Sequence Transpositions Restore Genes on the Highly Degenerated W Chromosomes of Songbirds


1
Department of Neurosciences and Developmental Biology, University of Vienna, 1090 Vienna, Austria
2
Department of Bioinformatics and Genetics, Swedish Museum of Natural History, 104 05 Stockholm, Sweden
3
MOE Laboratory of Biosystems Homeostasis & Protection, Life Sciences Institute, Zhejiang University, Hangzhou 310012, China
4
Center for Reproductive Medicine, The 2nd Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou 310012, China
*
Author to whom correspondence should be addressed.
Genes 2020, 11(11), 1267; https://doi.org/10.3390/genes11111267
Submission received: 23 September 2020
/
Revised: 15 October 2020
/
Accepted: 22 October 2020
/
Published: 28 October 2020
(This article belongs to the Special Issue Role of Gene Conversion in the Evolution of Sex Chromosomes)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The female-specific W chromosomes of most Neognathae birds are highly degenerated and gene-poor. Previous studies have demonstrated that the gene repertoires of the Neognathae bird W chromosomes, despite being in small numbers, are conserved across bird species, likely due to purifying selection maintaining the regulatory and dosage-sensitive genes. Here we report the discovery of DNA-based sequence duplications from the Z to the W chromosome in birds-of-paradise (Paradisaeidae, Passeriformes), through sequence transposition. The original transposition involved nine genes, but only two of them (ANXA1 and ALDH1A1) survived on the W chromosomes. Both ANXA1 and ALDH1A1 are predicted to be dosage-sensitive, and the expression of ANXA1 is restricted to ovaries in all the investigated birds. These analyses suggest the newly transposed gene onto the W chromosomes can be favored for their role in restoring dosage imbalance or through female-specific selection. After examining seven additional songbird genomes, we further identified five other transposed genes on the W chromosomes of Darwin’s finches and one in the great tit, expanding the observation of the Z-to-W transpositions to a larger range of bird species, but not all transposed genes exhibit dosage-sensitivity or ovary-biased expression We demonstrate a new mechanism by which the highly degenerated W chromosomes of songbirds can acquire genes from the homologous Z chromosomes, but further functional investigations are needed to validate the evolutionary forces underlying the transpositions.
1. Introduction
Unlike mammals, birds have a female heterogametic (ZW) sex chromosome system, i.e., females have a female-specific W chromosome and a Z chromosome, whereas males have two Z chromosomes. Similar to the mammalian Y chromosomes, the avian W chromosomes are highly degenerated and gene-poor, except for palaeognathous birds [1,2], suggesting convergent sex chromosome degeneration [3]. The degeneration of the W chromosomes makes the Z chromosome hemizygous throughout most of its length in females, with only a handful of genes surviving on the W chromosomes (those homologous ZW gene-pairs are called gametologs) [4,5]. For instance, a typical Z chromosome of Neognathae birds has more than 700 genes, but the W chromosome has only ~40 intact genes; in other words, more than 90% of the original gene content has been eliminated from the W chromosomes [5,6].
Characterization of the survivor genes on the Y and W chromosomes, therefore, is useful to elucidate the evolutionary forces that govern the retention of genes on the decaying sex chromosomes. Previous studies on chicken [7] and songbird [6] W chromosomes suggest a common force that shapes the gene content of the W chromosome, that is, purifying selection retaining the dosage-sensitive or housekeeping genes. This is consistent with what has been found for the mammalian Y chromosomes [8] and snake W chromosomes [9], though other genetic constraints on the mammalian Y chromosome have been recently proposed [10]. For the avian W chromosome, however, the strength of purifying selection on dosage-sensitive genes is perhaps stronger because the global dosage compensation is lacking or incomplete in birds [11,12].
In many organisms, however, the gene content of the Y chromosomes is more than simply the outcome of degeneration from the proto-Y chromosome. For instance, most of the Drosophila Y-linked genes were duplicated from the autosomes and are testis-specific [13,14], through DNA-based transposition [15] or RNA-based retrotransposition [16], likely driven by male-specific selection. Recruitment of genes into the Y chromosome through retrotransposons [17,18,19] and X-linked transposition [20,21,22,23,24] have also been widely reported across mammalian species. The Y-borne genes often massively increase their gene copies through amplification, which has been documented in both mice [25] and Drosophila [26]. The amplification can occur independently in multiple mammalian and Drosophila lineages, resulting in notable differences in gene content of the Y chromosomes even between closely related species [27,28,29]. Additionally, the genetic information of the Y chromosomes can sometimes be influenced by X-Y gene conversion by which the coding sequence of the Y-linked genes can be rewritten using that of the X-linked copy, retarding the genetic decay of the Y-linked genes. This evolutionary mechanism has been observed across a broad range of mammalian species [30,31,32]. Finally, gene conversion can also happen between the inverted repeats (palindromic sequences) of the Y chromosome, and such intra-chromosome gene conversion has been frequently found to maintain the intactness of coding genes within the palindromes [33,34]. Those observations suggest the action of multiple evolutionary forces that impact the gene content of the Y chromosome, with the male-specific selection as the dominant one.
On the contrary, previous studies suggested that the avian W chromosomes have relatively more conserved and evolutionarily stable gene content across Neognathae [6,7], covering the majority of bird species. This is partly because Neognathae shares a large portion (~50%) of non-recombining regions along the sex chromosomes and the majority of original genes had been deleted prior to species radiations [6]. Despite the overall conservation, the alteration of gene content on the avian W chromosomes has also been observed in a similar way as in Drosophila and mammals, with some differences. For example, HINT1 gene has been amplified on the chicken W chromosomes [7], though the copy number is limited to two or three in songbirds [6], and its expression does not seem to be ovary-biased [5,7]. In a rare case, an autosome-derived retrotransposed gene NARF has been found on the W chromosome of a crow [6], but it is not a gene with female-biased expression. To date, while intra-chromosome gene conversion seems to have occurred in New World sparrows and blackbirds [35], no transposition or Z-W gene conversion has been documented in bird sex chromosomes. It has been demonstrated that female-specific selection can drive the expression upregulation of W-linked genes in chicken breeds [36], but it is unclear whether it can recruit new genes into the W chromosomes. In this study, we report transpositions from the Z to W chromosomes across three lineages of songbirds that resulted in the restoration of several once-lost gametologs onto the W chromosomes. We found the transposition of at least one gene (ANXA1) was likely favored by female-specific selection, and more than half of the transposed genes were predicted to be dosage-sensitive; therefore, their restoration onto the W chromosome may help resolve the gene dosage imbalance.
2. Materials and Methods
2.1. Identification of Z-Linked Transpositions
The genomic and resequencing data used in this study are listed in Supplementary Tables S1 and S2. For the 12 songbird genomes, genomic data are available for both sexes except for three species. We first used the published Z chromosome sequence of great tit [37] to identify and order the Z-linked sequences among the investigated species. To calculate the sequencing coverage, we mapped the reads to the reference genomes using BWA-MEM (0.7.16a-r1181) with default parameters. We used the “depth” function in samtools (1.9) [38] to calculate coverage for every nucleotide site, subsequently removed those sites with mapping quality (-Q) lower than 60 or depth 3 times higher than average. Then we calculated genomic coverage of every 50 kb sliding window by using the “bedtools map” function. Any windows with less than 60% of the region (30 kb) mapped by reads were excluded. We used the GATK (3.8.0) [39] pipeline (HaplotypeCaller) to call variants. Raw variants were filtered by these criteria:—window 10—cluster 2 “FS > 10.0”, “QD < 2.0”, “MQ < 50.0”, “SOR > 1.5”, “MQRankSum < −1.5”, “ReadPosamplenkSum < −8.0”. We further required the variants showing an allele frequency between 0.3 and 07 (the expected heterozygosity should be 0.5 for one individual, but we allowed for some variation). The SNP density was defined by the number of SNPs over a 50 kb window.
Because the transpositions were inferred to be evolutionarily young (based on the inferred ages) and the W-derived reads can perfectly map to the Z-linked sequences, we used the called variants (both SNP and indels) from female reads to create the pseudo-sequence of the W-linked ZTR. To do so, we applied the FastaAlternateReferenceMaker tool from the GATK package. The gene models on the W chromosomes were then predicted by genewise (2.4.1) [40] with the protein sequences of the Z-linked homologs. To remove potential chimeric W-derived alleles in the Z-linked regions (due to the collapse of genome assembly), if any, we used male sequencing reads to polish the Z-linked sequence using pilon (1.22) [41].
To verify the identified ZTR in the great tit, we randomly chose 10 sequence fragments containing multiple SNP sites for designing primers and performed PCR amplification. The information about PCR primers were listed in Supplementary Table S3. The PCR products were purified and sequenced at Macrogen (Macrogen, Amsterdam, The Netherlands, Europe B.V.).
2.2. Synonymous and Nonsynonymous Substitution Rate
We aligned the Z- and W-linked ZTR genes of interests and several outgroups species (Supplementary Table S1) using the GUIDANCE2 (2.02) [42] pipeline. PRANK (170427) [43] was used as the aligner. Then we used the codeml tool from the PAML package (4.9e) [44] to estimate the substitution rate. We used the codeml model that allowed the substitution rate to vary among the branches.
2.3. Haploinsufficiency Score
We measured the probability of haploinsufficiency of avian genes, based on the published haploinsufficiency scores [45] of their human orthologs. Haploinsufficiency score is defined as the sufficiency of one single copy of genes to accomplish normal gene functions. Huang et al. predicted haploinsufficiency score for each human gene, based on known haploinsufficient genes identified from disease studies, and haploinsufficient genes having copy number variations among healthy human individuals.
2.4. Gene Expression Analyses
We used the program RSEM [46] (1.3.0) pipeline to estimate gene expression levels. We downloaded the RNA-seq data from the great tit [37], blue tit [47], collared flycatcher [48], rock pigeon [49,50], guineafowl [51], goose [51], duck [51], turkey [51], chicken [52], Chilean tinamou [1] and green anole [53], across tissues of both sexes (gonad, brain, liver, spleen and heart) when available. The accession numbers are listed in the Supplementary Table S4. When biological replicates were present, we downloaded RNA-seq data of all replicates, and the expression levels were calculated as the mean over replicates. The details of the methods of RNA-seq read mapping and expression level quantification were described in [6].
2.5. Data Accessibility
The published genomes and sequencing reads are listed in Supplementary Tables S1, S2 and S4. Codes have been deposited at GitHub [54].
3. Results
3.1. Discovery and Characterization of Sequence Transposition in Birds-of-Paradise
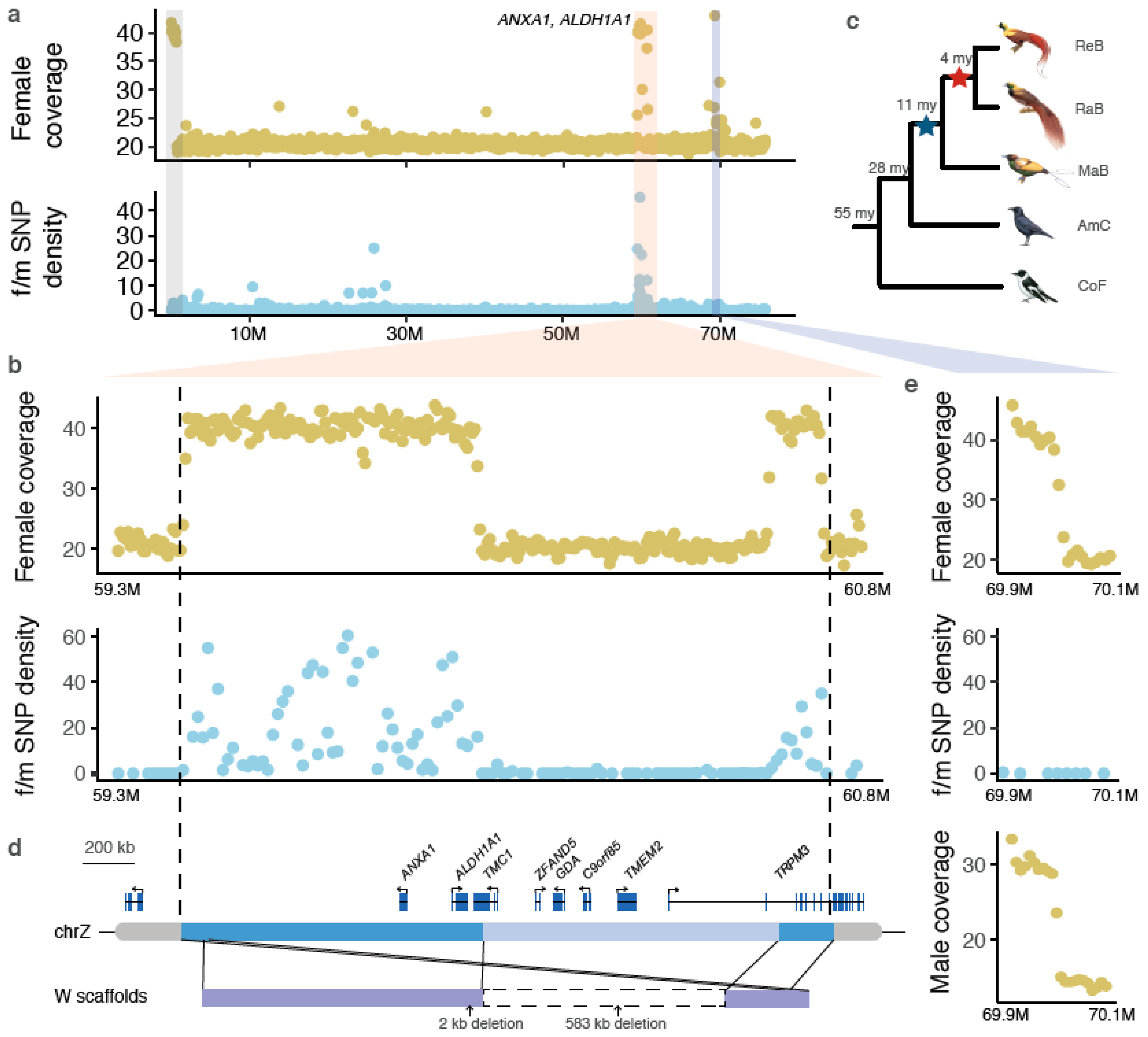
Because of the nearly complete degeneration of the W chromosomes in Neognathae birds (including songbirds), the Z chromosomes (except for the pseudoautosomal region, or PAR) are expected to be hemizygous and to have a reduced sequencing coverage in females. However, we found that one region residing at ~60 Mb of the Z chromosome of red bird-of-paradise (BOP, family Paradisaeidae, order Passeriformes) showed a female coverage level similar to that of autosomes or the PAR (Figure 1a,b). In this region, female reads also show a remarkably higher level of heterozygosity than male reads, indicating recent Z-W sequence divergence (Figure 1a,b, Supplementary Figure S1). This excludes the possibility of a Z-to-autosome translocation that might explain the autosome-like coverage pattern, because sex difference in heterozygosity is not expected for autosomes where the homologous chromosomes can freely recombine. We further excluded the possibility of Z-linked duplication, because we only found the doubled coverage in females but not in males (Supplementary Figure S2). Finally, the genomic scaffold that harbors this entire region shows highly conserved gene syntenies with the Z chromosome sequences of other songbirds, suggesting sequence assembly of this region is unlikely to be erroneous (Supplementary Figure S3). The remarkably high heterozygosity in females, equal coverage between males and females and the conserved synteny together suggest there is likely a new copy of this region recently transposed from the Z to the W chromosome followed by rapid Z-W divergence. Those patterns were observed in two Paradisaea BOP, but not in other BOP genera (Supplementary Figure S4), suggesting the origin of the Z-to-W transposition was prior to the divergence of Paradisaea BOP 4 million years ago (mya) (Figure 1c). Because the duplicated region involved a large genomic sequence spanning several complete genes (Figure 1d), the duplicative sequence movement should represent a result of DNA-based transposition rather than RNA-based retrotransposition. We named the W-linked region that is derived from the Z chromosome as the Z-transposed region (ZTR), following the naming of previously reported XTR (X-transposed region) in mammals.
It is noteworthy that our analysis also uncovered a recent duplication on the Z chromosome at ~70 Mb position (Figure 1a). It is different from ZTR in that male reads also showed increased coverage (Figure 1e). This inferred duplicated region (about 100 kb long) spans one gene YTHDC2 and exhibits similar levels of read coverage and heterozygosity between males and females (Figure 1e). Similar to the case of transpositions, only one copy was assembled with short-reads because of the low sequence divergence between the two duplicated copies. Therefore, we rely on the read coverage to infer the increase of copy number. A recent long-read genome assembly of a basal BOP (Lycocorax pyrrhopterus) [55] has successfully assembled the two copies of this sequence (data not shown), suggesting the existence of this duplication and dating the duplication event further back to all BOPs (Figure 1c).
The newly duplicated sequence on the W chromosome is subject to decay upon its arrival on the W because of the absence of recombination. The length of the W-linked ZTR is 0.7 Mb, shorter than the parental Z-linked sequence (1.3 Mb), primarily due to a large deletion (583 kb) (Figure 1d). This large deletion removed five complete (ZFAND5, TRPM3, TMEM2, GDA, C9orf85) and 2 partial (TMC1 and TRPM3) genes on the W-linked ZTR, accounting for 78% (7 out of 9) of the originally transposed genes (Figure 1d). Apart from that, we detected only two additional small sequence deletions (Supplementary Figure S5). The rapid sequence loss on the W chromosome is consistent with the general mode of sex chromosome degeneration at the early stage [6]. However, we did not detect any TE insertions on the W-linked ZTR.
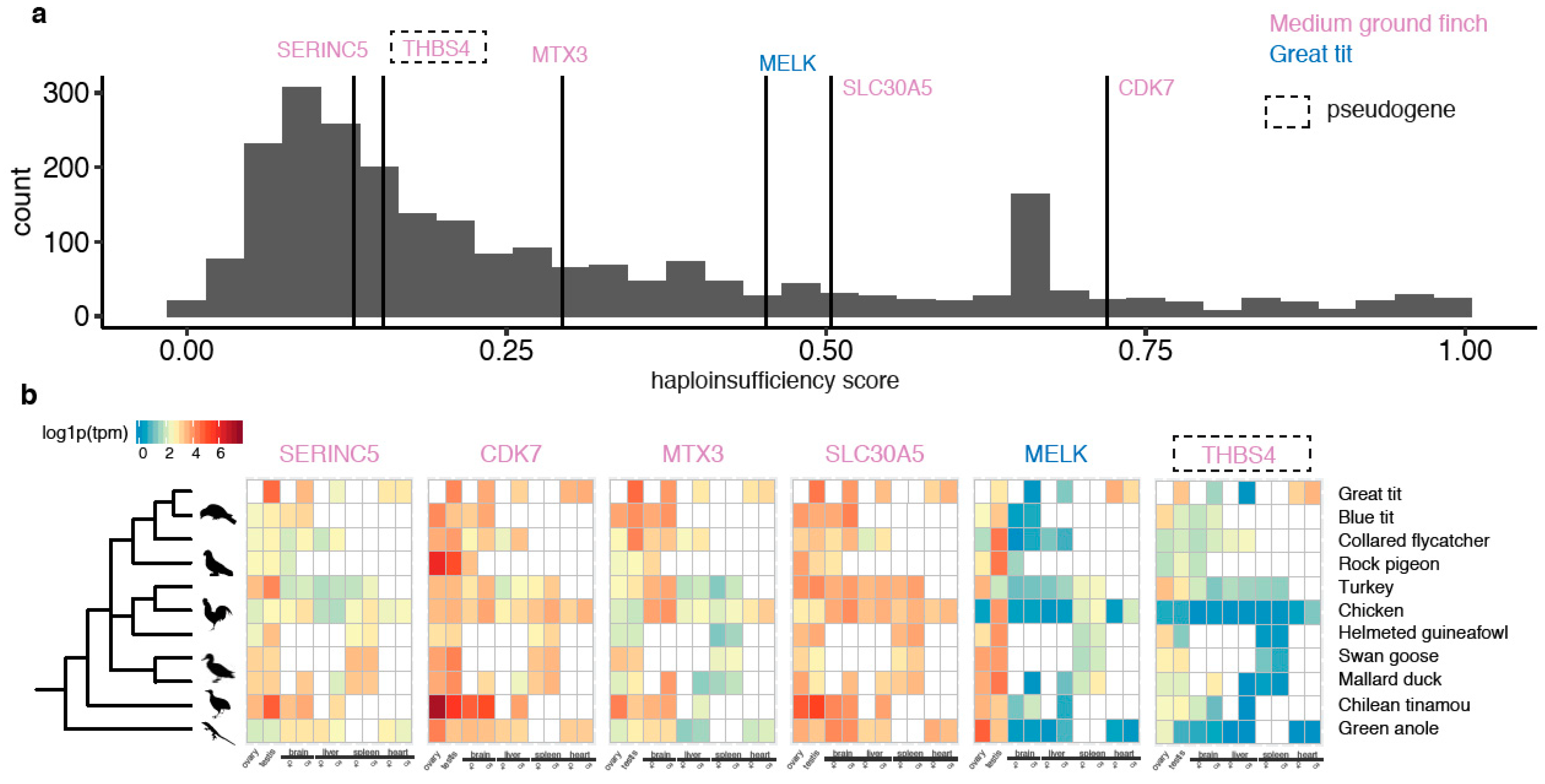
The retained W-linked ZTR contains only two intact genes: ANXA1 and ALDH1A1 (Figure 1d). Interestingly, both genes have high predicted haploinsufficiency scores (extrapolated from their human orthologs [45]), i.e., high dosage sensitivity (Figure 2a). While ALDH1A1 shows a relatively relaxed evolutionary constraint measured by the ratio of nonsynonymous over synonymous substitution rates, ANXA1 seems to be under strong purifying selection without a single nonsynonymous mutation in the W-linked copy (Figure 2b, Supplementary Table S5). In the absence of transcriptome data of BOPs, we used the RNA-seq data from other birds to infer the expression profiles of ANXA1 and ALDH1A1 prior to the transposition event in BOP. We found ALDH1A1 has a broad and non-sex-biased expression pattern, similar to other W-linked gametologs [6,7], but ANXA1 has a highly biased expression in ovaries, in all birds investigated here and a reptile (green anole lizard) (Figure 2c). Additionally, the ovary-biased expression of ANXA1 is only seen in later developmental stages of females (Supplementary Figure S6). Together, these results suggest the new copy of ANXA1 transposed onto the W chromosome is under strong purifying selection in females, potentially associated with its functional role in mature ovaries.
3.2. Identification of Sequence Transposition in Additional Songbird Lineages
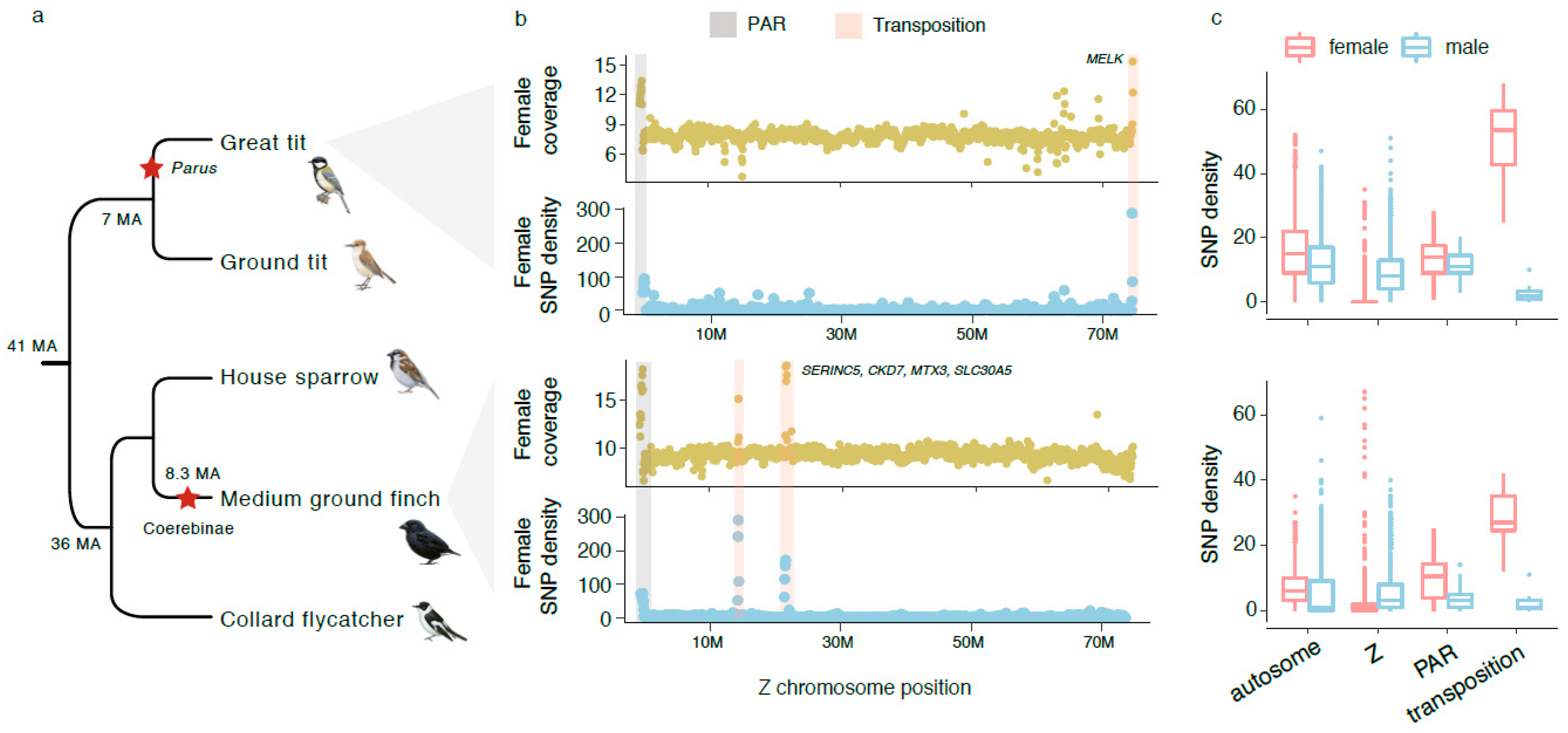
Having discovered the ZTR in BOPs, we set out to screen for signals of ZTR in other songbirds where both male and female sequencing reads are available and the Z-linked sequences have been previously identified [6]. Out of the five songbird species that we examined, we detected ZTRs in two species: medium ground finch and great tit (Figure 3). There are two ZTRs in medium ground finch, spanning about 100 and 200 kb of Z-linked homologous regions respectively, but only the second ZTR contains protein-coding genes (SERINC5, CKD7, MTX3, SLC30A5, THBS4) (Figure 3b). None of these five genes have been deleted on the W chromosome, but THBS4 has probably become a pseudogene due to frameshift mutations (Supplementary Figure S7). By examining other Darwin’s finches and their relatives, we inferred that the ZTRs occurred and were fixed at the ancestor of Coerebinae (Darwin’s finches and their closest relatives) 8.3 mya, because the ZTRs are present in all Coerebinae species but not in Sporophilinae (Supplementary Figure S8). In great tit, the ZTR is located at the end of the Z chromosome, about 50 kb long, and contains one single gene MELK (Figure 3b). This ZTR is also present in another Parus species (Supplementary Figure S9), so it is likely that it has been fixed at the common ancestor of Parus. To further assess whether the ZTR is fixed in the great tit population, we randomly genotyped 10 fragments with 7 in the ZTR and 3 in the nearby regions for 6 female and 6 male great tit individuals, and found in the ZTR the Z-W divergence sites were all heterozygous in females but homozygous in male (Supplementary Figure S10), consistent with the pattern of Z-W divergence between the ZTR and its parental Z-linked copy.
Together with the two survivor genes in BOPs, we discovered in total seven novel W-linked genes in songbirds due to Z-to-W transpositions. Like ANXA1 and ALDH1A1, CDK7 and SLC30A5 of medium ground finch and MELK of great tit are predicted to be dosage-sensitive relative to other Z-linked genes (Figure 4a). CDK7 and SLC30A5 are also broadly expressed at a relatively high level, but the expression of MELK seems to be biased in the gonads. The other two functional genes SERINC5 and MTX3 do not seem to be dosage-sensitive but are broadly expressed.
4. Discussion
In this study, we present the discovery and characterization of DNA-based sequence transpositions from the Z to the W chromosome in songbirds. In some other female heterogametic organisms including Lepidoptera [56] and plants [57,58], gene movement into the W chromosomes have been reported in recent years but not in birds. The bird genomes and chromosomes are characteristic for their evolutionary stasis with infrequent chromosomal rearrangements and gene movement [4,59]; consistently, the sex chromosomes and the gene content on the W chromosome remain stable over tens of million years’ evolution in birds [60]. The discovery of transpositions from the Z to W chromosome uncovers a new source from which the bird W chromosomes may regain once-lost genes.
Because population genetic data of BOPs is currently not available, we were only able to verify the transpositions using a molecular approach in the great tit. However, given the similar coverage and SNP density patterns exhibited by the ZTRs between the great tit and BOPs, it is reasonable to suggest the detected ZTR in BOPs is likely not an artifact. It needs to be noted that we have not directly assembled the sequences of W-linked ZTR genes, but because the transposition events are evolutionarily young (according to the ages we inferred) which allows the W-derived reads to map to the Z-linked sequences, we were able to infer the increase of copy number and the present of a new copy on the W chromosome. Our approach is not compatible with detecting ancient transposition events, if any. However, when the sequence of the W chromosome becomes available, the existence of ancient transposition can be directly tested. On the other hand, if ancient transposition events indeed existed, it raises the possibility that some of the gametologs might be the transposed (secondarily acquired) instead of the retained proto-W genes, particularly for those that are species- or lineage-specific. This can be detected through phylogenetic analyses on the Z and W gametologs [61,62], but it is beyond the scope of this study.
Undoubtedly, occasional transpositions are not expected to alter the evolutionary fate of the W chromosomes in Neognathae birds that are already gene-poor and highly degenerated, accompanied by massive accumulation of repeats [55,63]. The degenerative divergence of the W chromosome has made it distinct from the Z chromosome at both cytogenetic and sequence levels [4,6,64]. Additionally, when Z-to-W sequence transpositions occur, at a local scale, the newly arrived sequence onto the W chromosome is immediately subject to decay due to the lack of recombination. However, the duplicative transpositions give rise to intact genes on the W chromosomes, a result similar to gene conversion between the homologous sex chromosomes [30,31,32]. Importantly, when the transposed genes are beneficial to female individuals and when the female-specific selection is sufficiently strong, the transposed genes may survive in the “harsh” environment of the W chromosomes. Alternatively, the fixation of the transposed genes is simply because of genetic drift. Further investigations, including direct assembly of the W-linked ZTRs using long-read sequencing technology, and transcriptome and epigenetic profiling for the transposed genes in female organs, will provide more insights into the functional relevance of the ZTRs.
5. Conclusions
Among the 12 songbird genomes we investigated, five (in three lineages) exhibit signatures of sequence transposition from the Z to W chromosomes. The duplicative transposition is DNA based, and in all three bird lineages the transpositions led to regains of genes that had been deleted on the W chromosomes. Together, 14 different genes in the three lineages experienced Z-to-W duplications, with seven of them still functional on the W chromosomes. However, only one of them (ANXA1) seems to beneficial to females, therefore its fixation was likely associated with female-specific selection. The other transposed genes might be fixed in the female populations due to the need to restore gene-dosage balance.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4425/11/11/1267/s1, Figure S1: The elevated female SNP density of the ZTR. Figure S2: The coverage and SNP density in both female and male of red bird-of-paradise. Figure S3: Synteny of transposed sequences. Figure S4: Z-to-W transposition of red bird-of-paradise originated at the ancestor of Paradisaea. Figure S5: A W-linked 2-kb deletion in red bird-of-paradise transposed region. Figure S6: The expression of ANXA1 over different developmental stages of the gonads. Figure S7: Two frame shift mutations of THBS4 on the W-linked transposed sequence. Figure S8: Shared Z-to-W transposition across Darwin’s finches and their close relatives. Figure S9: The Z-to-W transposition in great tit is shared by green backed tit, but not ground tit. Figre S10: Verification of female-specific SNPs in the Z-transposed regions in great tit. Table S1: Accessions of NCBI genome assemblies. Table S2: Resequencing data of the Darwin’s finch and the related species. Table S3: PCR primers used to validate the ZTR in great tit. Table S4: RNA-seq datasets analyzed in this study. Table S5: Divergence rate of the transposed genes measured by pairwise synonymous (dS) and nonnsynonymous (dN) substitution rates between Z/W gametologs.
Author Contributions
Conceptualization, L.X. and Q.Z.; methodology, L.X., Q.Z.; validation, M.I.; formal analysis, L.X., Q.Z.; investigation, L.X.; writing—original draft preparation, L.X.; writing—review and editing, L.X., Q.Z.; supervision, Q.Z. All authors have read and agreed to the published version of the manuscript.
Funding
Q.Z. is supported by the National Natural Science Foundation of China (31671319, 31722050), the Natural Science Foundation of Zhejiang Province (LD19C190001) and the European Research Council Starting Grant (grant agreement 677696). M.I. is supported by the Swedish Research Council (grant number 621-2014-5113). L.X. is supported by the FWF Erwin Schrödinger Fellowship (J-4477).
Acknowledgments
We thank Fumin Lei and Yalin Chen for sharing their unpublished genomic sequencing data of the green-backed tit (Parus monticolus) for dating the transposition in great tit, Ulf Johansson for providing female and male samples of P. major.
Conflicts of Interest
The authors declare there are no competing interests.
References
- Xu, L.; Wa, S.S.Y.; Grayson, P.; Edwards, S.V.; Sackton, T.B. Evolutionary Dynamics of Sex Chromosomes of Paleognathous Birds. Genome Biol. Evol. 2019, 11, 2376–2390. [Google Scholar] [CrossRef] [Green Version]
- Yazdi, H.P.; Silva, W.T.A.F.; Suh, A. Why Do Some Sex Chromosomes Degenerate More Slowly Than Others? The Odd Case of Ratite Sex Chromosomes. Genes 2020, 11, 1153. [Google Scholar] [CrossRef] [PubMed]
- Ellegren, H. Sex-chromosome evolution: Recent progress and the influence of male and female heterogamety. Nat. Rev. Genet. 2011, 12, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Zhang, J.; Bachtrog, D.; An, N.; Huang, Q.; Jarvis, E.D.; Gilbert, M.T.P.; Zhang, G. Complex evolutionary trajectories of sex chromosomes across bird taxa. Science 2014, 346, 1246338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smeds, L.; Warmuth, V.; Bolivar, P.; Uebbing, S.; Burri, R.; Suh, A.; Nater, A.; Bureš, S.; Garamszegi, L.Z.; Hogner, S.; et al. Evolutionary analysis of the female-specific avian W chromosome. Nat. Commun. 2015, 6, 7330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Auer, G.; Peona, V.; Suh, A.; Deng, Y.; Feng, S.; Zhang, G.; Blom, M.P.K.; Christidis, L.; Prost, S.; et al. Dynamic evolutionary history and gene content of sex chromosomes across diverse songbirds. Nat. Ecol. Evol. 2019, 3, 834–844. [Google Scholar] [CrossRef]
- Bellott, D.W.; Skaletsky, H.; Cho, T.J.; Brown, L.; Locke, D.; Chen, N.; Galkina, S.; Pyntikova, T.; Koutseva, N.; Graves, T.; et al. Avian W and mammalian Y chromosomes convergently retained dosage-sensitive regulators. Nat. Genet. 2017, 49, 387–394. [Google Scholar] [CrossRef] [Green Version]
- Bellott, D.W.; Hughes, J.F.; Skaletsky, H.; Brown, L.G.; Pyntikova, T.; Cho, T.J.; Koutseva, N.; Zaghlul, S.; Graves, T.; Rock, S.; et al. Mammalian Y chromosomes retain widely expressed dosage-sensitive regulators. Nature 2014, 508, 494–499. [Google Scholar] [CrossRef]
- Bellott, D.W.; Page, D.C. Dosage-sensitive functions in embryonic development drove the survival of genes on sex-specific chromosomes in snakes, birds, and mammals. bioRxiv 2020. [Google Scholar] [CrossRef]
- Waters, P.D.; Ruiz-Herrera, A. Meiotic Executioner Genes Protect the Y from Extinction. Trends Genet. 2020, 36, 728–738. [Google Scholar] [CrossRef]
- Graves, J.A.M. Avian sex, sex chromosomes, and dosage compensation in the age of genomics. Chromosome Res. 2014, 22, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Walters, J.R. Evolution of sex chromosome dosage compensation in animals: A beautiful theory, undermined by facts and bedeviled by details. Genome Biol. Evol. 2017, 9, 2461–2476. [Google Scholar] [CrossRef] [Green Version]
- Bernardo Carvalho, A.; Koerich, L.B.; Clark, A.G. Origin and evolution of Y chromosomes: Drosophila tales. Trends Genet. 2009, 25, 270–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koerich, L.B.; Wang, X.; Clark, A.G.; Carvalho, A.B. Low conservation of gene content in the Drosophila Y chromosome. Nature 2008, 456, 949–951. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, A.B.; Vicoso, B.; Russo, C.A.M.; Swenor, B.; Clark, A.G. Birth of a new gene on the Y chromosome of Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 2015, 112, 12450–12455. [Google Scholar] [CrossRef] [Green Version]
- Tobler, R.; Nolte, V.; Schlötterer, C. High rate of translocation-based gene birth on the Drosophila Y chromosome. Proc. Natl. Acad. Sci. USA 2017, 114, 11721–11726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, R.; Brown, L.G.; Hawkins, T.; Alagappan, R.K.; Skaletsky, H.; Reeve, M.P.; Reijo, R.; Rozen, S.; Dinulos, M.B.; Disteche, C.M.; et al. The DAZ gene cluster on the human Y chromosome arose from an autosomal gene that was transposed, repeatedly amplified and pruned. Nat. Genet. 1996, 14, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.L.; Evans, J.M.; Noorai, R.E.; Starr-Moss, A.N.; Clark, L.A. Novel Y Chromosome Retrocopies in Canids Revealed through a Genome-Wide Association Study for Sex. Genes 2019, 10, 320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potrzebowski, L.; Vinckenbosch, N.; Marques, A.C.; Chalmel, F.; Jégou, B.; Kaessmann, H. Chromosomal gene movements reflect the recent origin and biology of therian sex chromosomes. PLoS Biol. 2008, 6, e80. [Google Scholar] [CrossRef] [PubMed]
- Page, D.C.; Harper, M.E.; Love, J.; Botstein, D. Occurrence of a transposition from the X-chromosome long arm to the Y-chromosome short arm during human evolution. Nature 1984, 311, 119–123. [Google Scholar] [CrossRef]
- Li, G.; Davis, B.W.; Raudsepp, T.; Pearks Wilkerson, A.J.; Mason, V.C.; Ferguson-Smith, M.; O’Brien, P.C.; Waters, P.D.; Murphy, W.J. Comparative analysis of mammalian Y chromosomes illuminates ancestral structure and lineage-specific evolution. Genome Res. 2013, 23, 1486–1495. [Google Scholar] [PubMed] [Green Version]
- Brashear, W.A.; Raudsepp, T.; Murphy, W.J. Evolutionary conservation of Y Chromosome ampliconic gene families despite extensive structural variation. Genome Res. 2018, 28, 1841–1851. [Google Scholar] [PubMed] [Green Version]
- Janečka, J.E.; Davis, B.W.; Ghosh, S.; Paria, N.; Das, P.J.; Orlando, L.; Schubert, M.; Nielsen, M.K.; Stout, T.A.E.; Brashear, W.; et al. Horse Y chromosome assembly displays unique evolutionary features and putative stallion fertility genes. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.C.; Yang, Y.; Retzel, E.F.; Liu, W.S. Male-specific region of the bovine Y chromosome is gene rich with a high transcriptomic activity in testis development. Proc. Natl. Acad. Sci. USA 2013, 110, 12373–12378. [Google Scholar] [PubMed] [Green Version]
- Soh, Y.Q.S.; Alföldi, J.; Pyntikova, T.; Brown, L.G.; Graves, T.; Minx, P.J.; Fulton, R.S.; Kremitzki, C.; Koutseva, N.; Mueller, J.L.; et al. Sequencing the mouse Y chromosome reveals convergent gene acquisition and amplification on both sex chromosomes. Cell 2014, 159, 800–813. [Google Scholar]
- Bachtrog, D.; Mahajan, S.; Bracewell, R. Massive gene amplification on a recently formed Drosophila Y chromosome. Nat. Ecol. Evol. 2019, 3, 1587–1597. [Google Scholar]
- Larson, E.L.; Kopania, E.E.K.; Good, J.M. Spermatogenesis and the Evolution of Mammalian Sex Chromosomes. Trends Genet. 2018, 34, 722–732. [Google Scholar]
- Mahajan, S.; Bachtrog, D. Convergent evolution of Y chromosome gene content in flies. Nat. Commun. 2017, 8, 785. [Google Scholar]
- Ellison, C.; Bachtrog, D. Recurrent gene co-amplification on Drosophila X and Y chromosomes. PLoS Genet. 2019, 15, e1008251. [Google Scholar]
- Peneder, P.; Wallner, B.; Vogl, C. Exchange of genetic information between therian X and Y chromosome gametologs in old evolutionary strata. Ecol. Evol. 2017, 7, 8478–8487. [Google Scholar] [PubMed]
- Slattery, J.P.; Pecon Slattery, J.; Sanner-Wachter, L.; O’Brien, S.J. Novel gene conversion between X-Y homologues located in the nonrecombining region of the Y chromosome in Felidae (Mammalia). Proc. Natl. Acad. Sci. USA 2000, 97, 5307–5312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwase, M.; Satta, Y.; Hirai, H.; Hirai, Y.; Takahata, N. Frequent gene conversion events between the X and Y homologous chromosomal regions in primates. BMC Evol. Biol. 2010, 10, 225. [Google Scholar]
- Rozen, S.; Skaletsky, H.; Marszalek, J.D.; Minx, P.J.; Cordum, H.S.; Waterston, R.H.; Wilson, R.K.; Page, D.C. Abundant gene conversion between arms of palindromes in human and ape Y chromosomes. Nature 2003, 423, 873–876. [Google Scholar] [PubMed]
- Cechova, M.; Vegesna, R.; Tomaszkiewicz, M.; Harris, R.S.; Chen, D.; Rangavittal, S.; Medvedev, P.; Makova, K.D. Dynamic evolution of great ape Y chromosomes. Proc. Natl. Acad. Sci. USA 2020. [Google Scholar] [CrossRef]
- Davis, J.K.; Thomas, P.J.; NISC; Comparative Sequencing Program; Thomas, J.W. A W-linked palindrome and gene conversion in New World sparrows and blackbirds. Chromosome Res. 2010, 18, 543–553. [Google Scholar]
- Moghadam, H.K.; Pointer, M.A.; Wright, A.E.; Berlin, S.; Mank, J.E. W chromosome expression responds to female-specific selection. Proc. Natl. Acad. Sci. USA 2012, 109, 8207–8211. [Google Scholar]
- Laine, V.N.; Gossmann, T.I.; Schachtschneider, K.M.; Garroway, C.J.; Madsen, O.; Verhoeven, K.J.F.; de Jager, V.; Megens, H.-J.; Warren, W.C.; Minx, P.; et al. Evolutionary signals of selection on cognition from the great tit genome and methylome. Nat. Commun. 2016, 7, 10474. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup the Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar]
- Birney, E.; Clamp, M.; Durbin, R. GeneWise and Genomewise. Genome Res. 2004, 14, 988–995. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Sela, I.; Ashkenazy, H.; Katoh, K.; Pupko, T. GUIDANCE2: Accurate detection of unreliable alignment regions accounting for the uncertainty of multiple parameters. Nucleic Acids Res. 2015, 43, W7–W14. [Google Scholar] [CrossRef] [Green Version]
- Löytynoja, A. Phylogeny-aware alignment with PRANK. Methods Mol. Biol. 2014, 1079, 155–170. [Google Scholar]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, N.; Lee, I.; Marcotte, E.M.; Hurles, M.E. Characterising and Predicting Haploinsufficiency in the Human Genome. PLoS Genet. 2010, 6, e1001154. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, J.C.; Kuhl, H.; Timmermann, B.; Kempenaers, B. Characterization of the genome and transcriptome of the blue titCyanistes caeruleus: Polymorphisms, sex-biased expression and selection signals. Mol. Ecol. Resour. 2016, 16, 549–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uebbing, S.; Künstner, A.; Mäkinen, H.; Backström, N.; Bolivar, P.; Burri, R.; Dutoit, L.; Mugal, C.F.; Nater, A.; Aken, B.; et al. Divergence in gene expression within and between two closely related flycatcher species. Mol. Ecol. 2016, 25, 2015–2028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacManes, M.D.; Austin, S.H.; Lang, A.S.; Booth, A.; Farrar, V.; Calisi, R.M. Widespread patterns of sexually dimorphic gene expression in an avian hypothalamic–pituitary–gonadal (HPG) axis. Sci. Rep. 2017, 7, 45125. [Google Scholar] [CrossRef] [Green Version]
- Gazda, M.A.; Andrade, P.; Afonso, S.; Dilyte, J.; Archer, J.P.; Lopes, R.J.; Faria, R.; Carneiro, M. Signatures of Selection on Standing Genetic Variation Underlie Athletic and Navigational Performance in Racing Pigeons. Mol. Biol. Evol. 2018, 35, 1176–1189. [Google Scholar] [CrossRef]
- Harrison, P.W.; Wright, A.E.; Zimmer, F.; Dean, R.; Montgomery, S.H.; Pointer, M.A.; Mank, J.E. Sexual selection drives evolution and rapid turnover of male gene expression. Proc. Natl. Acad. Sci. USA 2015, 112, 4393–4398. [Google Scholar]
- Uebbing, S.; Konzer, A.; Xu, L.; Backström, N.; Brunström, B.; Bergquist, J.; Ellegren, H. Quantitative Mass Spectrometry Reveals Partial Translational Regulation for Dosage Compensation in Chicken. Mol. Biol. Evol. 2015, 32, 2716–2725. [Google Scholar]
- Alföldi, J.; Di Palma, F.; Grabherr, M.; Williams, C.; Kong, L.; Mauceli, E.; Russell, P.; Lowe, C.B.; Glor, R.E.; Jaffe, J.D.; et al. The genome of the green anole lizard and a comparative analysis with birds and mammals. Nature 2011, 477, 587–591. [Google Scholar] [PubMed] [Green Version]
- ZWtransposition. Available online: https://github.com/lurebgi/ZWtransposition (accessed on 24 October 2020).
- Peona, V.; Blom, M.P.K.; Xu, L.; Burri, R.; Sullivan, S.; Bunikis, I.; Liachko, I.; Jønsson, K.A.; Zhou, Q.; Irestedt, M.; et al. Identifying the causes and consequences of assembly gaps using a multiplatform genome assembly of a bird-of-paradise. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Hof, A.E.V.; Van’t Hof, A.E.; Nguyen, P.; Dalíková, M.; Edmonds, N.; Marec, F.; Saccheri, I.J. Linkage map of the peppered moth, Biston betularia (Lepidoptera, Geometridae): A model of industrial melanism. Heredity 2013, 110, 283–295. [Google Scholar]
- Zhou, R.; Macaya-Sanz, D.; Carlson, C.H.; Schmutz, J.; Jenkins, J.W.; Kudrna, D.; Sharma, A.; Sandor, L.; Shu, S.; Barry, K.; et al. A willow sex chromosome reveals convergent evolution of complex palindromic repeats. Genome Biol. 2020, 21, 38. [Google Scholar]
- Tennessen, J.A.; Wei, N.; Straub, S.C.K.; Govindarajulu, R.; Liston, A.; Ashman, T.-L. Repeated translocation of a gene cassette drives sex-chromosome turnover in strawberries. PLoS Biol. 2018, 16, e2006062. [Google Scholar]
- Ellegren, H. Evolutionary stasis: The stable chromosomes of birds. Trends Ecol. Evol. 2010, 25, 283–291. [Google Scholar]
- Xu, L.; Zhou, Q. The Female-Specific W Chromosomes of Birds Have Conserved Gene Contents but Are Not Feminized. Genes 2020, 11, 1126. [Google Scholar] [CrossRef]
- Trombetta, B.; Sellitto, D.; Scozzari, R.; Cruciani, F. Inter- and intraspecies phylogenetic analyses reveal extensive X-Y gene conversion in the evolution of gametologous sequences of human sex chromosomes. Mol. Biol. Evol. 2014, 31, 2108–2123. [Google Scholar]
- Trombetta, B.; Cruciani, F.; Underhill, P.A.; Sellitto, D.; Scozzari, R. Footprints of X-to-Y gene conversion in recent human evolution. Mol. Biol. Evol. 2010, 27, 714–725. [Google Scholar] [CrossRef] [Green Version]
- Dechaud, C.; Volff, J.N.; Schartl, M.; Naville, M. Sex and the TEs: Transposable elements in sexual development and function in animals. Mob. DNA 2019, 10, 42. [Google Scholar] [CrossRef] [PubMed]
- Rutkowska, J.; Lagisz, M.; Nakagawa, S. The long and the short of avian W chromosomes: No evidence for gradual W shortening. Biol. Lett. 2012, 8, 636–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
A transposition from the Z to W chromosomes in birds-of-paradise. (a) the genomic region at ~60 Mb of the Z chromosome showing doubled female sequencing coverage and female-specific elevations of heterozygosities (“f/m SNP density”) was inferred as a recent Z-transposed region (ZTR), and is marked by a red vertical bar. Two genes that survived following ZTR were labeled close to the red vertical bar. PAR and Z-linked duplications (marked in gray and purple vertical bars) are not expected to show a female-specific elevation of SNP densities. (b) A zoom-in view of the ZTR, showing female coverage and female-to-male SNP density calculated in 50 kb windows. (c) The phylogeny of birds-of-paradise (BOP) showing the origin of the ZTR (red asterisk) and the Z-linked duplication (blue asterisk). ReB: red BOP, RaB: Raggiana BOP, Mab: magnificent BOP, AmC: American crow, CoF: collared flycatcher. All bird illustrations were ordered from https://www.hbw.com/. (d) The 1.3 Mb transposed sequence (vertically aligned with (B)) contains 8 genes, but 4 complete and 2 partial genes have been deleted due to a 583 kb long deletion. Only ANXA1 and ALDH1A1 were retained on the W after transposition. (e) A zoom-in view for the duplication that does not show elevated female-to-male SNP density and both female and male coverage are doubled.
Figure 1.
A transposition from the Z to W chromosomes in birds-of-paradise. (a) the genomic region at ~60 Mb of the Z chromosome showing doubled female sequencing coverage and female-specific elevations of heterozygosities (“f/m SNP density”) was inferred as a recent Z-transposed region (ZTR), and is marked by a red vertical bar. Two genes that survived following ZTR were labeled close to the red vertical bar. PAR and Z-linked duplications (marked in gray and purple vertical bars) are not expected to show a female-specific elevation of SNP densities. (b) A zoom-in view of the ZTR, showing female coverage and female-to-male SNP density calculated in 50 kb windows. (c) The phylogeny of birds-of-paradise (BOP) showing the origin of the ZTR (red asterisk) and the Z-linked duplication (blue asterisk). ReB: red BOP, RaB: Raggiana BOP, Mab: magnificent BOP, AmC: American crow, CoF: collared flycatcher. All bird illustrations were ordered from https://www.hbw.com/. (d) The 1.3 Mb transposed sequence (vertically aligned with (B)) contains 8 genes, but 4 complete and 2 partial genes have been deleted due to a 583 kb long deletion. Only ANXA1 and ALDH1A1 were retained on the W after transposition. (e) A zoom-in view for the duplication that does not show elevated female-to-male SNP density and both female and male coverage are doubled.

Figure 2.
Transposed genes in birds-of-paradise are likely under evolutionary constraint (a) The distribution of predicted haploinsufficiency scores of human orthologs of all Z-linked genes, marked with ZTR genes ALDH1A1 and AXA1 by vertical dashed lines. (b) The phylogenetic tree with the branch lengths scaled by the ratios of nonsynonymous substitution rate to synonymous substitution rate (dN/dS). No nonsynonymous substitutions have been detected for ANXA1. (c) The expression of ALDH1A1 and ANXA1 across four different tissues of both male and female in nine birds and one reptile species. The expression levels were log1p (log 1 + TPM) transformed. Species that do not have the tissue transcriptome are denoted with blank tiles.
Figure 2.
Transposed genes in birds-of-paradise are likely under evolutionary constraint (a) The distribution of predicted haploinsufficiency scores of human orthologs of all Z-linked genes, marked with ZTR genes ALDH1A1 and AXA1 by vertical dashed lines. (b) The phylogenetic tree with the branch lengths scaled by the ratios of nonsynonymous substitution rate to synonymous substitution rate (dN/dS). No nonsynonymous substitutions have been detected for ANXA1. (c) The expression of ALDH1A1 and ANXA1 across four different tissues of both male and female in nine birds and one reptile species. The expression levels were log1p (log 1 + TPM) transformed. Species that do not have the tissue transcriptome are denoted with blank tiles.

Figure 3.
ZTRs identified in another two songbird lineages. (a) We detected ZTRs in great tit (Parus) and medium ground finch (Coerebinae). The red asterisks denote the origins of ZTRs. (b) The ZTR regions are highlighted by the vertical red bars, showing double female coverage and increased female SNP density. The grey bars highlight the PARs which have double female coverage but do not have increased female SNP density. The intact genes in the ZTRs are labelled near the red bars. (c) The ZTRs (transpositions) have significantly higher (Wilconxon rank sum test, p < 3.8 × 105) female SNP density than the rest of the Z chromosome and autosomes, or than the male SNP density, for both species.
Figure 3.
ZTRs identified in another two songbird lineages. (a) We detected ZTRs in great tit (Parus) and medium ground finch (Coerebinae). The red asterisks denote the origins of ZTRs. (b) The ZTR regions are highlighted by the vertical red bars, showing double female coverage and increased female SNP density. The grey bars highlight the PARs which have double female coverage but do not have increased female SNP density. The intact genes in the ZTRs are labelled near the red bars. (c) The ZTRs (transpositions) have significantly higher (Wilconxon rank sum test, p < 3.8 × 105) female SNP density than the rest of the Z chromosome and autosomes, or than the male SNP density, for both species.

Figure 4.
The predicted dosage-sensitivity and expression profile of the transposed genes. (a) The distribution of predicted haploinsufficiency scores of human orthologs of all Z-linked genes, marked with ZTR genes. MELK, SLC30A5 and CDK7 have relatively higher dosage-sensitivity (measured by haploinsufficiency scores) than the rest Z-linked genes. (b) The expression of the ZTR genes across tissues of both male and female in birds and a reptile. The expression levels were log1p (log 1 + TPM) transformed. The pseudogene was denoted by the dashed square.
Figure 4.
The predicted dosage-sensitivity and expression profile of the transposed genes. (a) The distribution of predicted haploinsufficiency scores of human orthologs of all Z-linked genes, marked with ZTR genes. MELK, SLC30A5 and CDK7 have relatively higher dosage-sensitivity (measured by haploinsufficiency scores) than the rest Z-linked genes. (b) The expression of the ZTR genes across tissues of both male and female in birds and a reptile. The expression levels were log1p (log 1 + TPM) transformed. The pseudogene was denoted by the dashed square.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Xu, L.; Irestedt, M.; Zhou, Q. Sequence Transpositions Restore Genes on the Highly Degenerated W Chromosomes of Songbirds. Genes 2020, 11, 1267. https://doi.org/10.3390/genes11111267
AMA Style
Xu L, Irestedt M, Zhou Q. Sequence Transpositions Restore Genes on the Highly Degenerated W Chromosomes of Songbirds. Genes. 2020; 11(11):1267. https://doi.org/10.3390/genes11111267
Chicago/Turabian StyleXu, Luohao, Martin Irestedt, and Qi Zhou. 2020. "Sequence Transpositions Restore Genes on the Highly Degenerated W Chromosomes of Songbirds" Genes 11, no. 11: 1267. https://doi.org/10.3390/genes11111267
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.