Mouse Models of Human Pathogenic Variants of TBC1D24 Associated with Non-Syndromic Deafness DFNB86 and DFNA65 and Syndromes Involving Deafness

 , , ,
, , ,  , , and
, , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Ethic Approval and Clinical Evaluation
2.2. Whole Exome Sequencing (WES) and Bioinformatic Analyses
2.3. Mouse Models of Human TBC1D24-Associated Deafness
2.4. ABR and DPOAE Measurements of Hearing Ability
2.5. Immunofluorescence Staining in Celloidin Sectionsof Human Cochleae
2.6. In Situ Hybridization and Immunohistochemistry Using Mouse Cochleae
2.7. scRNA-Seq
2.8. Computational Modeling
2.9. Molecular Dynamic Simulations
3. Results
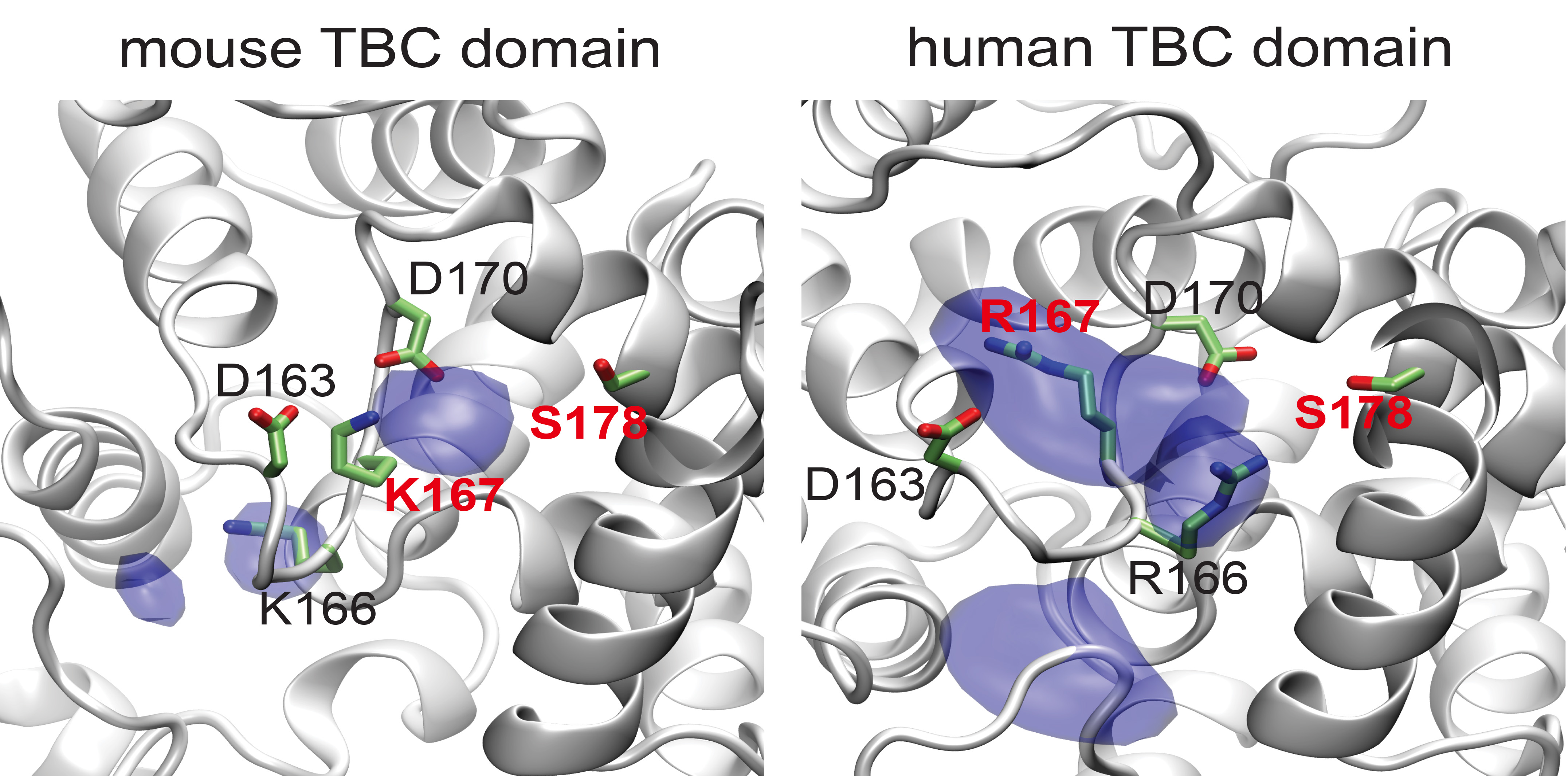
3.1. Novel Splice Variant of Human TBC1D24 Associated with Deafness and Seizures
3.2. TBC1D24 Protein Localization in Human and Mouse Temporal Bone and Tbc1d24 mRNA Expression in Wild Type Mouse Cochlea
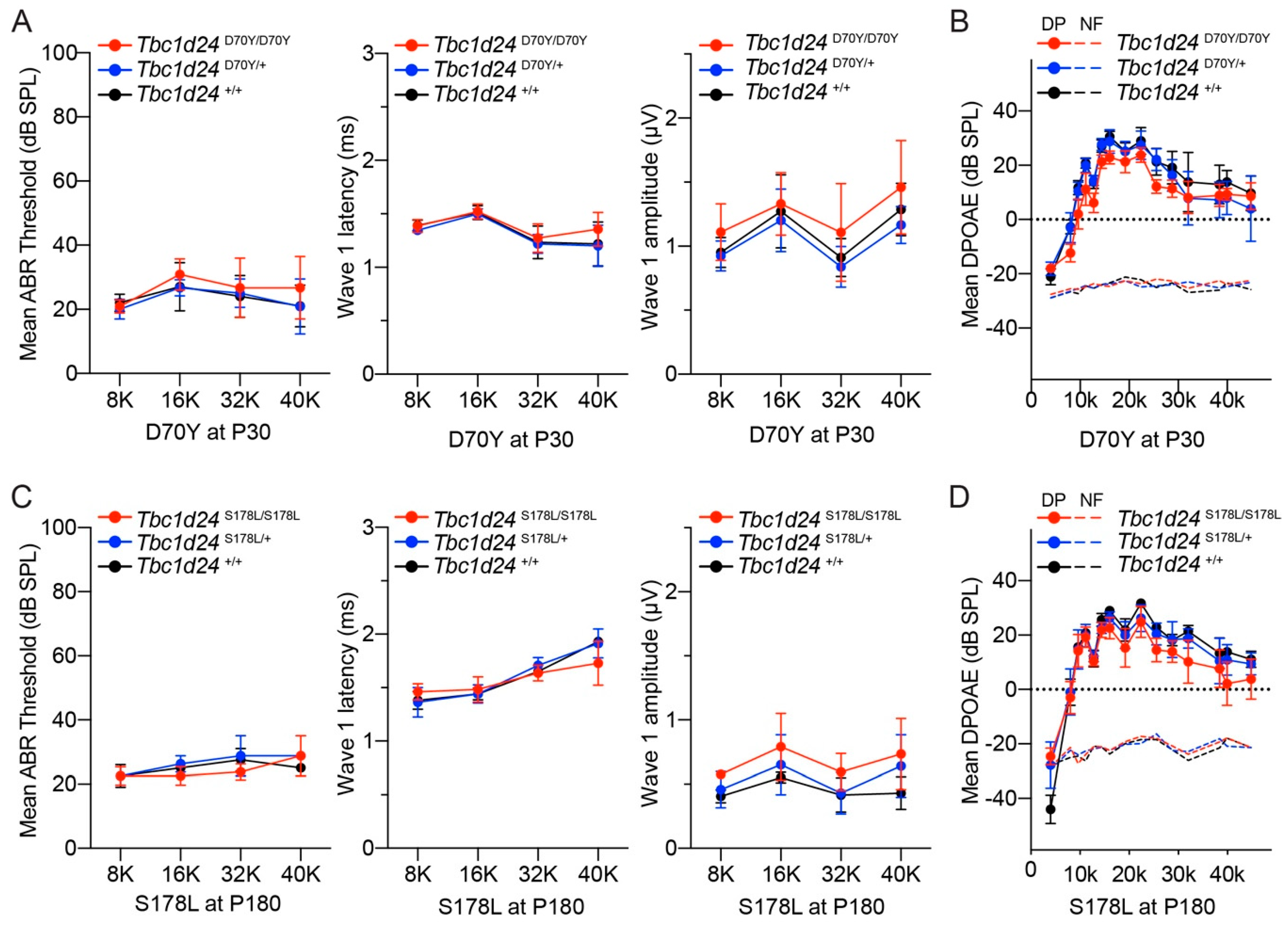
3.3. Auditory Function in the Mouse Models of DFNB86 and DFNA65
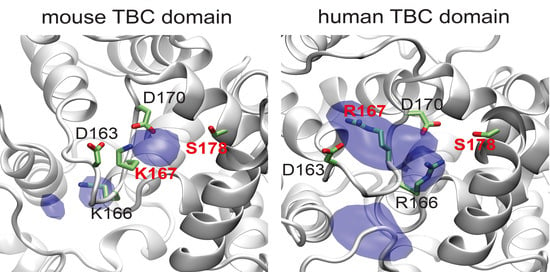
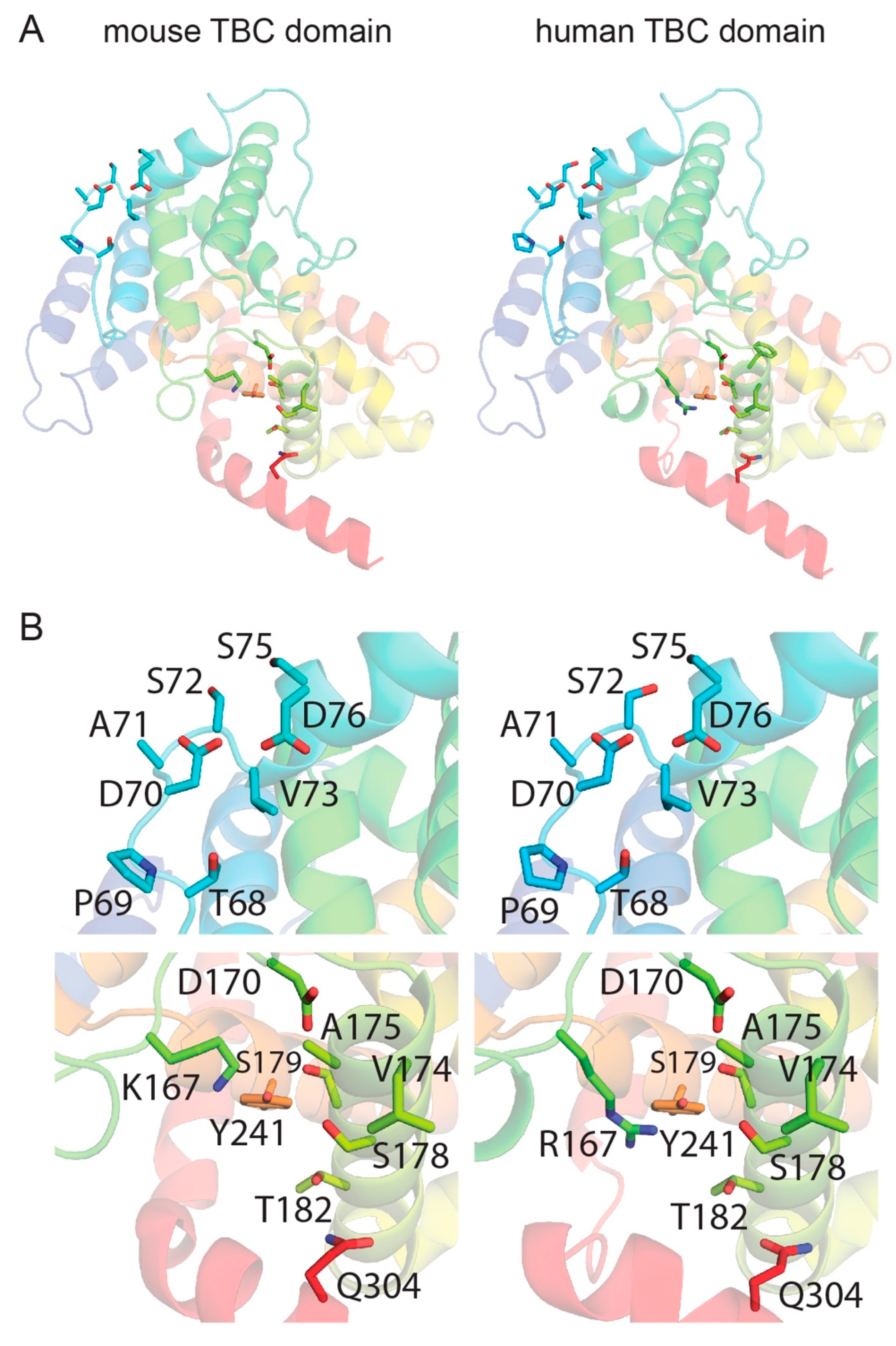
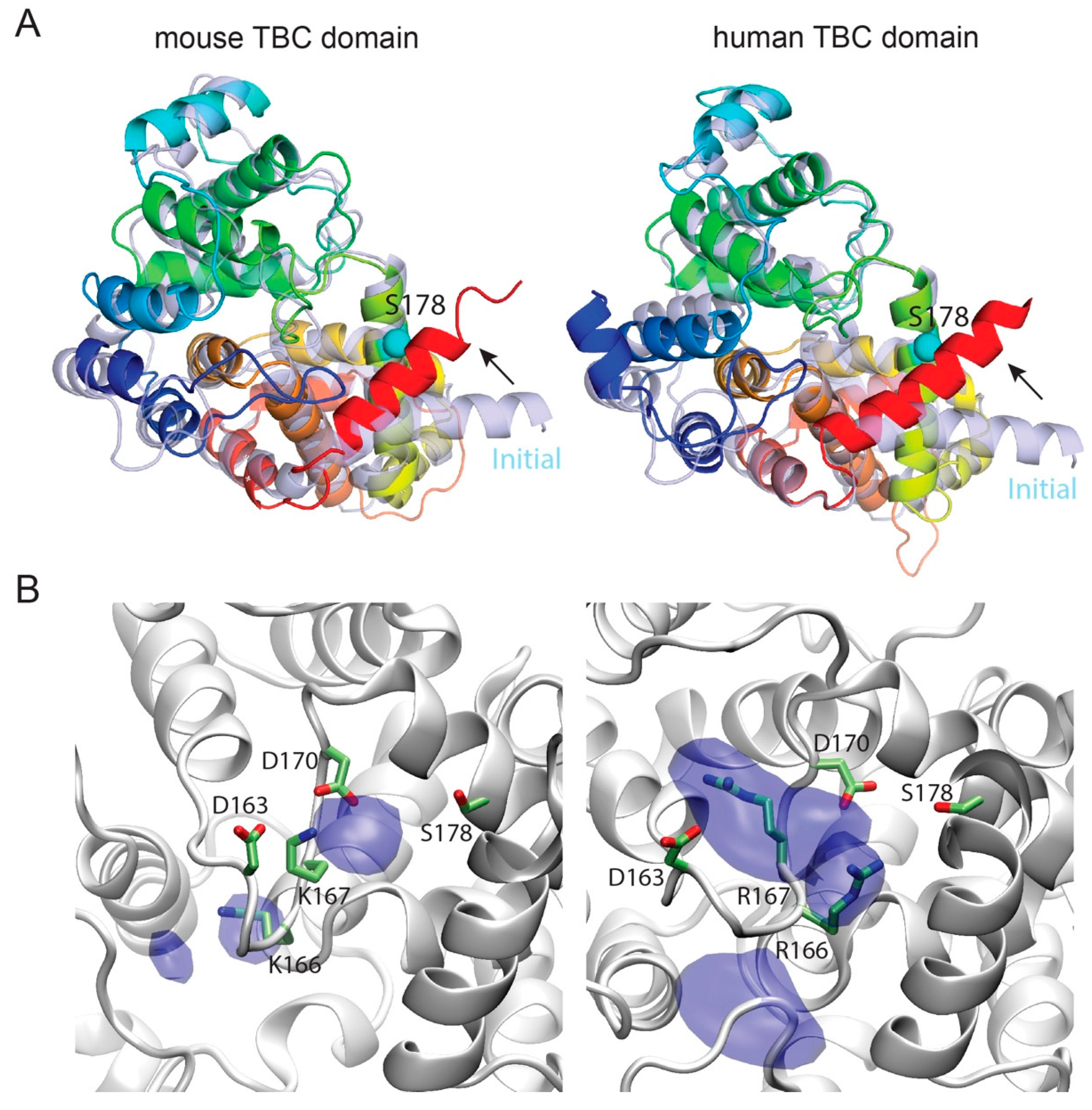
3.4. Template-Based Models of Mouse and Human TBC1D24
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barbi, M.; Binda, S.; Caroppo, S.; Ambrosetti, U.; Corbetta, C.; Sergi, P. A wider role for congenital cytomegalovirus infection in sensorineural hearing loss. Pediatr. Infect. Dis. J. 2003, 22, 39–42. [Google Scholar] [CrossRef]
- Brock, P.R.; Knight, K.R.; Freyer, D.R.; Campbell, K.C.; Steyger, P.S.; Blakley, B.W.; Rassekh, S.R.; Chang, K.W.; Fligor, B.J.; Rajput, K.; et al. Platinum-induced ototoxicity in children: A consensus review on mechanisms, predisposition, and protection, including a new International Society of Pediatric Oncology Boston ototoxicity scale. J. Clin. Oncol. 2012, 30, 2408–2417. [Google Scholar] [CrossRef] [Green Version]
- Hilgert, N.; Smith, R.J.; Van Camp, G. Function and expression pattern of nonsyndromic deafness genes. Curr. Mol. Med. 2009, 9, 546–564. [Google Scholar] [CrossRef]
- Griffith, A.J.; Friedman, T.B. Hereditary Hearing Loss, 18th ed.; Wackym, P.A., Snow, J.B., Eds.; People’s Medical Publishing House-USA: Shelton, CT, USA, 2016. [Google Scholar]
- Rehman, A.U.; Friedman, T.B.; Griffith, A.J. Unresolved questions regarding human hereditary deafness. Oral. Dis. 2017, 23, 551–558. [Google Scholar] [CrossRef]
- Toriello, H.V.; Smith, S.D. Hereditary Hearing Loss and ITS Syndromes; Oxford University Press: Oxford, UK, 2013. [Google Scholar]
- Liu, X.Z.; Walsh, J.; Tamagawa, Y.; Kitamura, K.; Nishizawa, M.; Steel, K.P.; Brown, S.D. Autosomal dominant non-syndromic deafness caused by a mutation in the myosin VIIA gene. Nat. Genet. 1997, 17, 268–269. [Google Scholar] [CrossRef]
- Weil, D.; Blanchard, S.; Kaplan, J.; Guilford, P.; Gibson, F.; Walsh, J.; Mburu, P.; Varela, A.; Levilliers, J.; Weston, M.D.; et al. Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature 1995, 374, 60–61. [Google Scholar] [CrossRef]
- Weil, D.; Küssel, P.; Blanchard, S.; Lévy, G.; Levi-Acobas, F.; Drira, M.; Ayadi, H.; Petit, C. The autosomal recessive isolated deafness, DFNB2, and the Usher 1B syndrome are allelic defects of the myosin-VIIA gene. Nat. Genet. 1997, 16, 191–193. [Google Scholar] [CrossRef]
- Rehman, A.U.; Santos-Cortez, R.L.; Morell, R.J.; Drummond, M.C.; Ito, T.; Lee, K.; Khan, A.A.; Basra, M.A.; Wasif, N.; Ayub, M.; et al. Mutations in TBC1D24, a gene associated with epilepsy, also cause nonsyndromic deafness DFNB86. Am. J. Hum. Genet. 2014, 94, 144–152. [Google Scholar] [CrossRef] [Green Version]
- Azaiez, H.; Booth, K.T.; Bu, F.; Huygen, P.; Shibata, S.B.; Shearer, A.E.; Kolbe, D.; Meyer, N.; Black-Ziegelbein, E.A.; Smith, R.J. TBC1D24 mutation causes autosomal-dominant nonsyndromic hearing loss. Hum. Mutat. 2014, 35, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Hu, L.; Chai, Y.; Pang, X.; Yang, T.; Wu, H. A dominant mutation in the stereocilia-expressing gene TBC1D24 is a probable cause for nonsyndromic hearing impairment. Hum. Mutat. 2014, 35, 814–818. [Google Scholar] [CrossRef]
- Tona, R.; Chen, W.; Nakano, Y.; Reyes, L.D.; Petralia, R.S.; Wang, Y.X.; Starost, M.F.; Wafa, T.T.; Morell, R.J.; Cravedi, K.D.; et al. The phenotypic landscape of a Tbc1d24 mutant mouse includes convulsive seizures resembling human early infantile epileptic encephalopathy. Hum. Mol. Genet. 2019, 28, 1530–1547. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, M. TBC proteins: GAPs for mammalian small GTPase Rab? Biosci. Rep. 2011, 31, 159–168. [Google Scholar] [CrossRef]
- Pan, X.; Eathiraj, S.; Munson, M.; Lambright, D.G. TBC-domain GAPs for Rab GTPases accelerate GTP hydrolysis by a dual-finger mechanism. Nature 2006, 442, 303–306. [Google Scholar] [CrossRef]
- Finelli, M.J.; Sanchez-Pulido, L.; Liu, K.X.; Davies, K.E.; Oliver, P.L. The Evolutionarily Conserved Tre2/Bub2/Cdc16 (TBC), Lysin Motif (LysM), Domain Catalytic (TLDc) Domain Is Neuroprotective against Oxidative Stress. J. Biol. Chem. 2016, 291, 2751–2763. [Google Scholar] [CrossRef] [Green Version]
- Finelli, M.J.; Oliver, P.L. TLDc proteins: New players in the oxidative stress response and neurological disease. Mamm. Genome 2017, 28, 395–406. [Google Scholar] [CrossRef] [Green Version]
- Guven, A.; Tolun, A. TBC1D24 truncating mutation resulting in severe neurodegeneration. J. Med. Genet. 2013, 50, 199–202. [Google Scholar] [CrossRef]
- Campeau, P.M.; Kasperaviciute, D.; Lu, J.T.; Burrage, L.C.; Kim, C.; Hori, M.; Powell, B.R.; Stewart, F.; Félix, T.M.; van den Ende, J.; et al. The genetic basis of DOORS syndrome: An exome-sequencing study. Lancet Neurol. 2014, 13, 44–58. [Google Scholar] [CrossRef]
- Stražišar, B.G.; Neubauer, D.; Paro Panjan, D.; Writzl, K. Early-onset epileptic encephalopathy with hearing loss in two siblings with TBC1D24 recessive mutations. Eur. J. Paediatr. Neurol. 2015, 19, 251–256. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Varshney, G.K.; Pei, W.; LaFave, M.C.; Idol, J.; Xu, L.; Gallardo, V.; Carrington, B.; Bishop, K.; Jones, M.; Li, M.; et al. High-throughput gene targeting and phenotyping in zebrafish using CRISPR/Cas9. Genome Res. 2015, 25, 1030–1042. [Google Scholar] [CrossRef] [Green Version]
- Richardson, C.D.; Ray, G.J.; DeWitt, M.A.; Curie, G.L.; Corn, J.E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 2016, 34, 339–344. [Google Scholar] [CrossRef]
- Wang, H.; Yang, H.; Shivalila, C.S.; Dawlaty, M.M.; Cheng, A.W.; Zhang, F.; Jaenisch, R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013, 153, 910–918. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.R.; Erway, L.C.; Cook, S.A.; Willott, J.F.; Zheng, Q.Y. A major gene affecting age-related hearing loss in C57BL/6J mice. Hear. Res. 1997, 114, 83–92. [Google Scholar] [CrossRef]
- Keithley, E.M.; Canto, C.; Zheng, Q.Y.; Fischel-Ghodsian, N.; Johnson, K.R. Age-related hearing loss and the ahl locus in mice. Hear. Res. 2004, 188, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Morozko, E.L.; Nishio, A.; Ingham, N.J.; Chandra, R.; Fitzgerald, T.; Martelletti, E.; Borck, G.; Wilson, E.; Riordan, G.P.; Wangemann, P.; et al. ILDR1 null mice, a model of human deafness DFNB42, show structural aberrations of tricellular tight junctions and degeneration of auditory hair cells. Hum. Mol. Genet. 2015, 24, 609–624. [Google Scholar] [CrossRef] [Green Version]
- Lopez, I.A.; Ishiyama, G.; Hosokawa, S.; Hosokawa, K.; Acuna, D.; Linthicum, F.H.; Ishiyama, A. Immunohistochemical techniques for the human inner ear. Histochem. Cell Biol. 2016, 146, 367–387. [Google Scholar] [CrossRef] [Green Version]
- Kolla, L.; Kelly, M.C.; Mann, Z.F.; Anaya-Rocha, A.; Ellis, K.; Lemons, A.; Palermo, A.T.; So, K.S.; Mays, J.C.; Orvis, J.; et al. Characterization of the development of the mouse cochlear epithelium at the single cell level. Nat. Commun. 2020, 11, 2389. [Google Scholar] [CrossRef]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- Fischer, B.; Lüthy, K.; Paesmans, J.; De Koninck, C.; Maes, I.; Swerts, J.; Kuenen, S.; Uytterhoeven, V.; Verstreken, P.; Versées, W. Skywalker-TBC1D24 has a lipid-binding pocket mutated in epilepsy and required for synaptic function. Nat. Struct. Mol. Biol. 2016, 23, 965–973. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [Green Version]
- Wallner, B.; Elofsson, A. Can correct protein models be identified? Protein Sci. 2003, 12, 1073–1086. [Google Scholar] [CrossRef] [Green Version]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Finelli, M.J.; Aprile, D.; Castroflorio, E.; Jeans, A.; Moschetta, M.; Chessum, L.; Degiacomi, M.T.; Grasegger, J.; Lupien-Meilleur, A.; Bassett, A.; et al. The epilepsy-associated protein TBC1D24 is required for normal development, survival and vesicle trafficking in mammalian neurons. Hum. Mol. Genet. 2019, 28, 584–597. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Morozenko, A.; Leontyev, I.V.; Stuchebrukhov, A.A. Dipole Moment and Binding Energy of Water in Proteins from Crystallographic Analysis. J. Chem. Theory Comput. 2014, 10, 4618–4623. [Google Scholar] [CrossRef]
- Bennett, C.H. Efficient estimation of free energy differences from Monte Carlo data. J. Comp. Phys. 1976, 22, 245–268. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; Mackerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [Green Version]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D., Jr.; Pastor, R.W. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef] [Green Version]
- Bakhchane, A.; Charif, M.; Salime, S.; Boulouiz, R.; Nahili, H.; Roky, R.; Lenaers, G.; Barakat, A. Recessive TBC1D24 Mutations Are Frequent in Moroccan Non-Syndromic Hearing Loss Pedigrees. PLoS ONE 2015, 10, e0138072. [Google Scholar] [CrossRef]
- Gow, A.; Davies, C.; Southwood, C.M.; Frolenkov, G.; Chrustowski, M.; Ng, L.; Yamauchi, D.; Marcus, D.C.; Kachar, B. Deafness in Claudin 11-null mice reveals the critical contribution of basal cell tight junctions to stria vascularis function. J. Neurosci. 2004, 24, 7051–7062. [Google Scholar] [CrossRef] [Green Version]
- Kitajiri, S.; Miyamoto, T.; Mineharu, A.; Sonoda, N.; Furuse, K.; Hata, M.; Sasaki, H.; Mori, Y.; Kubota, T.; Ito, J.; et al. Compartmentalization established by claudin-11-based tight junctions in stria vascularis is required for hearing through generation of endocochlear potential. J. Cell Sci. 2004, 117, 5087–5096. [Google Scholar] [CrossRef] [Green Version]
- Kitajiri, S.I.; Furuse, M.; Morita, K.; Saishin-Kiuchi, Y.; Kido, H.; Ito, J.; Tsukita, S. Expression patterns of claudins, tight junction adhesion molecules, in the inner ear. Hear. Res. 2004, 187, 25–34. [Google Scholar] [CrossRef]
- Liu, W.; Schrott-Fischer, A.; Glueckert, R.; Benav, H.; Rask-Andersen, H. The Human “Cochlear Battery”—Claudin-11 Barrier and Ion Transport Proteins in the Lateral Wall of the Cochlea. Front. Mol. Neurosci. 2017, 10, 239. [Google Scholar] [CrossRef]
- Melcher, J.R.; Kiang, N.Y. Generators of the brainstem auditory evoked potential in cat. III: Identified cell populations. Hear. Res. 1996, 93, 52–71. [Google Scholar] [CrossRef]
- Eilers, M.; Patel, A.B.; Liu, W.; Smith, S.O. Comparison of helix interactions in membrane and soluble α-bundle proteins. Biophys. J. 2002, 82, 2720–2736. [Google Scholar] [CrossRef] [Green Version]
- Ngoh, A.; Bras, J.; Guerreiro, R.; McTague, A.; Ng, J.; Meyer, E.; Chong, W.K.; Boyd, S.; MacLellan, L.; Kirkpatrick, M.; et al. TBC1D24 Mutations in a Sibship with Multifocal Polymyoclonus. Tremor Other Hyperkinet Mov. (N. Y.) 2017, 7, 452. [Google Scholar] [CrossRef]
- Ragona, F.; Castellotti, B.; Salis, B.; Magri, S.; DiFrancesco, J.C.; Nardocci, N.; Franceschetti, S.; Gellera, C.; Granata, T. Alternating Hemiplegia and Epilepsia Partialis Continua: A new phenotype for a novel compound TBC1D24 mutation. Seizure 2017, 47, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Balestrini, S.; Milh, M.; Castiglioni, C.; Lüthy, K.; Finelli, M.J.; Verstreken, P.; Cardon, A.; Stražišar, B.G.; Holder, J.L., Jr.; Lesca, G.; et al. TBC1D24 genotype-phenotype correlation: Epilepsies and other neurologic features. Neurology 2016, 87, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Falace, A.; Buhler, E.; Fadda, M.; Watrin, F.; Lippiello, P.; Pallesi-Pocachard, E.; Baldelli, P.; Benfenati, F.; Zara, F.; Represa, A.; et al. TBC1D24 regulates neuronal migration and maturation through modulation of the ARF6-dependent pathway. Proc. Natl. Acad. Sci. USA 2014, 111, 2337–2342. [Google Scholar] [CrossRef] [Green Version]
- Falace, A.; Filipello, F.; La Padula, V.; Vanni, N.; Madia, F.; De Pietri Tonelli, D.; de Falco, F.A.; Striano, P.; Dagna Bricarelli, F.; Minetti, C.; et al. TBC1D24, an ARF6-interacting protein, is mutated in familial infantile myoclonic epilepsy. Am. J. Hum. Genet. 2010, 87, 365–370. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.; Hwang, Y.S.; Lee, M.; Sun, J.; Cho, H.J.; Knapik, L.; Daar, I.O. TBC1d24-ephrinB2 interaction regulates contact inhibition of locomotion in neural crest cell migration. Nat. Commun. 2018, 9, 3491. [Google Scholar] [CrossRef]
- Bunton-Stasyshyn, R.K.A.; Wells, S.; Teboul, L. When all is not lost: Considering genetic compensation in laboratory animals. Lab. Anim. (N. Y.) 2019, 48, 282–284. [Google Scholar] [CrossRef]
- Sittig, L.J.; Carbonetto, P.; Engel, K.A.; Krauss, K.S.; Barrios-Camacho, C.M.; Palmer, A.A. Genetic Background Limits Generalizability of Genotype-Phenotype Relationships. Neuron 2016, 91, 1253–1259. [Google Scholar] [CrossRef] [Green Version]
- Frasa, M.A.; Koessmeier, K.T.; Ahmadian, M.R.; Braga, V.M. Illuminating the functional and structural repertoire of human TBC/RABGAPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 67–73. [Google Scholar] [CrossRef]
- DiFiglia, M. An early start to Huntington’s disease. Science 2020, 369, 771–772. [Google Scholar] [CrossRef]
- Vonsattel, J.P. Huntington disease models and human neuropathology: Similarities and differences. Acta Neuropathol. 2008, 115, 55–69. [Google Scholar] [CrossRef] [Green Version]
- Kuo, Y.M.; Li, Z.; Jiao, Y.; Gaborit, N.; Pani, A.K.; Orrison, B.M.; Bruneau, B.G.; Giasson, B.I.; Smeyne, R.J.; Gershon, M.D.; et al. Extensive enteric nervous system abnormalities in mice transgenic for artificial chromosomes containing Parkinson disease-associated α-synuclein gene mutations precede central nervous system changes. Hum. Mol. Genet. 2010, 19, 1633–1650. [Google Scholar] [CrossRef]
- Tian, C.; Gagnon, L.H.; Longo-Guess, C.; Korstanje, R.; Sheehan, S.M.; Ohlemiller, K.K.; Schrader, A.D.; Lett, J.M.; Johnson, K.R. Hearing loss without overt metabolic acidosis in ATP6V1B1 deficient MRL mice, a new genetic model for non-syndromic deafness with enlarged vestibular aqueducts. Hum. Mol. Genet. 2017, 26, 3722–3735. [Google Scholar] [CrossRef] [Green Version]
- Peng, J. Gene redundancy and gene compensation: An updated view. J. Genet. Genom. 2019, 46, 329–333. [Google Scholar] [CrossRef]
- Morell, R.J.; Olszewski, R.; Tona, R.; Leitess, S.; Wafa, T.T.; Taukulis, I.; Schultz, J.M.; Thomason, E.J.; Richards, K.; Whitley, B.N.; et al. Noncoding Microdeletion in Mouse Hgf Disrupts Neural Crest Migration into the Stria Vascularis, Reduces the Endocochlear Potential, and Suggests the Neuropathology for Human Nonsyndromic Deafness DFNB39. J. Neurosci. 2020, 40, 2976–2992. [Google Scholar] [CrossRef]
- Riazuddin, S.; Castelein, C.M.; Ahmed, Z.M.; Lalwani, A.K.; Mastroianni, M.A.; Naz, S.; Smith, T.N.; Liburd, N.A.; Friedman, T.B.; Griffith, A.J.; et al. Dominant modifier DFNM1 suppresses recessive deafness DFNB26. Nat. Genet. 2000, 26, 431–434. [Google Scholar] [CrossRef]
- Yousaf, R.; Ahmed, Z.M.; Giese, A.P.; Morell, R.J.; Lagziel, A.; Dabdoub, A.; Wilcox, E.R.; Riazuddin, S.; Friedman, T.B.; Riazuddin, S. Modifier variant of METTL13 suppresses human GAB1-associated profound deafness. J. Clin. Investig. 2018, 128, 1509–1522. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant | Genotype | Human Phenotype | Mouse Phenotype |
|---|---|---|---|
| p.Ser178Leu | p.Ser178Leu/p.Ser178Leu | not reported | normal hearing, no seizures |
| p.Ser178Leu/+ | progressive hearing loss [11,12] | normal hearing, no seizures | |
| p.Asp70Tyr | p.Asp70Tyr/p.Asp70Tyr | congenital profound hearing loss [10] | normal hearing, no seizures |
| p.Asp70Tyr/+ | no clinical phenotype | normal hearing, no seizures | |
| p.His336Glnfs*12 | p.His336Glnfs*12/ p.His336Glnfs*12 | not reported | embryonic lethality |
| p.His336Glnfs*12/ p.Asp11Gly | seizures with deafness [20] | not reported | |
| p.His336Glnfs*12/ c.1206 + 5G > A | DOORS syndrome [20] | not reported | |
| p.His336Glnfs*12/ p.Ser324Thrfs*3 | not reported | seizures, postnatal death ~P20, normal hearing |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tona, R.; Lopez, I.A.; Fenollar-Ferrer, C.; Faridi, R.; Anselmi, C.; Khan, A.A.; Shahzad, M.; Morell, R.J.; Gu, S.; Hoa, M.; et al. Mouse Models of Human Pathogenic Variants of TBC1D24 Associated with Non-Syndromic Deafness DFNB86 and DFNA65 and Syndromes Involving Deafness. Genes 2020, 11, 1122. https://doi.org/10.3390/genes11101122
Tona R, Lopez IA, Fenollar-Ferrer C, Faridi R, Anselmi C, Khan AA, Shahzad M, Morell RJ, Gu S, Hoa M, et al. Mouse Models of Human Pathogenic Variants of TBC1D24 Associated with Non-Syndromic Deafness DFNB86 and DFNA65 and Syndromes Involving Deafness. Genes. 2020; 11(10):1122. https://doi.org/10.3390/genes11101122
Chicago/Turabian StyleTona, Risa, Ivan A. Lopez, Cristina Fenollar-Ferrer, Rabia Faridi, Claudio Anselmi, Asma A. Khan, Mohsin Shahzad, Robert J. Morell, Shoujun Gu, Michael Hoa, and et al. 2020. "Mouse Models of Human Pathogenic Variants of TBC1D24 Associated with Non-Syndromic Deafness DFNB86 and DFNA65 and Syndromes Involving Deafness" Genes 11, no. 10: 1122. https://doi.org/10.3390/genes11101122