1. Introduction
Severe equine asthma (sEA) is an incurable obstructive respiratory condition affecting approximately 10% to 15% of horses in temperate climates [
1]. When exposed to airborne antigens, mainly during hay feeding, affected horses develop the characteristic signs of sEA exacerbation: neutrophilic airway inflammation, bronchial hyperreactivity, bronchoconstriction, and mucus hypersecretion. These chronic or recurrent exacerbations manifest with coughing, exercise intolerance, and increased respiratory effort. Marked clinical improvement with phases of disease remission are observed with decreased exposure to allergens and irritants in stable dust [
2]. However, complete avoidance of the offending agents is often impossible, and medical treatment with corticosteroids and bronchodilators becomes necessary to relieve acute asthma symptoms. Chronic changes and residual airway obstruction related to airway remodeling persist in disease remission [
3]. sEA is currently incurable and its consequences range from substantial welfare concerns to negative economic effects on equestrian sports and horse breeding [
4]. Elucidation of the underlying pathophysiological mechanisms of asthma in horses is needed to improve prevention and treatment of this condition. Importantly, such advances may also improve our understanding of asthma in humans, as immunological and pathophysiological characteristics of sEA show important parallels with allergic as well as severe neutrophilic human asthma [
5].
Asthma in humans has a prevalence of around 4.3% globally, and not only poses a significant threat to human health, but also presents a massive economic burden [
6,
7]. Much of the pathophysiological mechanisms of asthma are studied in experimental animal models (mostly rodents). However, sEA offers one of the few spontaneous models of human asthma [
4]. In order to improve the management of sEA in horses as well as discover novel comparative information relevant to human asthma, specific molecular pathways and mechanisms at the transcriptomic level need to be better understood. The current evidence regarding the immunological basis of sEA is conflicting. There are data that support both aspects of type I and type III hypersensitivities as well as involvement of the major immunological pathways including Th1, Th2, and Th17 [
8,
9,
10,
11]. An immediate-phase response is lacking in sEA, but affected horses characteristically show a delayed-phase allergic reaction to antigen challenge with neutrophil recruitment to the bronchial lumen and mixed Th-cell responses [
12]. Several studies have reported a predominant Th2-type immune response (characterized by increased IL-4, IL-5, and IL-13 expression) [
13,
14,
15] and evidence from genome-wide association studies (GWAS)tentatively supports
IL4R as a possible candidate gene [
16], while other investigations reported a mixed Th1–Th2-type response [
17,
18,
19].
In human asthma, which is now recognized as a complex, heterogeneous disease group, the intricate and intertwined gene–environment interactions [
20] are being categorized and assigned to sub-conditions with different clinical presentations (phenotypes) and diverse underlying pathophysiological mechanisms (endotypes) [
5,
21]. Recently, transcriptomic analyses of bronchial epithelium and of stimulated peripheral blood mononuclear cells (PBMC) have been performed in sEA [
9,
22]. Differentially expressed genes, pathways, and networks were identified in the airway epithelium of affected horses (e.g., MMP1, IL8, TLR4, and MMP9; neutrophil chemotaxis, immune, and inflammatory responses; circadian rhythm dysregulation; and the sonic Hedgehog pathway) that were also found to contribute to human asthma. Furthermore, novel differentially expressed genes (e.g.,
CXCL13, which is predominantly produced by Th17 cells) as well as dysregulated cellular pathways including immune cell trafficking, neutrophil chemotaxis, immune and inflammatory responses, cell cycle regulation, and apoptosis were discovered in allergen-stimulated peripheral blood mononuclear cells from horses with sEA [
9]. MicroRNAs (miRNA) orchestrate many biological and developmental processes and the top differentially expressed serum miRNAs in horses suggest that dysregulated pathways are involved in airway remodeling in sEA (e.g., CD4+ T cell function, and a Th2/Th17-type immune response) [
11].
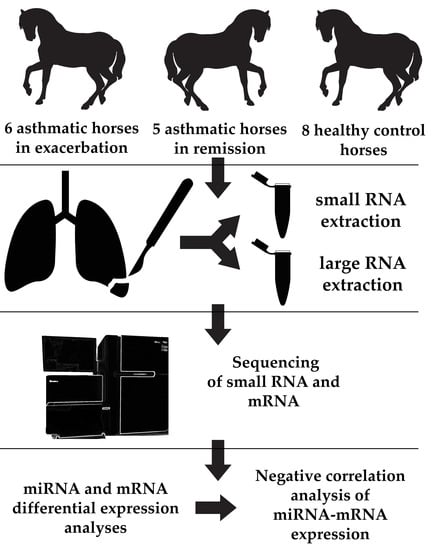
The present study aimed to investigate miRNA and mRNA expression changes in an integrative analysis in the lung tissue of sEA-affected horses in exacerbation, in remission as well as in healthy controls. We report dysregulated miRNA-mRNA networks in sEA and interpret the findings in light of molecular mechanisms described in human asthma and experimental animal models.
4. Discussion
In this study, we identified molecular mechanisms and pathways that may play important roles in the pathogenesis of sEA and offer novel comparative data for human asthma research based on comprehensive global miRNA and mRNA expression profiling in lung tissue samples of sEA-affected horses in exacerbation, in remission, and in healthy controls. To the best of our knowledge, this is the first study using an integrative approach combining miRNA and mRNA expression profiles in a spontaneous model of asthma. Dysregulated miRNA-mRNA networks were identified, revealing novel and striking similarities at the transcriptomic level between sEA and human asthma pheno- and endotypes (particularly severe neutrophilic asthma) based on in-depth literature research (
Supplementary Table S10).
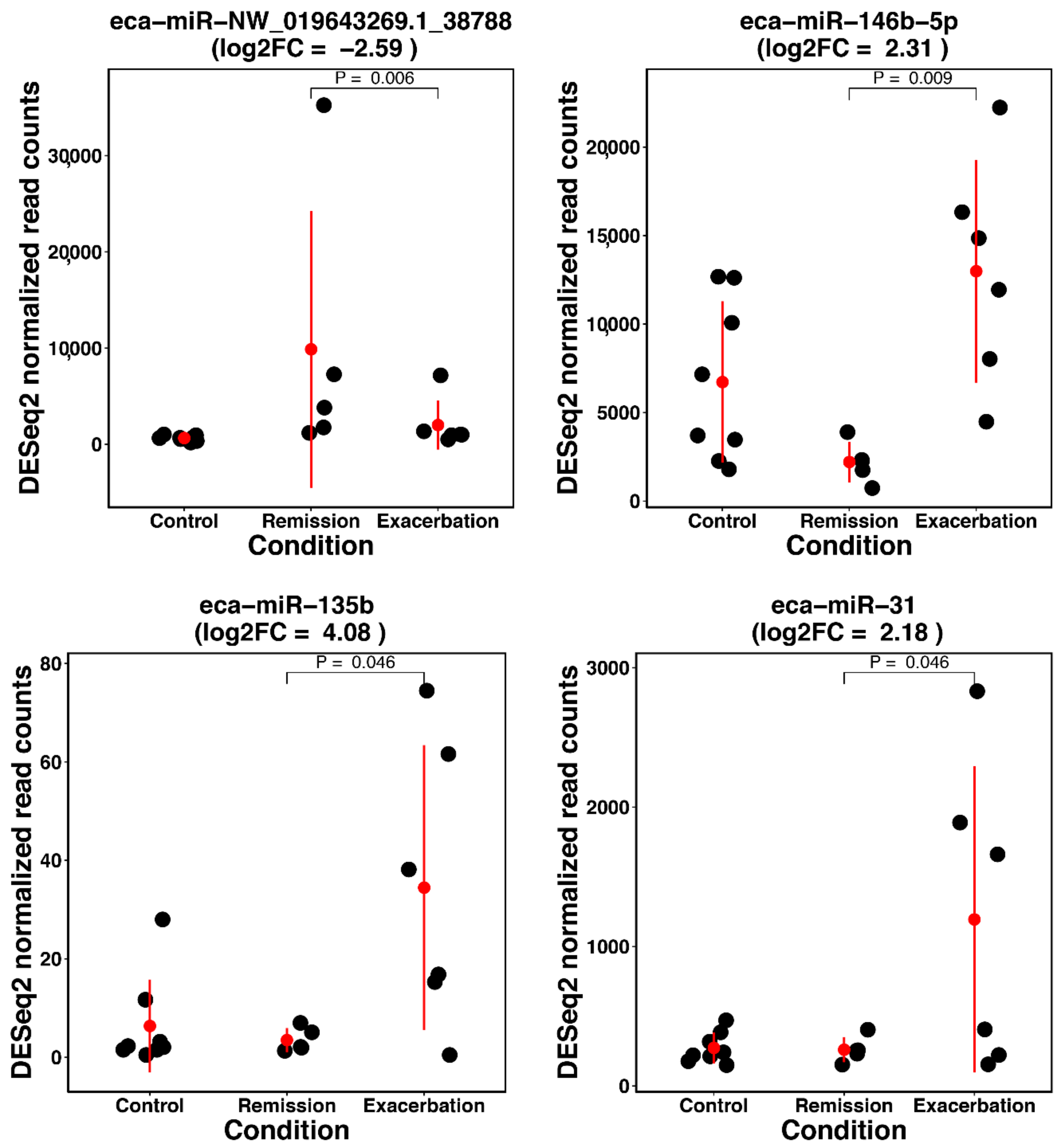
Some of the most prominent findings relate to inflammatory processes documented in severe neutrophilic asthma of humans. We decided to focus on the three miRNAs (miR-142-3p, miR-142-5p, and miR-223) that were found to be upregulated in both comparisons, asthmatic horses in exacerbation versus healthy controls and horses in remission versus controls (
Figure 2). Two of these miRNAs, namely miR-142-3p and miR-223, play a central role in the molecular pathology of human severe neutrophilic asthma by promoting airway inflammation and obstruction [
38]. It was not only shown that these two miRNAs are overexpressed in the sputum of humans with severe neutrophilic asthma, but also that their overexpression is strongly associated with inflammatory parameters, airway obstruction, and proinflammatory cytokine levels [
38]. Validated target genes of these miRNAs are implicated in several inflammatory pathways including Toll-like receptor signaling, NOD-like receptor signaling, and mitogen-activated protein kinase (MAPK) signaling, leading to increased levels of IL-1β, IL-6, and IL-8 [
38]. There was a significant increase in the corresponding IL-1β and IL-8 levels in patients with severe asthma [
38]. Furthermore, the expression levels were correlated with the neutrophil counts in the sputum samples. Several studies reporting increased expression levels of IL-1β and IL-8 in humans with neutrophilic asthma agree with these results [
39,
40]. Interestingly, these pathways and signature genes also are consistent with findings in horses with sEA, including recent transcriptomic analyses of the bronchial epithelium of affected horses relative to healthy horses in response to antigen challenge [
22] and several other studies reporting overexpressed IL-1β and IL-8 [
17,
41,
42,
43,
44].
Although the exact molecular mechanism of how these two miRNAs affect the pathophysiology of severe neutrophilic asthma remains unknown, recent findings point towards their role in the development of other important disorders: in the lungs of tuberculosis patients, miR-223 upregulates miR-142-3p expression as part of the ‘miR-223-CEBP-β-LMO2-miR-142’ pathway [
45] and controls neutrophil recruitment as well as neutrophil-driven inflammation [
39]. Furthermore, miR-223 plays an essential role in autoimmune inflammation, where it mediates the myeloid dendritic cell induced pathologic Th17 response [
46]. Its targeting of the inflammatory mediator ICAM1 [
47] and its effect on macrophage polarization towards the M2 type [
48] suggest additional important functions of miR-223 in neutrophilic asthma. Furthermore, miR-142-3p is implicated in aberrant WNT signaling during airway remodeling [
49], in pro-inflammatory processes via monocyte-derived dendritic cells (while suppressing Treg expansion) [
50], and in the suppression of Th1 cytokines by dendritic cells [
51]. Additionally, miR-142-3p is overexpressed in Lipopolysaccharide (LPS)-stimulated macrophages [
52] and impairs antigen processing [
53]. Thus, dysregulated miR-223 and miR-142-3p might affect sEA pathophysiology by promoting airway remodeling, positively regulating neutrophil recruitment and airway inflammation, and possibly affecting macrophage polarization. MiR-223, together with miR-31, showed an interesting pattern of a rather high within-group variation for horses in exacerbation (
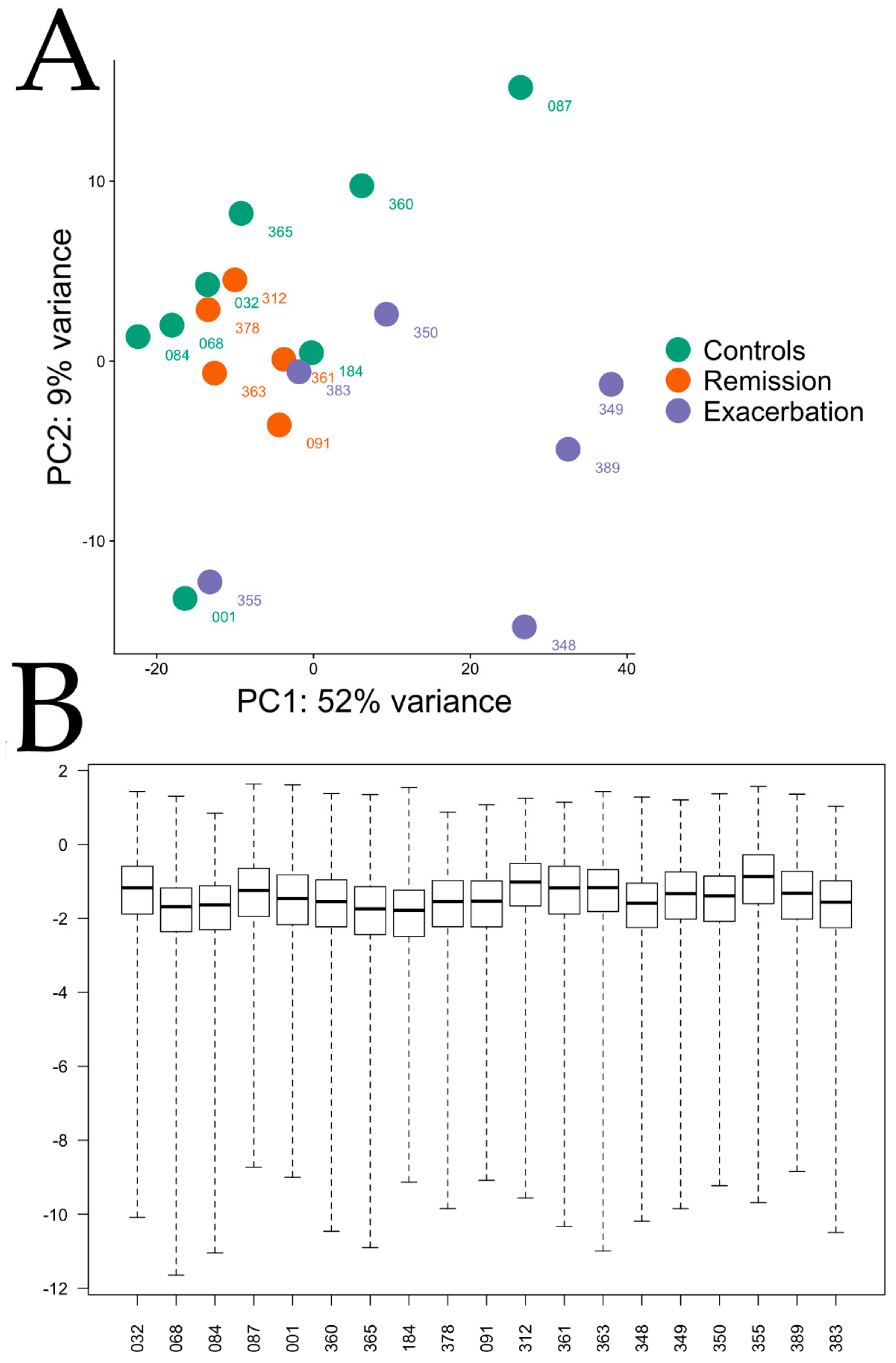
Appendix A Figure A5). While this might be expected owing to the symptomatic stage of the disease, it might also reflect the presence of potential unidentified endotypes of sEA.
Several differentially expressed miRNAs detected in the lungs of horses in exacerbation compared to controls are linked with the positive regulation of the Th17 pathway. Indeed, downregulated miR-26a leads to increased expression of IL-17 and IL-6 [
54] and affects Th17/Treg balance towards Th17 cells [
55]. Additionally, miR-31 drives Th17 cell differentiation through targeting IL-25, while suppressing Treg differentiation through FOXP3 [
56]. Moreover, miR-212 promotes Th17 cell differentiation likely by targeting Bcl-6 [
57], while miR-223 regulates the Th17 immune response in a myeloid dendritic cell-driven manner [
46]. The potential involvement of an increased Th17-type immune response is further supported by multiple dysregulated miRNAs in the second contrast comparing sEA-affected horses in remission to healthy horses (miR-363, miR-379, miR-193a-3p, miR-135b [
58,
59,
60,
61,
62]). Th17 cells recruit neutrophils through releasing Th17 cytokines (IL-17A, IL17F, and IL-22) and epithelial-derived neutrophilic chemokines [
63]. Interestingly, the miRNA–mRNA networks that we expanded with regulatory transcription factor interactions support the hypothesis of a dysregulated Th17 cell differentiation pathway (
Supplementary Figures S1 and S2). The expanded network suggests an important role for the IL-17 signaling pathway and for STAT3, which is a key transcription factor in Th17 cellular development. Taken together, these findings suggest an important role of the Th17 pathway in the pathophysiology of sEA, especially in disease exacerbation. The dysregulated miRNAs presented in this study might affect sEA pathology by leading to increased recruitment and differentiation of Th17 cells in the lung and increased Th17 cell differentiation. The Th17-mediated inflammation then in turn potentially leads to increased neutrophilic airway inflammation and increased airway remodeling [
63]. This agrees with our previous findings in stimulated PBMC and those of other groups in mediastinal lymph nodes, BALF, and epithelial cells of sEA-affected horses [
9,
11,
64,
65,
66]. However, previous findings in sEA also suggest that Th2-type immune responses consistent with allergic-type asthma play a role in sEA [
12,
13,
14], and some of our present results support this pathway.
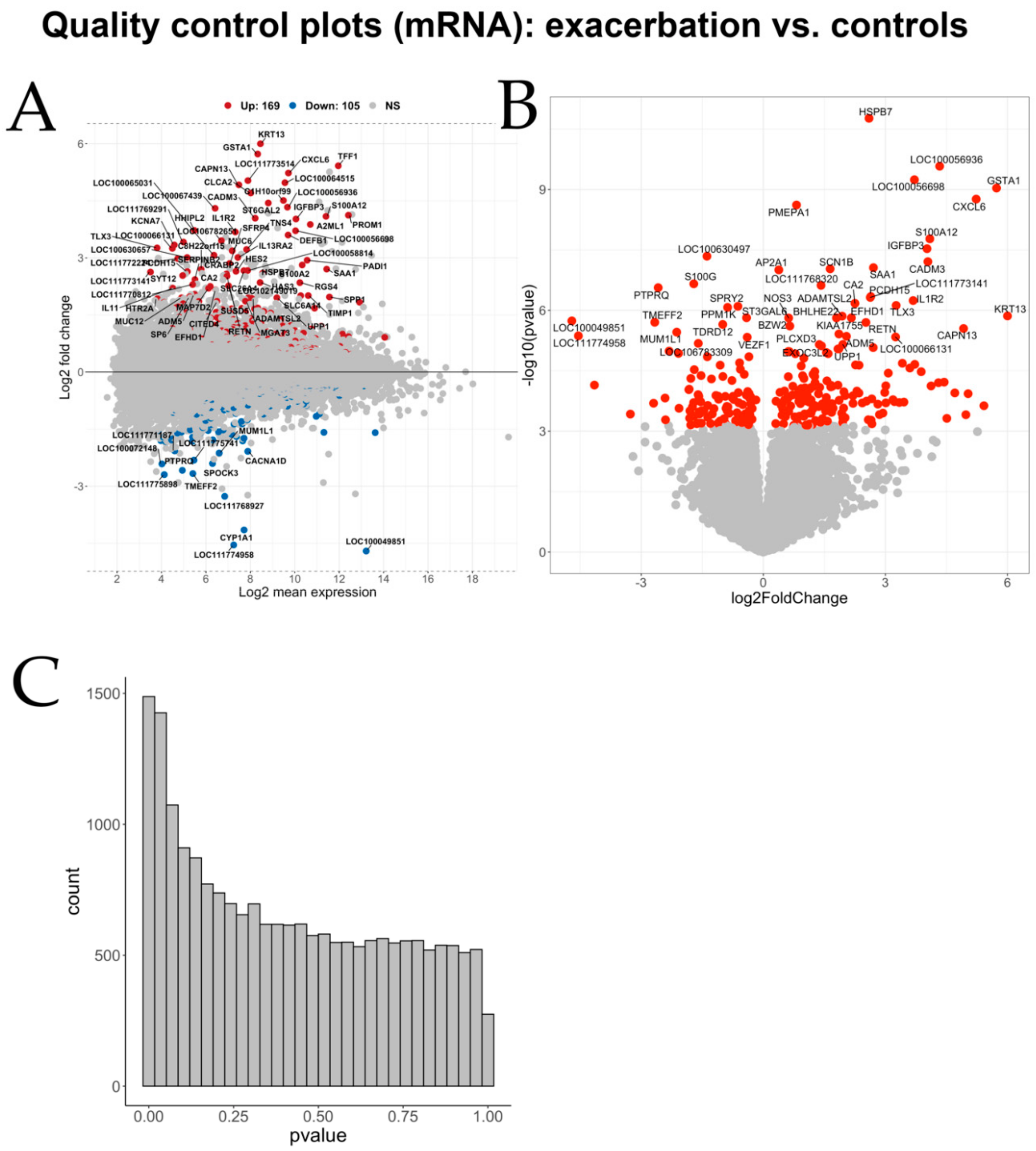
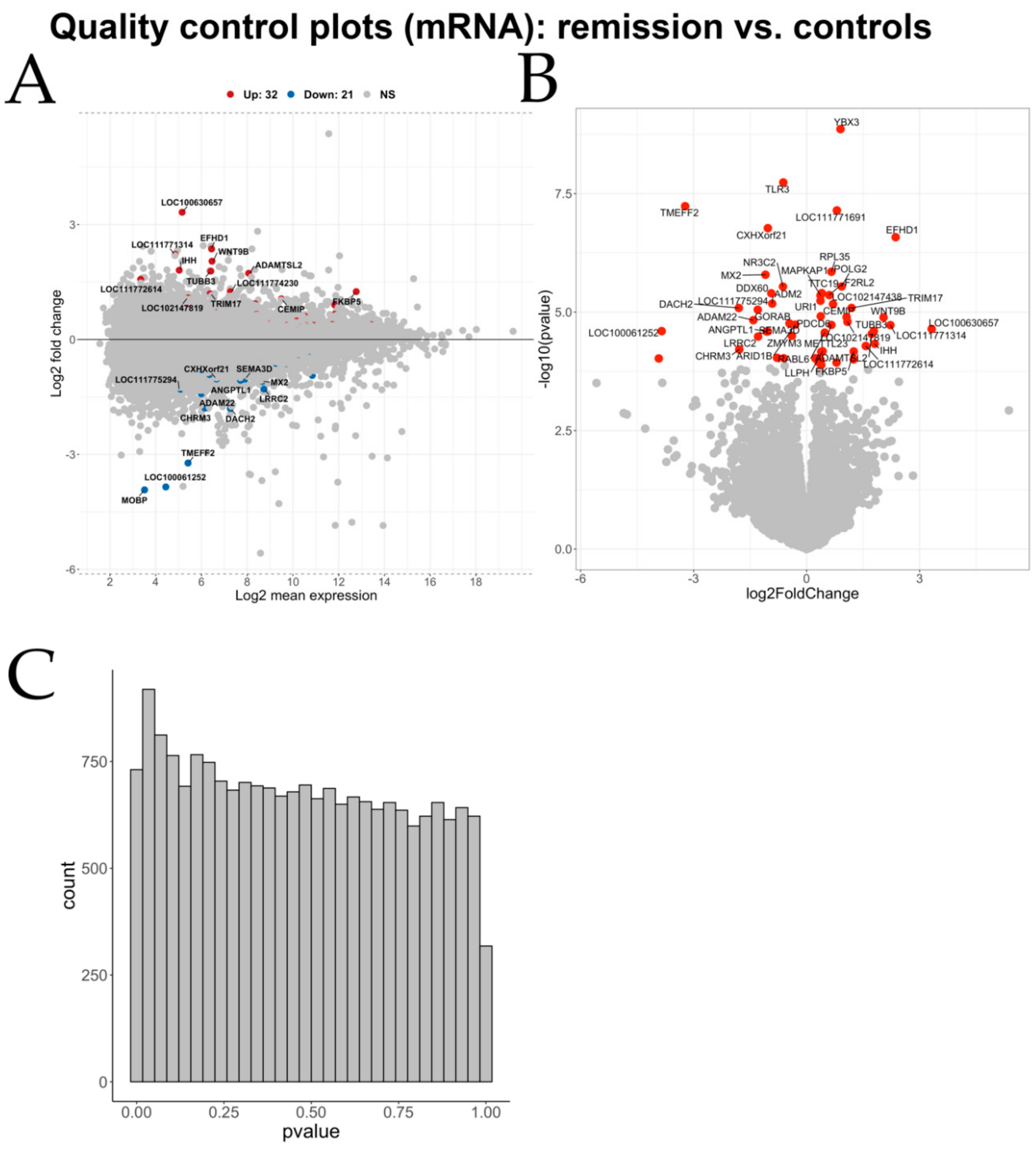
Differential gene expression analysis showed hundreds of genes to be significantly differentially expressed between the three group comparisons.
IL4R was among the significantly differentially expressed genes of the first comparison (exacerbation against controls) and is also on the list of genes previously associated with human asthma (
Supplementary Table S6).
IL4R, which codes for the cytokine receptor binding the Th2 cytokines IL-4 and IL-13, was previously identified by our group as a candidate gene for sEA in a GWAS experiment [
67]. Our network analysis, expanded with annotated pathways (
Supplementary Figure S2), also supports involvement of the IL-4 signaling pathway in sEA. Of note, our integrative analysis of miRNA–mRNA pairs further substantiates the role of the Th2-type response during sEA exacerbation. Carbonic anhydrase II (
CA2), which was negatively correlated with miR-26a-5p and significantly upregulated in exacerbated horses, is known to be upregulated by Th2 cytokines like IL-4 and IL-13, and reportedly promotes airway hyperresponsiveness [
68,
69]. Moreover, RGS4 (also negatively correlated with miR-26a-5p) plays a role in modulating airway hyper-responsiveness and airway obstruction [
70,
71].
Our results suggest additional dysregulated processes in sEA exacerbation including alternative macrophage activation (miR-26a, miR-142-5p, miR-212, miR-223 [
48,
52]) and NF-kB signaling (miR-224 [
72]). The latter being additionally supported by our expanded transcription factor–gene interaction network analysis (
Supplementary Figure S1) and by earlier studies investigating sEA [
11,
73,
74]. Importantly, our miRNA–mRNA integrative analysis also indicates that NF-kB plays an important role in sEA:
CYP1A1 was negatively correlated with miR-142-3p. This NF-kB controlled gene has been associated with asthma and lung function deficits [
75,
76]. Additionally, SIAH-1 (targeted by miR-142-3p) regulates NF-kB activity via TNF-α and has been shown to interact with many other genes involved in the mammalian immune response [
77].
Counteracting these deleterious processes, we also identified anti-inflammatory pathways taking place during asthma exacerbation, but not in remission. MiR-146-5p targets inflammatory mediators like
COX2, IL-1β, KIT, NFKB1, TLR4, TRAF6, and
UHRF1 [
78] and has been proposed as a predictor for asthma exacerbations in childhood asthma [
79,
80]. The anti-inflammatory miR-135b acts by targeting
FOXN3 and
RECK, an inhibitor of MMP2, and the known sEA pathogenesis factor MMP9 [
81,
82,
83]. These anti-inflammatory actions might be the immunological counterbalance, and thus the defense mechanism of the lung against acute inflammation (likely Th17-mediated, as outlined above) during disease exacerbation.
Paralleling the pathology of severe human asthma [
84], the significantly dysregulated miRNAs (miR-363, miR-379, miR-193a-3p) found in sEA-affected horses in remission (when compared with healthy horses) are likely to enhance TGF-β signaling [
58,
59,
60]. This is further supported by the expanded network analysis including annotated pathways (
Supplementary Figure S2). Thus, these miRNAs may be key players in airway remodeling during disease remission, prolonging and amplifying effects of miRNAs that were significantly upregulated in exacerbation. Specifically, miR-26a, miR-142-3p, and miR-142-5p promote airway remodeling (in parts by positively affecting TGF-β pathways) [
55,
85].
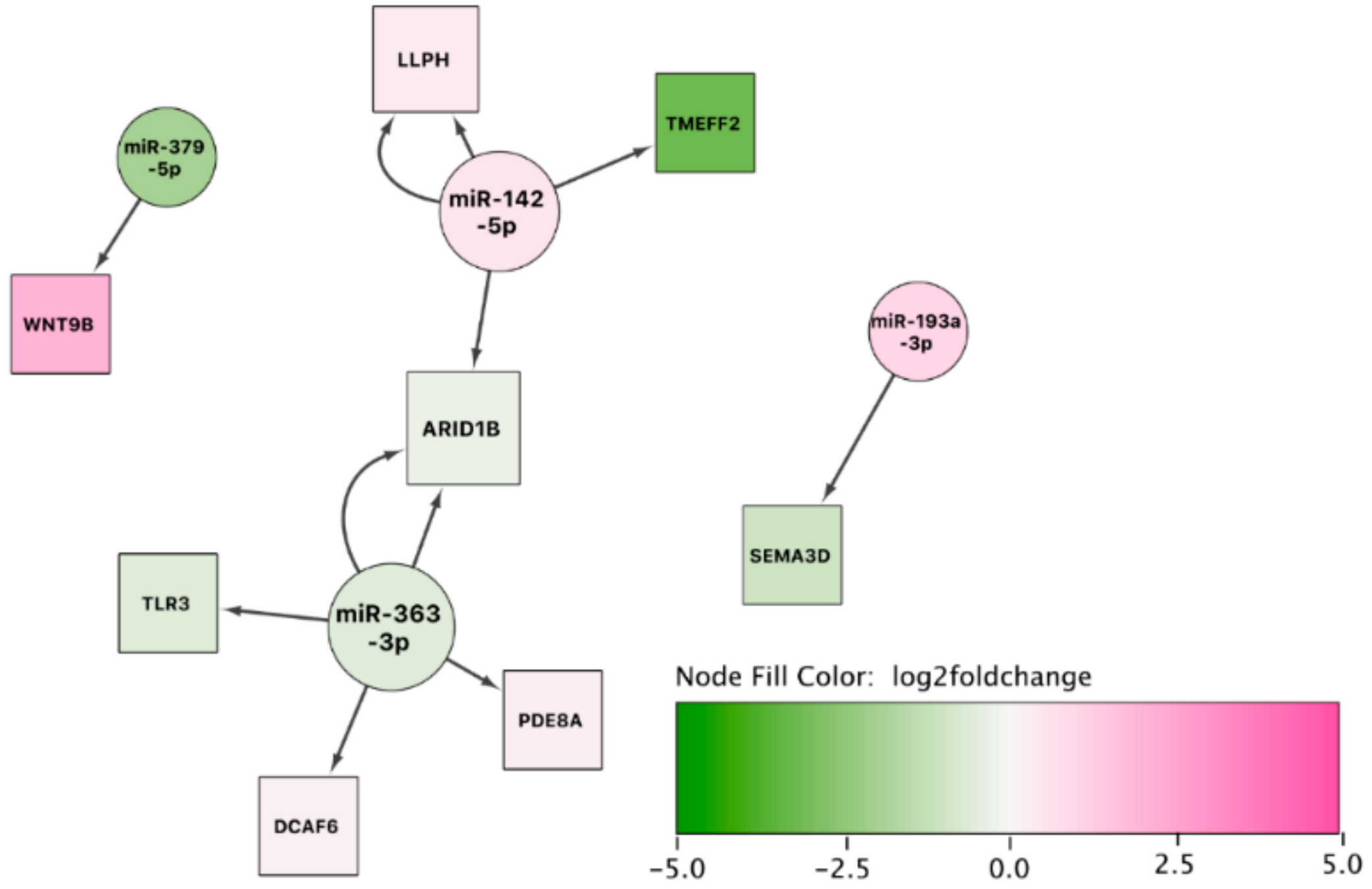
Besides the miRNA–mRNA pairs discussed above, our integrative miRNA–mRNA expression analysis revealed numerous negatively correlated pairs implicated in sEA. Many of these have not yet been investigated regarding a potential role in asthma pathology. Because we detected the miRNA–mRNA pair ADAM22-miR-142-3p in both comparisons (exacerbation vs. controls and remission vs. controls), it might be a key player in the pathophysiology of sEA that not only affects exacerbation of the disease, but also disease remission. Additionally, we constructed miRNA–mRNA networks as an alternative approach. This visual representation helped to put some of the negatively correlated miRNA–mRNA pairs in a bigger perspective. Besides the pair ADAM22-miR-142-3p, the genes CCNT2 (paired with miR-142-5p and miR-142-3p), SIAH1, RGS4 (negatively correlated with miR-26a-5p), and SYDE2 (paired with miR-31-5p) represent promising starting points for future research to validate the role of these dysregulated pathways in functional experiments.
Additionally, future research could be applied to validate our findings in an independent and larger study cohort, as well as by applying different technical approaches (e.g., RT-qPCR). While differential expression analyses with six or more samples per group are considered robust [
86], an increased sample size can increase statistical power, and thus the ability to detect more differentially expressed genes and miRNAs.
In conclusion, we found important similarities between sEA (at the transcriptomic level) and severe neutrophilic asthma in humans. Specifically, our findings suggest that the dysregulated miRNAs and mRNAs might lead to a predominantly Th17-mediated immune response in exacerbation and remission of sEA. This Th17-mediated inflammation could potentially explain the prominent role of neutrophils in sEA. Other main findings also show parallels with allergic asthma (Th2-type response), and present potential pathways that promote airway remodeling. These results expand our understanding of the molecular mechanisms that underlie airway hyper-responsiveness and bronchospasm during disease exacerbation and airway remodeling during disease remission.

 ,
, 



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}