Coloboma/Microphthalmia: Identification of Novel and Recurrent Variants in Seven Genes" Genes 12, no. 1: 65. https://doi.org/10.3390/genes12010065